Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Aleksandr Shikalov | -- | 5452 | 2024-01-16 09:24:25 | | | |
| 2 | Lindsay Dong | + 4 word(s) | 5456 | 2024-01-17 01:57:40 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Shikalov, A.; Koman, I.; Kogan, N.M. Targeted Glioma Therapy. Encyclopedia. Available online: https://encyclopedia.pub/entry/53871 (accessed on 26 July 2026).
Shikalov A, Koman I, Kogan NM. Targeted Glioma Therapy. Encyclopedia. Available at: https://encyclopedia.pub/entry/53871. Accessed July 26, 2026.
Shikalov, Aleksandr, Igor Koman, Natalya M. Kogan. "Targeted Glioma Therapy" Encyclopedia, https://encyclopedia.pub/entry/53871 (accessed July 26, 2026).
Shikalov, A., Koman, I., & Kogan, N.M. (2024, January 16). Targeted Glioma Therapy. In Encyclopedia. https://encyclopedia.pub/entry/53871
Shikalov, Aleksandr, et al. "Targeted Glioma Therapy." Encyclopedia. Web. 16 January, 2024.
Copy Citation
Glioblastoma multiforme (GBM) is the most common type of glioma, with a median survival of 14.6 months post-diagnosis. Understanding the molecular profile of such tumors allowed the development of specific targeted therapies toward GBM, with a major role attributed to tyrosine kinase receptor inhibitors and immune checkpoint inhibitors. Targeted therapeutics are drugs that work by specific binding to GBM-specific or overexpressed markers on the tumor cellular surface and therefore contain a recognition moiety linked to a cytotoxic agent, which produces an antiproliferative effect.
targeted therapy
anticancer
glioma
brain tumors
1. Introduction
Glioma is the term referring to brain tumors that arise from glial or precursor cells. This group of oncological conditions includes astrocytoma, oligodendroglioma, ependymoma, oligoastrocytoma, and a few rare histologies. In medical practice, gliomas account for nearly 25% of all primary brain tumors [1]. According to the most recent WHO guideline for the classification of central nervous system (CNS) tumors, molecular, genetic, and epigenetic markers are used [2]; however, it is also possible to subgroup gliomas into a grading scale: low-grade gliomas (LGG, WHO grades 1–2) and high-grade gliomas (HGG, WHO grades 3–4).
The most common glioma is glioblastoma (GBM, WHO grade 4 astrocytoma), accounting for 14.3% of all primary brain and central nervous system neoplasms and 49.1% of all malignant brain tumors [1], with a poor prognosis for patients: the 5-year survival rate is at 6.8% [1] and it has a median survival of 14.6 months after diagnosis [3][4]. The patient’s prognosis remains weak despite conventional standard-of-care therapy that includes maximal tumor resection followed by radiotherapy and adjuvant temozolomide (TMZ) administration [3].
To better understand the molecular basis of GBM, extensive genomic [5] and transcriptomic [6] analyses were performed. In 2010, these allowed the classification of glioblastomas into subtypes according to their genetic expression profiles: classical (EGFR overexpression, CDKN2A deletion, and a lack of TP53 mutations), mesenchymal (altered NF1, PTEN mutations, a high activity of MET and CD44, and MERTK genes), proneural (altered PDGFRA, mutated TP53, point mutations in IDH1 and the increased expression of OLIG2), and neural (expression of neural markers such as NEFL, GABRA1, SYT1, and SLC12A5) [5]. Median survival rates of 11.5, 14.7 and 17.0 months for the mesenchymal, classical and proneural subtypes were determined.
2. Conventional Standard of Care for Glioma Therapy
For glioblastoma patients, surgical resection of the tumor to the maximal feasible extent followed by focal tumor radiotherapy and concomitant temozolomide chemotherapy combined with additional irradiation therapy should be taken as the standard treatment (Stupp treatment) [3]. However, it is known that all glioblastomas will eventually relapse or progress [7].
For low-grade gliomas, the optimal treatment involves immediate surgical resection of the tumor to avoid further cancer progression and allow precise molecular characterization of the tumor [8], which is indeed crucial for the development of a treatment plan for any glioma [2][9][10]. For high-risk cohorts of low-grade-glioma-diagnosed patients (age > 40 years; patients who do not undergo gross total resection surgery [11]), postoperative care consists of 50–54 Gy radiotherapy followed by adjuvant therapy with DNA-alkylating or cytostatic agents. Lomustine, a DNA-alkylating agent, is usually preferred due to its tolerable toxicity and blood-brain barrier (BBB) permeation properties [12]. Taking the disease progression from low- to high-grades and the recurrent nature of gliomas [7], treatment options require constant attention and rapid development.
