Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Michał Fedoryński | -- | 2519 | 2024-01-16 13:16:40 | | | |
| 2 | Lindsay Dong | -2 word(s) | 2517 | 2024-01-17 02:23:29 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Mąkosza, M.; Fedoryński, M. Chlorine in an Organic Molecule. Encyclopedia. Available online: https://encyclopedia.pub/entry/53899 (accessed on 24 July 2026).
Mąkosza M, Fedoryński M. Chlorine in an Organic Molecule. Encyclopedia. Available at: https://encyclopedia.pub/entry/53899. Accessed July 24, 2026.
Mąkosza, Mieczysław, Michał Fedoryński. "Chlorine in an Organic Molecule" Encyclopedia, https://encyclopedia.pub/entry/53899 (accessed July 24, 2026).
Mąkosza, M., & Fedoryński, M. (2024, January 16). Chlorine in an Organic Molecule. In Encyclopedia. https://encyclopedia.pub/entry/53899
Mąkosza, Mieczysław and Michał Fedoryński. "Chlorine in an Organic Molecule." Encyclopedia. Web. 16 January, 2024.
Copy Citation
Due to the electronic configuration of the atom and charge of the nucleus, the chlorine in organic molecules can exert a variety of effects. It can depart as a chloride anion in the process of substitution and elimination, facilitates the abstraction of protons and stabilizes generated carbanions, exerts moderate stabilizing effect of carbenes, carbocations and radicals. There are frequent cases where chlorine substituent promotes more than one transformation.
phase transfer catalysis
γ-halocarbanions
dichlorocarbene
vicarious nucleophilic substitution
nucleophilic substitution of chlorine
base induced β-elimination
α-halocarbanions
Darzens reaction
chlorine as electrophilic center
1. Introduction
Due to the electronic configuration of the chlorine atom and the charge of the nucleus, the chlorine in organic molecules can exert a variety of effects. Due to the polarity and relatively moderate energy of the carbon–chlorine bond, it can depart from organic molecules as a chloride anion in the process of substitution and elimination. Due to electron-withdrawing effect and low laying 3d orbitals, chlorine facilitates the abstraction of a proton and stabilizes generated carbanions. It also exerts a moderate stabilizing effect of carbenes, carbocations and radicals.
The substitution by a nucleophile proceeds as a synchronous process according to the SN2 mechanism, or via initial dissociation of the carbon–chlorine bond, resulting in formation of chloride anion and carbocation, followed by coupling of the carbocation with a nucleophile, SN1. Furthermore, when chlorine is connected with the electron-deficient π-system as in the case of acid chlorides, chloronitroarenes, β-chlorovinyl ketones, etc., the addition of nucleophiles to the carbon atom connected with chlorine, followed by departure of chloride anion from the intermediate adducts, takes place.
Substitution of halogen atom, connected to aromatic ring, with highly basic nucleophiles can proceed via elimination-addition process with the aryne intermediacy.
Nucleophilic substitution reactions of halogen in aromatic haloarenes initiated by transfer of single electron to chloroarenes, followed by dissociation of the produced anion radical (SRN1), are also known.
The elimination can proceed as the spontaneous departure of the chloride anion from the negatively charged carbon—α-elimination to form carbenes, and three variants of β-elimination. The departure of the chloride anion occurs synchronously with the abstraction of a proton from vicinal carbon (E2), the spontaneous dissociation of the carbon–chlorine bond to form chloride anion and carbocation, subsequently losing a proton (E1) and the departure of the chloride anion promoted by the generation of carbanion on β-carbon, E1cB.
Chlorine can also act as an electrophilic center when connected with an electron-deficient carbon atom.
There are frequent cases where a chlorine substituent promotes more than one transformation of a molecule.