There are several FDA-approved drugs and one medical device for glioma management aside from temozolomide (TMZ) [13]: lomustine [14], intravenous carmustine [15], carmustine wafer implants [16], bevacizumab [17], and tumor treatment fields [18]. These drugs and devices are mainly approved for the management of recurrent high-grade gliomas with only TMZ, carmustine wafer implants, and tumor treatment fields suitable for application in de novo diagnoses. Except for bevacizumab, FDA-approved drugs for gliomas represent the class of DNA-alkylating agents—their effect is not targeted on tumor cells; therefore, their application is linked with systemic adverse effects.
Vascular endothelial growth factor (VEGF) was shown to be one of the key regulators of malignant angiogenesis in glioblastoma cells, while its inhibition with the help of antisense oligonucleotides correlated positively with reduced tumor growth and, indeed, diminished vasculature formation [19]. This discovery led to the FDA approval of bevacizumab, a monoclonal antibody that targets VEGF-A on the surface of endothelial cells and blocks its interaction with its receptors VEGFR1 and VEGFR2, becoming the first FDA-approved targeting therapy for use in recurrent GBM. Although bevacizumab demonstrated enhanced progression-free survival (PFS), it did not provide a benefit to overall survival (OS). This suggests that solely targeting the VEGFR axis is inadequate for suppressing tumor progression [20][21].
3. Delivery of Therapeutic Drugs to the Brain and Associated Difficulties
3.1. Blood–Brain Barrier as an Obstacle for Drug Delivery
The blood–brain barrier poses a significant challenge in treating conditions within the central nervous system (CNS) and brain tumors are not an exception. The components of BBB include endothelial cells with tight junctions (specific for CNS), smooth muscle cells, microglial cells, astrocytes and pericytes [22]. Tight junctions in endothelial cells create a nearly impenetrable barrier, restricting the passage of small and lipid-soluble molecules through the paracellular route.
Relevant to drug delivery, the BBB allows the free passage of hydrophilic molecules with a molecular weight of up to 150 Da and hydrophobic molecules with a molecular weight of 400–600 Da [23], while the penetration of large or hydrophilic drugs is limited. In order to travel through the BBB, the drug may utilize receptor-mediated transcytosis mechanism (e.g., with transferrin and insulin receptors [24]). P-glycoprotein-1 (P-gp), which can pump out more than 60% of the marketed drugs, makes it more difficult to penetrate the BBB [25].
The transcellular route of drug transport through the BBB is favorable for molecules with a low molecular weight (<500 Da) and high lipophilicity [26]. The relationship between the polar surface area and brain permeability is proved to be inverse—drug candidates that have >80 Å of polar surface and a tendency to form multiple H-bonds require higher free energy to penetrate lipophilic membranes of the BBB cells [26][27][28]. However, the predominant usage of less polar or highly lipophilic compounds also has its drawbacks as lipophilic molecules would aggregate bound to plasma proteins, effectively decreasing the amount of available active compound [29].
For the treatment of high-grade gliomas [30], only a small number of cytotoxic drugs are being used, such as TMZ, which allows about 20% penetration through the BBB at the administered systemic dose [31]. Other well-studied drugs such as paclitaxel (PTX), doxorubicin, and cisplatin, are not included in the standard-of-care treatment for GBM due to their poor BBB permeability, even though preclinical in vitro studies have shown an inspiring decrease in the viability of cancer cells using these drugs [32][33][34].
3.2. Targeted Drug Delivery to the Brain
3.2.1. Passive Targeting
Passive targeting is a major strategy in drug development to achieve therapeutic effects while using systemic administration, particularly for drugs that need to reach the brain. This strategy takes advantage of the abnormal vessel formation in tumors, resulting in enlarged pore sizes in the vasculature endothelium at the tumor site. As a result, compounds that usually cannot cross the blood–brain barrier can be delivered to the tumor site with a higher probability [35][36]. This condition is known as the Enhanced Permeation and Retention (EPR) effect. Pore cutoff sizes differ drastically in healthy (4–25 nm) and brain tumor blood vessels (380–780 nm). Taking that into account, as well as the fact that lymphatic drainage is also impaired in brain tumors, drug delivery nanosystems that are designed to utilize the EPR effect for reaching the tumor site may be up to several hundred nanometers in diameter and could stay in the targeted area for longer periods of time [37]. The size of the nanostructures is a variable parameter in drug design and could be changed to increase the tumor permeation rate and decrease systemic toxicity [38]. Dual-modality nanosystems, designed for both the diagnostics and therapeutics of gliomas, such as QSC-LP liposomes loaded with quantum dots, supramagnetic iron oxide and cytotoxic peptide cliengitide, have shown promising results in vivo by significantly prolonging overall survival and diminishing tumor size in a xenograft mouse model [39].
3.2.2. Mechanical Targeting (Local Delivery)
Local application of the therapeutic agents to the tumor site, which scholars will refer to as ‘mechanical’ targeting, could be achieved via intracerebroventricular injection, intratumoral injection, convection-enhanced delivery, and adding anti-cancer compounds intraoperatively into the post-lesion cavity, such as carmustine wafers [17][40]. In this case, as there is no need to pass the BBB and no application of drugs occurs in the circulation, therefore, the systemic toxic effects are significantly lower than in the case with intravenous application, as well as there being increased therapeutic efficacy of the active compounds as they are delivered locally to the site of the tumor [41][42][43][44].
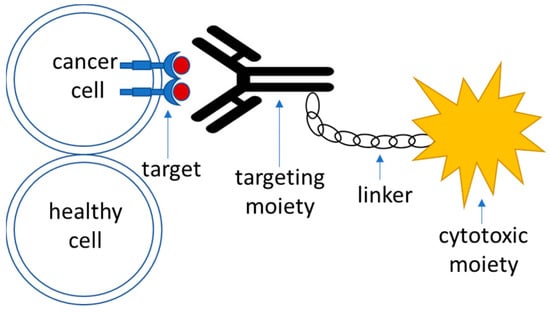
3.2.3. Active Targeting
Active targeting is a method of drug delivery that utilizes ligands (monoclonal antibodies and their derivatives, peptides, etc.) for tumor-specific recognition. Upon recognition of tumor-specific biomarkers which are represented by overexpressed or mutated surface proteins, the drug promotes its cytotoxic effect either directly or indirectly based on a type of drug system and its corresponding specifications. As an example, contrary to passive drug delivery through the EPR effect described above, targeting BBB-overexpressed glioma markers with corresponding ligands showed successful delivery to the tumor site in a phase I trial of the peptide–drug conjugate GRN1005 [45], as well as nanoparticles in preclinical in vitro [46] and in vivo studies [47][48][49][50].
4. Drug Delivery Systems for Active Targeting of Brain Tumors
4.1. General Scheme for Drug Delivery Systems That Use Active Targeting
Targeted drug delivery systems that utilize active targeting strategy are generally composed of the following parts (Figure 1):
-
Target—this is a moiety that is specific to the cell of interest—in this case, a tumor cell. It is required to be expressed or significantly overrepresented in the tumor over healthy cells; high-level expression is advantageous, although not necessary [51]. It could also represent a molecule within an altered biochemical pathway that is specific to a tumor cell.
-
Recognition moiety—a part of the drug delivery system that recognizes the target specifically and allows address delivery to a specified tumor site with minimal toxicity and off-target effects.
-
Linker—an engineered connective unit that ties the recognition part of the drug delivery system to the payload.
-
Payload—a cytotoxic agent that acts upon delivery to the tumor cells either directly or indirectly through the mediation of the host’s immune response.
Figure 1. A schematic outline of the components of an active targeting drug delivery system.
4.2. Targets
Targeted glioma therapies commonly aim for the recognition/inhibition of the tumor-overexpressed variants of the receptor tyrosine kinase (RTK) family of transmembrane proteins that are responsible for cell growth and proliferation signaling. RTKs represent the most abundant and clinically relevant group of surface targets for GBM drug development [6]. In glioma initiation, several subclasses of RTKs are prone to alterations such as amplification and point mutations [6][52].
From a structural point of view, RTKs contain an extracellular domain required for ligand binding, a transmembrane domain, and an intracellular domain with tyrosine residues required for receptor activation upon phosphorylation [53]. The binding of receptor-specific ligands triggers dimerization of the RTK and subsequent autophosphorylation at intracellular tyrosine residues (activation sites). Activated residues act as landing pads for small G-proteins whose recruitment is necessary for downstream signaling through the RAF/MEK/ERK and PI3K/AKT pathways, which control tumor formation, cell survival, proliferation, and invasion [52]. In the context of GBM, alteration in the RTK/PI3K/AKT pathway, which is responsible for constitutive RTK signaling, is considered to be the most elevated core signaling pathway [54]. Alterations (mutations/deletions) in oncosuppressor genes such as TP53, NF1, and PTEN additionally promote RTK activity, which accelerates malignant progression and plays an important role in tumor initiation [54][55].