2. Nucleophilic Substitution of Chlorine
2.1. In Aliphatic Compounds
The nucleophilic substitution of chlorine (or bromine) in organic molecules is perhaps the most frequently used reaction in organic chemistry. A variety of nucleophiles—inorganic anions, uncharged nucleophiles (amines, phosphines, etc.), organic anions, particularly carbanions—can afford the substitution. The reaction was thoroughly studied and mechanistic questions are clarified in detail. Of particular interest is nucleophilic substitution by carbanions, commonly referred to as alkylation, because it is the general way of construction of carbon skeletons. In these reactions, the key problem is efficient and economical way of generation of carbanions.
The first example of the application of this catalytic system was, as elaborated by us, the synthesis and manufacturing of 2-phenylbutyronitrile via the alkylation of phenylacetonitrile with ethyl chloride in the presence of 50% aqueous NaOH and benzyltriethylammonium chloride, TEBA (Scheme 1) [1][2].
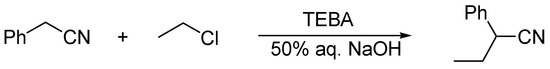
Scheme 1. Ethylation of phenylacetonitrile in the presence 50% aqueous sodium hydroxide and TEBA catalyst.
Under identical conditions, a variety of CH acids (arylacetonitriles, acidic hydrocarbons, esters, ketones, sulfones, etc.) and OH, NH acids were efficiently alkylated with alkyl chlorides and bromides. Some examples are given in Scheme 2.
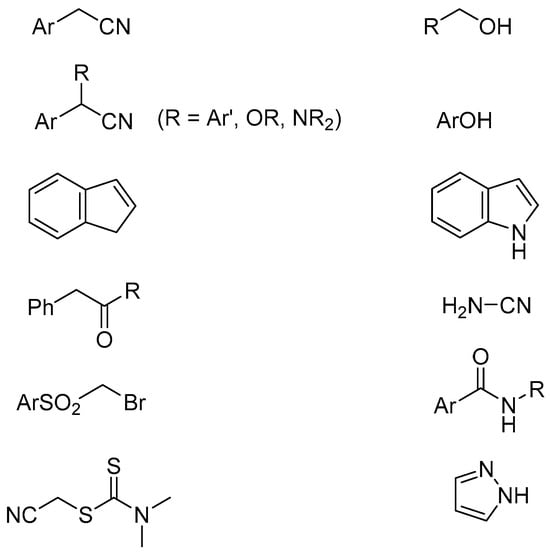
Scheme 2. Examples of CH, OH and NH acids alkylated in PTC system.
The PTC procedure is also applicable for the synthesis of chiral amino acids via the enantioselective alkylation of protected glycine and other amino acids, catalyzed by properly chosen chiral tetraalkylammonium salts, which, due to the secondary interactions, can differentiate the free energies of diastereoisomeric transition states, and thus assure a high induction of enantioselectivity [3][4][5][6].
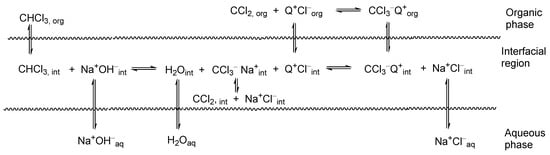
Under conditions described above, the reaction is carried out in a system of two immiscible phases. The organic phase is a mix of the reagent–carbanion precursor, an alkylating agent and, when necessary, a small amount of a solvent—whereas the inorganic phase consists of the concentrated aqueous solution of sodium hydroxide. The boundary between two immiscible liquid phases A and B (see Figure 1) is in fact a region in which, due to thermal motion, components of both phases can mix to form a kind of interfacial region, in which there is gradient of the concentration of the components of phases A and B. Due to short residence time, only very fast reactions, such as deprotonation, between these components can proceed. The interfacially generated carbanions are in low concentration, and cannot migrate into the organic phase being associated with hydrated sodium cations.
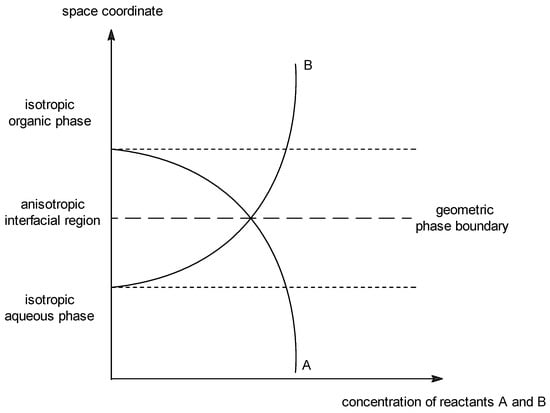
Figure 1. Definition of the interfacial region. Representation of the interfacial region between two immiscible phases, in which there is a gradient of concentrations. Curves A and B represent concentrations of components of organic and aqueous phases, respectively.
When a lipophilic TAA salt is added to such a system, an ion exchange takes place in the interfacial region between the TAA salt and the carbanion, with the formation of a lipophilic ion pair: carbanion/TAA cation. This ion pair migrates from the interfacial region to the organic phase, where carbanion reacts with the chloroalkane, with the formation of the alkylation product, whereas liberated TAA salt enters the catalytic cycle (Scheme 3).
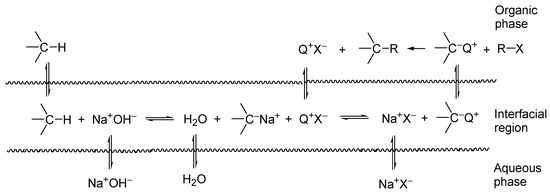
Scheme 3. Interfacial mechanism of the catalytic generation of carbanions in two-phase systems. Q+ denotes quaternary ammonium cation.
2.2. Substitution of Chlorine in Aromatic Rings
Chlorine connected with the π-system, as for instance in vinyl chloride or chlorobenzene, does not enter SN2 nucleophilic substitution. The situation is changed when the π-system become electron deficient, as for instance in p-chloronitrobenzene, β-chloroacrylonitrile or acyl chlorides. In such compounds, chlorine is readily replaced by nucleophiles via an addition–elimination mechanism. Of particular importance is the substitution of chlorine in activated positions of nitroarenes with a variety of nucleophiles. The reactions with carbanions proceeds satisfactorily, provided they are not in the form of tight ion pairs with alkali metal cations in nonpolar solvents. Scholars have applied PTC conditions for the nitroarylation of carbanions of arylalkanenitriles by o- and p-chloronitroarenes. These conditions assure excellent results of substitution in methinic carbanions. Methylenic carbanions, e.g., phenylacetonitrile, upon nitroarylation form highly acidic phenyl nitroarylacetonitriles that are immediately deprotonated and exist in the reaction system in the form of ion pairs with TAA cations; thus, the catalytic process is arrested. A low nucleophilic activity of highly stabilized carbanion prevents its further reactions with chloronitrobenzene, thus bis-nitroarylation (Scheme 4) [7].
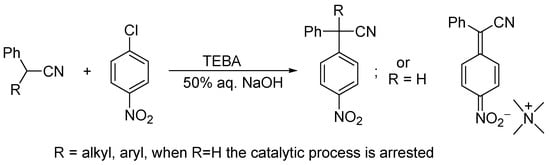
Scheme 4. Catalytic reactions of arylacetonitriles with halonitrobenzenes in the presence of aqueous NaOH.
3. β-Elimination
The base-induced β-elimination of hydrogen halide from haloalkane is an important way of synthesis of alkenes. It requires the use of a strong, non-nucleophilic base, such as a hydroxide anion, for the abstraction of proton, with the synchronous departure of the halide anion from the vicinal carbon atom (Scheme 5).
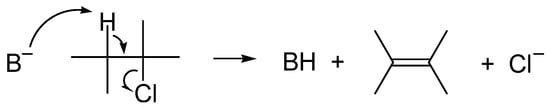
Scheme 5. Base-induced β-elimination of hydrogen halide from haloalkane.