4.3. Recognition Moiety
Recognition moieties in the development of targeted drug delivery systems are represented by several classes of molecules. As mentioned before, the recognition particle of the drug should be tumor-specific, with minimal off-target effects as well as no reactivity with healthy tissues [56]. Recognition moieties utilized in drug delivery systems include
-
Monoclonal antibodies and their derivatives (such as antibody fragments (Fabs), single chain variable fragments (scFvs), and bispecific antibodies) [57]. The most frequently used format for the development of antibody–drug conjugates is IgG1 immunoglobulin, as it is both easy to manufacture as well as the fact that it shows a strong cytotoxic effect [58].
-
Aptamers could also be used as a recognition unit of a targeted drug delivery system. Aptamers are short nucleic acid sequences that could be selected based on their affinity towards tumor cells via the systematic evolution of ligands by an exponential enrichment (SELEX) process [64]. I
4.4. Linker
For antibody- and peptide-conjugated drugs, linkers could be classified into cleavable and non-cleavable subgroups.
Cleavable linkers in conjugated drugs are developed for the cytotoxic drugs to be released upon cellular uptake of the drug conjugate, which reduces off-target drug release with associated unwanted toxicity in healthy tissues [65][66]. In general, there are three main triggers for linker cleavage and drug release for conjugated drugs: acid-cleavable linkers undergo degradation and drug release due to the usage of the difference between pH levels in plasma (approx. pH 7.4) and endosomal/lysosomal compartments of the tumor cells [67][68] that could be as low as pH 4.5–4.7 [69]; enzyme-cleavable linkers are being cleaved upon internalization in the endosomal or lysosomal compartment of the cell based on the linker structure, with help of intracellular cathepsin B and beta-glucuronidase enzymes [70][71][72][73], and redox-sensitive disulphide linkers undergo reductive cleavage based on the difference in the number of reducing equivalents in the form of glutathione between the plasma (2–20 μM/ L) and cytoplasm (0.5–10 mM) [74].
Non-cleavable linkers in the conjugated drugs require retention of the drug activity upon degradation of the recognition moiety within the endosomal compartment of the cells while the linker or its part stays intact. The advantage of using such types of drugs is associated with a decreased chance of off-target effects, as the drug is more likely to transform to its active form only upon internalization to the cells of interest [75][76][77].
4.5. Payload (Cytotoxic Agent)
Cytotoxic payloads that are commonly used in the arsenal of targeted therapeutics include
-
Microtubule assembly inhibitors: auristatins that inhibit tubulin polymerase and promote cell cycle arrest [82] such as monomethyl auristatin E (MMAE) [83] and monomethyl auristatin F (MMAF) [84], or maytansines and their derivatives that bind to tubulin and therefore interfere with the assembly of microtubules [85]. Common examples include mertansine (DM1) [86][87][88] or ravtansine (DM4) [89][90] as parts of the cytotoxic agents in targeted therapeutic drugs.
-
Bacterial toxins—toxic compounds produced by Pseudomonas aeruginosa (Pseudomonas Exotoxin A) and Corynebacterium diphtheria (Diphtheria toxin) are the most commonly used toxins of bacterial origin that are utilized for cancer therapies [91]. Both toxins act upon binding and irreversibly modify the eukaryotic EF2 elongation factor, which results in impaired protein synthesis and cell death [92][93].
-
Radioligands fused with peptides or antibodies with tumor specificity are used as a cytotoxic moiety for the targeted delivery of drugs to the tumor sites [94][95][96][97], which allows tumor-specific distribution of the radioactivity as compared to standard beam irradiation in the standard-of-care Stupp protocol [3].
-
Photodynamic therapy (PDT) is a therapeutic approach that requires the incorporation of non-toxic and inactive photosensitizer molecules into the cells of interest with subsequent light irradiation, in which the therapeutic molecules become activated. Upon irradiation, photosensitizer molecules transfer light energy and excitate molecular oxygen present in the surrounding tissues to a triplet or singlet state. In the triplet state, oxygen is capable of the generation of reactive oxygen species that react with molecules containing double bonds, leading to their damage and subsequent cell death [98]. In glioma studies and treatment, PDT is implemented via placing fiberoptics at the site of the tumor in order to irradiate cells within the brain that have obtained photosensitizer molecules [99][100].
-
Oncolytic virotherapy is a novel approach for the targeted therapy of neoplasms that utilizes viruses for the elimination of tumor cells. While the exact mechanisms and the nature of tropism for such viruses might still be unknown, the main theories involve either direct killing of the targeted cells or indirect modulation of the host’s immune system response in order to mediate immunogenic cytotoxicity [101][102].