The application of PTC methodology for this β-elimination is limited, because it requires the transfer of hydroxide anions into the organic phase. However, ion exchange equilibrium between the organic and aqueous phase of the hydroxide and chloride anions is unfavorable for hydroxide anions. This problem was solved by the use of cocatalysts–organic acids (YHs) that can be deprotonated in the interfacial region with aqueous NaOH, producing lipophilic anions Y− of high basicity, but low nucleophilicity. Such anions can be transferred into the organic phase as ion pairs with TAA cations and abstract protons from haloalkanes to give products of β-elimination (Scheme 6).
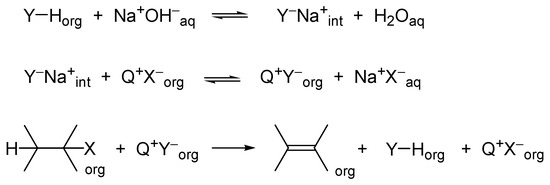
Scheme 6. Concept of cocatalysis in PTC β-elimination reactions.
Using the model β-elimination reaction of hydrogen bromide from bromocyclohexane, scholars tested a variety of YHs and have shown that the most efficient cocatalyst is mesitol, with a reasonable OH acidity and basicity of its anion, whereas steric bulkiness prevents substitution, so this readily available compound is the most recommended cocatalyst for β-elimination (Scheme 7) [8][9].
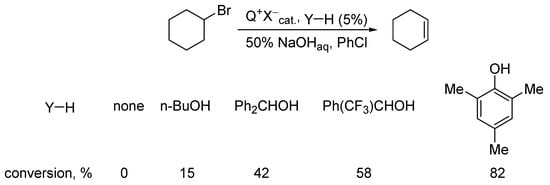
Scheme 7. Search for efficient cocatalyst in PTC β-elimination.
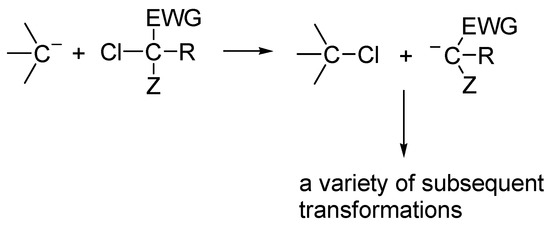
4. Chlorine Substituent as Promotor of CH Acidity
4.1. Generation and Reactions of Dihalocarbenes
Due to strong electron-withdrawing effect and presence of unoccupied 3d orbitals, chlorine connected with a carbon atom exerts a moderate carbanion-stabilizing effect, thus facilitating the abstraction of the proton. Depending on the structure of the molecule, this effect ranges from 3 to 4 pKa units.
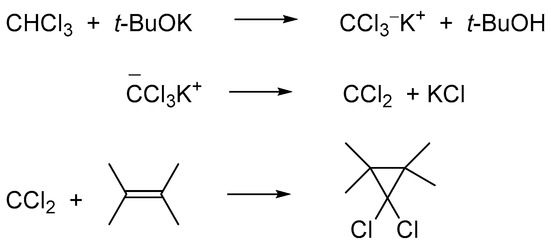
The acidity of chloromethane is negligible, and the estimated pKa of dichloromethane is around 30; on the other hand, chloroform is relatively acidic, estimated pKa ~15. In the α-chlorocarbanions, generated via the deprotonation of methylene chloride and chloroform, a negative charge is located on chloro substituents; thus, they are very unstable and undergo rapidly α-elimination of the chloride anion to form mono- and dichlorocarbenes, respectively. The produced carbenes are very active electrophiles; they add to alkenes with the formation of mono- and gem-dichlorocyclopropanes, respectively. Due to facile deprotonation of chloroform and subsequent α-elimination, dichlorocarbene becomes an active agent widely used for the synthesis of cyclopropanes. The carbene is highly sensitive to water and other nucleophiles; thus, the known procedures required the use of t-BuOK under strictly anhydrous conditions (Scheme 8) [10][11][12].
Scheme 8. Generation of dichlorocarbene under action of t-BuOK on CHCl3 in a nonpolar solvent is an irreversible process.
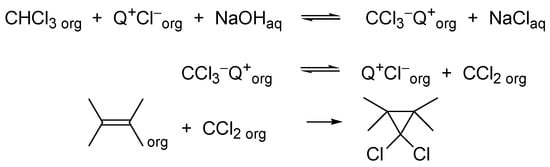
However, it found that dichlorocarbene (DCC) could be generated and added efficiently to alkenes by the treatment of a mixture of chloroform and alkenes with a concentrated aqueous sodium hydroxide solution in the presence of the TAA catalyst [13]. This procedure is much simpler and usually assures good results (Scheme 9).
Scheme 9. Generation of dichlorocarbene in PTC conditions is a reversible process.
Hundreds of examples of the syntheses of dihalocyclopropanes from PTC-generated dihalocarbenes and unsaturated compounds are now described, including adducts of alkenes, unconjugated and conjugated di- and polyenes, allenes, cumulenes, and alkenes substituted with different kinds of substituents (haloalkenes; unsaturated ethers, acetals and ketals; esters of unsaturated alcohols; alkenes substituted by sulfur, nitrogen, silicon atoms; and some unsaturated carbonyl compounds and many others) [11][12][14].
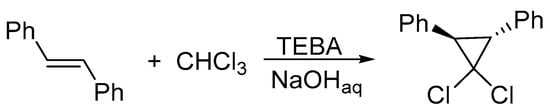
There are many observations that DCC generated under PTC conditions is more efficient in reactions with partners of low activities—addition to alkenes of low nucleophilicity or insertion into the CH bond. For instance, PTC-generated DCC adds to trans-stilbene to form a dichlorocyclopropane derivative with a high yield (Scheme 10), whereas under classical conditions, the addition practically does not proceed [15].
Scheme 10. Under PTC conditions, the addition of dichlorocarbene to trans-stilbene proceeds efficiently.
This substantial difference in efficiency cannot be explained by differences in activities, because the carbenes are kinetically free species and should not “remember” ways of their generation.
On the other hand, under PTC conditions, chloroform is deprotonated in the interfacial region and trichloromethyl anions enter the organic phase as soluble ion pairs with TAA cations. Further reaction: the dissociation of trichloromethyl anion is a reversible process, because all components are soluble in the reaction medium; thus, when the desired reaction of DCC is not a fast process, the carbene adds a chloride anion and is kept “ready for use” for a longer time (Scheme 11).
Scheme 11. Interfacial processes during PTC generation of dichlorocarbene.
This simple explanation of the high efficiency of PTC-generated DCC is unambiguously confirmed, because it was shown that the rate of consumption of chloroform is a function of nucleophilic activity of alkenes, in other words, the rate of consumption of DCC, thus the removal of the carbene from the equilibrating system [16].
4.2. Generation of α-Halocarbanions and Their Addition to Activated Carbon–Carbon Multiple Bonds
The introduction of chlorine in position α- to the functional groups of nitriles, esters, sulfones, etc., increases the CH acidity of these compounds. Thus, α-chloronitriles, esters, sulfones, etc., undergo relatively readily deprotonation to form α-chlorocarbanions. These carbanions are unable to enter α-elimination, because the negative charge is located mostly on the functional groups, but are unstable for other reasons.
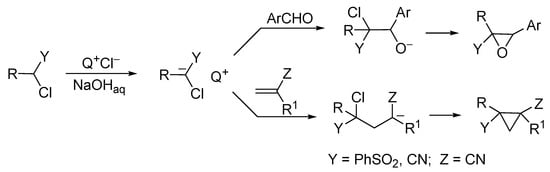
Because of the presence of chlorine substituents α- to the functional group of aliphalic nitriles, esters, sulfones, etc., they become sufficiently strong CH acids; hence, the generation of α-chlorocarbanions can be effected under the action of the concentrated aqueous NaOH solution in the presence of TAA catalysts. Therefore, a simple PTC methodology can be applied for the synthesis of a variety of oxiranes [17][18][19][20] and cyclopropanes (Scheme 12) [21][22][23][24].