5. Strategies in Clinical Approaches towards the Treatment of GBM
5.1. Targeting Receptor Tyrosine Kinases
5.1.1. EGFR
Epidermal growth factor receptor is a member of the ErbB family of receptors [103]. Downstream signaling pathways for EGFR regulate DNA synthesis and since inclusion criteria for clinical trials differ between the studies, it is impossible to uniformly assess the success or failure of the tested compounds (e.g., survival parameters obtained in de novo diagnosed patient cohorts are different from survival data obtained in the studies for patients who have failed in trials for the other lines of treatment).
Glioblastoma somatic genomic landscaping revealed that 57% of GBM cells harbor EGFR genetic alterations [6]. EGFR amplification and overexpression were identified in 40 and 60% of primary glioblastomas, respectively, often leading to aggressive GBM tumors resistant to treatment, and enhanced proliferation, invasion, and survival [104][105][106][107]. Point mutations and deletions are also observed among GBM patients with around 20% of diagnosed cases [54] harboring a deletion of exons 2–7, resulting in a truncated mutant variant III (EGFRvIII) [108].
Depatuximab–mafodotin, which contains an internalizing EGFR/EGFRvIII-specific antibody linked to the cytotoxic agent monomethyl auristatin F via a non-cleavable maleimidocaproyl (MCC) linker, did not show increased survival rates when tested in combination with conventional radiotherapy and temozolomide in NCT02573324 [109] or as a monotherapy versus conventional treatment in NCT02590263 and NCT02343406 [110][111], as well as it not having an impact on the quality of life of the patients with recurrent glioblastoma [112]; however, preclinical evaluation in vivo showed total tumor regression in mice bearing U87MGde2–7 model tumors, as well as it showing significant tumor growth reduction in patient-derived xenograft models [113].
AMG 595, composed of the maytansinoid DM1 toxin attached to a highly selective anti-EGFRvIII antibody via a non-cleavable linker, showed promising cytotoxic effects in vitro and in vivo [86] and was tested in a phase I trial (NCT01475006) on a group of 32 patients with recurrent glioma, in which 2 patients showed a partial response, 15 showed stable disease and 15 showed progressive disease outcomes [114].
The D2C7-IT dual-specific immunotoxin, comprising an EGFR wild-type and mutant-specific (EGFRvIII) monoclonal antibody (mAb) fragment linked to a genetically engineered form of the Pseudomonas exotoxin A via PCR, is currently being investigated in two active clinical trials, NCT04547777 and NCT05734560, with predicted completion in 2025 and 2028, respectively. Preliminary results summarized in a study abstract by [115] show that this drug might be promising for patients diagnosed with malignant glioma.
The first proposed antibody for EGFR targeting was the chimeric human–murine monoclonal antibody Cetuximab. Unfortunately, phase II clinical trials for this drug did not show therapeutic benefit for patients with relapsed GBM, both as monotherapy, showing median PFS of 1.9 months and OS of 5.06 months [116], and in combination with bevacizumab (a monoclonal antibody targeting neoplastic angiogenesis) and irinotecan (DNA synthesis blocker), showing median PFS of 16 weeks and OS of 30 weeks [117].
The first fully human monoclonal antibody targeting EGFR, panitumumab, did not show promising results in a phase II clinical trial combined with the administration of irinotecan; the study was terminated due to not showing efficacy (NCT01017653).
Nimotuzumab, a humanized anti-EGFR antibody, was tested in a phase III clinical trial (NCT00753246) comparing its efficacy in a treatment combined with a standard Stupp protocol (tumor resection followed by radiotherapy and temozolomide), in which no significant prolongation of the OS and PFS was observed for patients with both recurrent and primary glioblastomas [118].
5.1.2. PDGFR
Platelet-derived growth factor receptors are a family of six subunit homo- and heterodimers that form tyrosine kinase receptors, whose physiological function involves the regulation of hematopoiesis, embryogenesis, and glial cell development, and the protection of neuronal tissue [119]. PDGFR alteration and genetic mutations play a driving role in GBM cell proliferation and malignancy, including the dedifferentiation of glial cells and epithelial-to-mesenchymal transition, as well as the effects of intratumoral vessel formation and immunosuppression [120][121].
In GBM patients, amplification and genetic alterations in PDGFR are found in 13% of diagnosed cases, overexpression of this RTK is related to poor prognosis [6]. Activation of PDGFR signaling in glioma cells induces malignant vascularization, cell survival, and growth [122][123], and dysregulation of normal PDGFR activity in tumors leads to oncotransformation of healthy glia and GBM growth through autocrine signal transduction [124][125].
MEDI-575 is a PDGFRα specific monoclonal antibody that was tested in a Phase II study (NCT01268566) and is a monotherapeutic agent. It showed disappointing clinical outcomes as the median PFS for patients was only at the 1.4 months level with median overall survival at 9.7 months [126].
Olaratumab—a PDGFRα specific monoclonal antibody—was tested as a monotherapeutic agent in a phase II NCT00895180 clinical trial and showed median overall survival at 34.3 weeks level, with 7.5% of patients from the recruitment cohort reaching 6 months progression-free survival endpoint and only 2.5% of the olaratumab treated cohort achieving response from the treatment.
5.1.3. VEGF/VEGFR
Neoplastic vessel formation is one of the hallmarks of malignant tumor growth and progression, with VEGF receptors and ligands signaling pathways being a potent driver of angiogenesis [127]. Extensive vascularization as a result of angiogenesis is one of the tumor hallmarks, indeed present in glioblastoma with VEGF being detected in 64% of diagnosed GBM cases [128] and up to 17% of diagnosed cases showing amplification of the VEGFR2 gene [129]. Several reports outline that VEGF/VEGFR axis members are strongly upregulated in glioblastoma with a direct correlation to the level of malignancy of the tumor and therefore VEGF/VEGFR amplification could serve as a marker of a poor prognosis [130][131]. VEGF/VEGFR signaling in the context of tumor plays a role in tumor growth [132] and proliferation of the cancer stem cells [133] as well as it is responsible for tumor immunoevasive microenvironment [134].
Since 2009, when bevacizumab (VEGF-A specific monoclonal antibody) was approved by the FDA the for treatment of recurrent glioblastoma [135], it has become a treatment of reference for combinatorial clinical trials within recurrent GBM. Two major lines of clinical investigation exist in targeting VEGF/VEGFR axis, similar to PDGFR treatment strategies—usage of monoclonal antibodies and small molecule inhibitors.
The combination of bevacizumab with irinotecan (DNA topoisomerase-I inhibitor) showed a promising trend in increased progression-free survival for the patients [135][136] and was a basis for combinatorial trials of bevacizumab with standard-of-care therapy, radiotherapy, and conventional chemotherapeutics (outlined in the table below). Surprisingly, in the majority of the studies that involve bevacizumab as a monotherapy or a part of a combinatorial treatment, only progression-free survival is increased significantly while no therapeutic benefit is observed for the overall survival of the patients [20][137][138][139][140].
CT-322, an investigational peptide drug that is comprised of a modified extracellular domain of fibronectin 10th type 3 with a specificity towards VEGFR-2 was tested in a phase II clinical trial NCT00562419 as a monotherapy or in combination with irinotecan.
CT-322 acts as a high-affinity blocker of VEGFR-2 preventing its binding with VEGF ligands, most importantly, VEGF-A, therefore blocking VEGF-A induced dimerization of the receptor and subsequent MAPK signaling [141].
While showing adequate tolerability and side effects profile, this compound did not reach the trial’s prespecified efficacy measures and was terminated [142].
5.1.4. c-MET Pathway
Another example of a receptor with tyrosine kinase activity that is altered in GBM cells is c-MET. According to glioblastoma molecular landscaping, 1.6–13.1% of diagnosed cases harbor c-MET overexpression, with gains in MET found in 47% of primary and 44% of recurrent GBM cases [6][143] and up to 4% of cases present amplification of MET [54][144].
Since it was shown that inhibition of angiogenesis through blockade of the VEGF/VEGFR axis stimulates c-MET activation and leads to an invasive form of glioblastoma [145], it is important to target the c-MET pathway to effectively treat tumors. In clinical trials, c-MET pathway is targeted with monoclonal antibodies with specificity towards c-MET receptor or its ligand HGF [146][147].
Onartuzumab, a monoclonal antibody targeting c-MET, was investigated in a clinical study NCT01632228 and showed a trend towards reducing tumor growth, however, no significant increase in median overall survival and progression-free survival were observed in patients undergoing combinatorial therapy with onartuzumab and bevacizumab as compared to bevacizumab plus placebo [148].
Rilotumumab, an anti-HGF monoclonal antibody, was tested in a NCT01113398 clinical study in combination with bevacizumab pretreatment. Unfortunately, this investigational drug did not show a single-agent antitumor activity in the cohort of patients enrolled for the study [149].
5.1.5. FGFR
FGFR family of receptor tyrosine kinases is comprised of four transmembrane receptors named FGFR1–4 [150]. Upon activation of FGFRs, downstream signaling includes the ERK/MAPK pathway, which in turn is responsible for the regulation of cell survival, proliferation, angiogenesis, development, and differentiation [151][152][153][154]. Even though only 3.2% of glioblastoma patients harbor amplification or point mutation in FGFR genes [6], it was shown that GBM cells could evade EGFR and MET inhibition through FGFR-mediated bypass, therefore pointing FGFR inhibitors as possible potentiators of EGFR or MET-targeted therapies [155].