Scheme 12. Synthesis of oxirane and cyclopropane derivatives in PTC system.
4.3. Reactions of α-Chlorocarbanions with Electron-Deficient Arenes
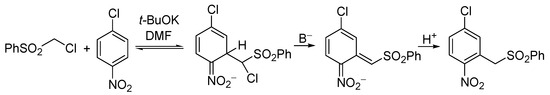
An interesting general reaction that scholars discovered was between α-chlorocarbanions and electron-deficient arenes, particularly nitroarenes. For instance, the carbanion of chloromethyl phenyl sulfone adds to nitrobenzene at positions o- and p- to form adducts (σH adducts). These adducts are short-lived species; nevertheless, they undergo the base-induced β-elimination of HCl at the expense of the ring hydrogen to form anionic products, that upon protonation gave a mixture of o- and p-nitrobenzyl phenyl sulfones. Interestingly, the carbanion of chloromethyl phenyl sulfone adds to p-chloronitrobenzene at the position ortho to the nitro group, giving σH adducts, that upon β-elimination and protonation gave 2-nitro-5-chlorobenzyl phenyl sulfone. Substitution of the ring chlorine did not proceed (Scheme 13) [25].
Scheme 13. The first example of Vicarious Nucleophilic Substitution, VNS.
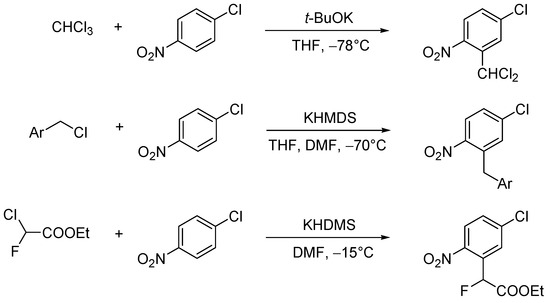
This unprecedented pathway of conversion of the adducts of α-chlorocarbanions to nitroaromatic rings is of general character. For this new reaction, scholars proposed the name the Vicarious Nucleophilic Substitution of hydrogen, VNS [26]. Further examples of this reaction are shown in scheme (Scheme 14) [27][28][29][30].
Scheme 14. Examples of VNS reactions.
Interestingly, carbanions without a chlorine substituent react with o- and p-chloronitrobenzene via the replacement of chlorine, whereas α-chlorocarbanions replace hydrogen ortho to the nitro group. Similarly, reactions of α-chlorocarbanions with o-chloronitrobenzene result in the substitution of hydrogen para into the nitro group. These seemingly controversial unusual observations suggest that the addition of carbanions at positions ortho and para of nitrobenzene is a fast and reversible process. Initially formed σH adducts of α-chlorocarbanions to o- and p-chloronitrobenzene undergo fast further conversion via the base-induced β-elimination of HCl. On the other hand, σH adducts of carbanions, which do not have such a possibility, dissociate, and further, the slower addition of the carbanions at the position occupied by chlorine results in nucleophilic substitutions of chlorine.
4.4. γ-Halocarbanions
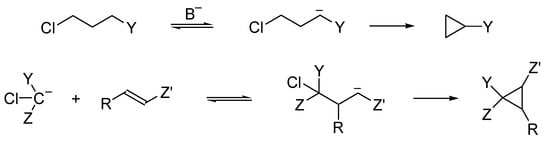
Being engaged in the chemistry of α-chlorocarbanions, scholars were looking for unprecedented reactions of β-, γ- and δ-chlorocarbanions. The thorough analysis of β-chlorocarbanions indicate that the elimination of the chloride anion to form alkenes, the E1cB elimination of HCl, is a very fast process; thus, the only observed intramolecular process is protonation, which can be monitored as isotope exchange.