Currently, the research in the field of targeted FGFR inhibitors is mostly preclinical with limited information available for clinical trials in the context of GBM. One clinical trial evaluated the effect of pan-FGFR inhibitor infigratinib in patients with recurrent glioblastoma (NCT01975701) after this drug showed potent inhibition of FGFRs and cell growth on a panel of cancer cell lines in vitro [156], resulting in progression-free survival of 1.7 months and median overall survival of 6.7 months, Unfortunately, infigratinib as a therapeutic compound was outlicenced since then and no further trials and investigations were performed [147].
5.1.6. Multikinase Inhibitors
Imatinib is the first multikinase inhibitor whose targets are PDGFRα/β, BCR-Abl, and c-Kit. The initial trial of this drug in combination with hydroxyurea did not show an increase in survival for patients [157]; however, it was tested in trials after that, returning unsatisfactory or clinically insignificant results both in combination with hydroxyurea [158] and as monotherapy [159].
Dasatinib, an inhibitor of PDGFRβ, EPHA2, BCR-Abl, c-Kit, and SRC, was tested in clinical trials on patients with recurrent glioblastoma as a monotherapeutic agent and in combination with chemotherapy, bevacizumab or the conventional standard-of-care Stupp protocol. The monotherapeutic application of dasatinib failed in a clinical trial NCT00423735 as the study did not pass the efficacy preset and was terminated [160]. The combination of dasatinib with lomustine proved to be ineffective due to the efficacy endpoints not reaching the measures of historical controls [161].
Sunitinib, an inhibitor of PDGFRα/β, c-Kit, VEGFR1/2/3, FLT3, and RET, also showed upsetting results in clinical trials with no significant antitumor activity for patients with recurrent glioblastomas [162][163], while in one study a sub-cohort of patients with high c-Kit expression undergoing monotherapy with sunitinib showed median PFS of 16.0 months and median OS of 46.9 months, with the rest of the tested cohorts showing PFS and OS endpoints at 2.2 and 9.4 months, respectively [164]. This observation might prove valuable that treatment with sunitinib needs to be fine-tuned to a specific cohort of glioma patients.
Ponatinib, a multikinase inhibitor that targets BCR-Abl, PDGFRα, VEGFR2, FGFR1, and Src [165] but also RET, c-Kit, and FLT1, is under investigation in an NCT02478164 clinical trial as a monotherapy for patients with bevacizumab-refractory GBM. Preliminary results show a median OS of 98 days and PFS of 28 days, with minimal activity of the investigational drug in the selected group of patients [166].
5.2. Targeting Immune Checkpoints
5.2.1. PD-1/PD-L1
Clinical trials that have investigated the effect of blockade of PD-1 on host T-lymphocytes with the PD-L1 ligand through the use of monoclonal antibodies in glioblastoma were performed for a variety of developed investigational drugs.
Pembrolizumab, an anti-PD-1 monoclonal antibody, was tested in the NCT02337686 phase II trial for patients with recurrent glioblastoma. The investigation tested the effect of the adjuvant or neoadjuvant application of a checkpoint inhibitor for patients undergoing tumor resection. When compared between adjuvant and neoadjuvant cohorts, the addition of pembrolizumab significantly prolongs overall survival (13.7 vs. 7.5 months for neoadjuvant vs. adjuvant therapy) as well as a progression-free survival rate (3.3 vs. 2.4 months for neoadjuvant vs. adjuvant application).
In the monotherapy cohort for patients treated with pembrolizumab (NCT02054806), a durable anti-tumor trend was observed with a median overall survival rate of 13.1 months and median PFS rate of 2.8 months, suggesting that single-agent application of this drug might be beneficial for a certain subpopulation of GBM patients. However, as the authors suggest, the increase in OS might be unclear as the baseline characteristic of patient records used as a historical cohort in that study might be non-uniformed and, therefore, this result should be taken with caution [167].
Another example of an anti-PD-1 monoclonal antibody investigated in the clinical setting for GBM patients is nivolumab. It was tested in patients with recurrent glioblastoma in a phase III NCT02017717 [168] trial as a monotherapy vs. bevacizumab monotherapy, as well as in two phases III trials in newly diagnosed glioblastoma patients with methylated ([169], NCT02667587) and unmethylated MGMT promoter ([170], NCT02617589). MGMT-related studies compared combinatorial treatment with nivolumab and concomitant radiotherapy with temozolomide compared to RT/TMZ alone.