There are two main methods of the generation of γ-chlorocarbanions: the deprotonation of appropriate precursors—CH acids and addition of α-chlorocarbanions to Michael acceptors. Precursors of γ-chlorocarbanions can be prepared in advance or generated in situ, as shown in Scheme 15.
Scheme 15. γ-Chlorocarbanions enter 1,3-intramolecular substitution to form cyclopropane derivatives.
5. Chlorine as Electrophilic Center
Chlorine connected with an electron-withdrawing carbon center exhibits an electrophilic character; thus, it can react with nucleophiles, particularly carbanions, affording electrophilic chlorination. The most readily available sources of electrophilic chlorine are CCl4 and hexachloroethane (Scheme 16).
Scheme 16. Chlorine as electrophilic center.
References
- Urbański, T.; Bełżecki, C.; Lange, J.; Makaruk, H.; Mąkosza, M.; Serafinowa, B. Sposób Wytwarzania Kwasu α-Fenylomasłowego do Celów Farmaceutycznych (Production of α-Phenylbutyric Acid for Pharmaceutical Purposes). Polish Patent 46,030, 19 June 1962.
- Mąkosza, M.; Serafinowa, B. Reakcje anionów organicznych. I. Katalityczne etylowanie fenyloacetronitrylu w środowisku wodnym (Reactions of organic anions. I. Catalytic ethylation of phenylacetonitrile in aqueous medium). Rocz. Chem. 1965, 39, 1223–1231.
- O’Donnell, M.J. Benzophenone Schiff bases of glycine derivatives: Versatile starting materials for the synthesis of amino acids and their derivatives. Tetrahedron 2019, 75, 3667–3696.
- Maruoka, K. (Ed.) Asymmetric Phase Transfer Catalysis; Wiley-VCH: Weinheim, Germany, 2008.
- Shirakawa, S.; Maruoka, K. Recent developments in asymmetric phase-transfer reactions. Angew. Chem. Int. Ed. 2013, 52, 2–39.
- Denmark, S.E.; Gould, N.D.; Wolf, L.M. A systematic investigation of quaternary ammonium ions as asymmetric phase-transfer catalysts. Application of quantitative structure activity/selectivity relationships. J. Org. Chem. 2011, 76, 4337–4357.
- Mąkosza, M. Reactions of organic anions. XVI. Catalytic nitroarylation of phenylacetonitrile derivatives in aqueous medium. Tetrahedron Lett. 1969, 10, 673–676.
- Mąkosza, M.; Chesnokov, A. Cocatalysis in phase-transfer catalyzed base induced β-elimination. Model studies of dehydrobromination of bromocyclohexane. Tetrahedron 2000, 56, 3553–3558.
- Mąkosza, M.; Fedoryński, M. Co-catalysis in phase transfer catalyzed reactions (a concept paper). Arkivoc 2006, 4, 7–17.
- Parham, W.E.; Schweizer, E.E. Halocyclopropanes from halocarbenes. Org. React. 1963, 13, 55–90.
- Fedoryński, M. Syntheses of gem-dihalocyclopropanes and their use in organic synthesis. Chem. Rev. 2003, 103, 1099–1132.
- Jończyk, A.; Fedoryński, M. Houben-Weyl Methods of Organic Chemistry; de Meijere, A., Ed.; Georg Thieme Verlag: Stuttgart, Germany, 1997; Volume E17a, pp. 589–734.
- Mąkosza, M.; Wawrzyniewicz, M. Reactions of organic anions. XXIV. Catalytic method for preparation of dichlorocyclopropane derivatives in aqueous medium. Tetrahedron Lett. 1969, 10, 4659–4662.
- Dehmlow, E.V. Houben-Weyl Methoden der Organischen Chemie; Regitz, M., Ed.; Georg Thieme Verlag: Stuttgart, Germany, 1989; Volume E19, pp. 1467–1505, 1521–1600, 1608–1622, and 1625–1627.
- Dehmlow, E.V.; Schönefeld, J. Umsetzung von Olefinen mit Dihalogencarbenen (Reactions of alkenes with dihalocarbenes). Liebigs Ann. Chem. 1971, 744, 42–50.
- Dehmlow, E.V.; Lissel, M.; Heider, J. Anwendungen der phasentransfer-katalyse—4: Halogenaustausch und reaktivitäten bei dibrom-, chlorbrom- und dichlorcarben (Applications of phase transfer catalysis—4: Halogen interchange and reactivities of dibromo-, chlorobromo- and dichlorocarbenes). Tetrahedron 1977, 33, 363–366.
- Jończyk, A.; Bańko, K.; Mąkosza, M. Some carbanionic reactions of halomethyl aryl sulfones. J. Org. Chem. 1975, 40, 266–267.
- Jończyk, A.; Fedoryński, M.; Mąkosza, M. Reactions of organic anions. XLIII. Catalytic method for synthesis of glycidic nitriles in aqueous medium. Tetrahedron Lett. 1972, 13, 2395–2396.
- Goliński, J.; Mąkosza, M. Reactions of organic anions. LXXXIV. A convenient method for synthesis of 1-chloro-, 1,1-dichloro-, and 1,2-epoxyalkanesulfonamides. Synthesis 1978, 1978, 823–825.
- Jończyk, A.; Zomerfeld, T. Convenient synthesis of t-butyl Z-3-substituted glycidates under conditions of phase-transfer catalysis. Tetrahedron Lett. 2003, 44, 2359–2361.
- Jończyk, A.; Mąkosza, M. Reactions of organic anions. LXVII. Catalytic synthesis of cyclopropane derivatives in aqueous medium. Synthesis 1976, 1976, 387–388.
- Russell, G.A.; Mąkosza, M.; Hershberger, J. Synthesis of nitrocyclopropanes by cyclization of γ-chloro-γ-nitrocarboxylic esters and derivatives. J. Org. Chem. 1979, 44, 1195–1199.
- Kryshtal, G.V.; Kulganek, V.V.; Kucherov, V.F.; Yanovskaya, L.A. Phase-transfer catalysis of the Michael addition to α,β-unsaturated aldehydes. Synthesis 1979, 1979, 107–109.
- Kryshtal, G.V.; Serebryakov, E.P. Regio- and stereoselectivity in the addition reactions of CH-acids under the conditions of phase-transfer catalysis. Russ. Chem. Bull. 1995, 44, 1785–1804.
- Mąkosza, M.; Winiarski, J. Vicarious nucleophilic substitution of hydrogen. Acc. Chem. Res. 1987, 20, 282–289.
- Goliński, J.; Mąkosza, M. “Vicarious” nucleophilic substitution of hydrogen in aromatic nitro compounds. Tetrahedron Lett. 1978, 19, 3495–3498.
- Mąkosza, M.; Owczarczyk, Z. Dihalomethylation of nitroarenes via vicarious nucleophilic substitution of hydrogen with trihalomethyl carbanions. J. Org. Chem. 1989, 54, 5094–5100.
- Kisiel, K.; Brześkiewicz, J.; Loska, R.; Mąkosza, M. Transition metal free nucleophilic benzylation of nitroarenes. Umpolung of the Friedel Crafts reaction. Adv. Synth. Catal. 2019, 361, 1641–1646.
- Czaban-Jóźwiak, J.; Loska, R.; Mąkosza, M. Synthesis of α-fluoro-α-nitroarylacetates via vicarious nucleophilic substitution of hydrogen. J. Org. Chem. 2016, 81, 11751–11757.
- Khutorianskyi, V.V.; Klepetářová, B.; Beier, P. Vicarious nucleophilic chloromethylation of nitroarenes. Org. Lett. 2019, 21, 5443–5446.
More
Information
Subjects:
Chemistry, Organic
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
982
Revisions:
2 times
(View History)
Update Date:
17 Jan 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No