5.2.2. CTLA-4
Cytotoxic T-lymphocyte associated protein 4 (CTLA-4) is an immune checkpoint receptor that acts upon binding with ligands CD80 and CD86 present on antigen-presenting cells leading to inhibition of T-lymphocyte stimulatory signaling [171]. CTLA-4 expressing T-lymphocytes are usually present in the lymph nodes unlike PD-1-expressing cells in the tumor microenvironment [172].
Tremelimumab, an anti-CTLA-4 monoclonal antibody, was recently tested in a phase II NCT02794883 clinical trial as a monotherapeutic agent or combined with anti-PD-L1 monoclonal antibody durvalumab. A combination of these drugs showed contradictory results as compared with durvalumab alone; however, the combination of durvalumab with tremelimumab might be beneficial for increasing the PFS rate of the patients.
5.2.3. TIM-3
TIM-3 is an immune checkpoint that is widely expressed in glioblastoma and is an important regulator of the inflammatory response related to anti-PD-1 inhibition by therapeutic agents [173][174].
Thus, the combined anti-TIM3 inhibitor and other immunotherapies are getting promising. In preclinical investigation, the combination of anti-TIM3 therapy, anti-PD-1 therapy, and radiotherapy in animal models has reported promising efficacy—Kim et al. showed, in a murine orthotopic GL261 glioma model, that the addition of anti-TIM-3 therapy to stereotactic radiosurgery (SRS), as well as triple therapy with anti-TIM3, anti-PD-1, and stereotactic radiosurgery, significantly increases overall survival and therefore needs to be studied in detail for clinical implementation.
5.2.4. LAG-3
LAG-3 is a marker of active immune cells [175][176] with a similar mode of immune evasion in cancers such as PD-1 [177]. In tumors, LAG-3 is usually found within T cells that have lost their functions and, therefore, they could be a potential target for immune checkpoint inhibitors in oncology treatment [178][179].
In GBM, the LAG-3 expression profile correlates with the expression of the CD8+ T-cells CD8A [180]; therefore, targeting LAG-3 in glioblastomas with high levels of CD8+ cells in the tumor environment might be a beneficial clinical strategy. Currently, two phases I studies for the anti-LAG-3 monoclonal antibody are in the process of investigation (NCT02658981 and NCT03493932).
5.3. Targeting Extracellular Matrix (ECM) Components in Glioma Microenvironment
The extracellular matrix in brain tumors is a promising target for therapeutic treatment, as components of the brain ECM in pathologic conditions show tumor-specific behavior and ECM-dependent metabolic activity [181], which makes it possible to differentiate between targeting healthy tissue and the tumor environment in therapeutic development. The extracellular matrix in the brain tumor environment overexpresses several markers that are possible targets for glioma-specific therapies, such as tenascin-C, fibulin-3, fibronectin, and hyaluronan; as well as this it shows increased activity of metalloproteinase MMP9 [182].
Preclinical trials of the tenascin-C A1-targeting scFv IgE based antibody showed good accumulation in a tumor site in xenograft mice bearing U87 glioma cells, with comparable results to fibronectin-targeting antibody that is undergoing clinical trials at the moment [183].
Fibronectin is a promising target for delivering drugs to the extracellular compartment of solid tumors as it is a good marker of neovascularization and therefore could be targeted for eliminating the spread of the tumor tissue [184]. Preliminary in vivo studies showed that a radiolabeled scFv fragment of the L19 antibody that targets an ED-B domain of fibronectin significantly prolongs survival in mice with intracranially placed C6 glioma cells (22 vs. 16 days in mice bearing tumors of which the volume exceeded 150 mm3 [185]), as well as it showing preferential binding in the neovasculature region of the tumor, which is increased when antivasculature drugs are coadministered [290
6. Conclusions
Gliomas, as representatives of CNS tumors, are characterized by a vast variety of altered markers within tumor cells and the surrounding microenvironment [2], with glioblastoma diagnosis holding poor survival rates and prognosis [1][3][4]. Many efforts in preclinical and clinical research are being invested into developing anti-glioma and GBM drugs; however, in many cases, these attempts are proving to be ineffective due to factors such as tumor location in the brain that is protected from main circulation due to presence of the BBB [186], as well as inter- and intratumoral heterogeneity [5][6][187][188][189], often resulting in acquired therapy resistance. Nonetheless, accumulating data on unsuccessful trials plays a great role in enlarging the understanding of gliomas from a fundamental science point of view and helps to elucidate the mechanisms of drug resistance for future improvements in therapeutic strategies.
References
- Ostrom, Q.T.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014–2018. Neuro Oncol. 2021, 23, 1–105.
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro Oncol. 2021, 23, 1231–1251.
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996.
- Tran, B.; Rosenthal, M.A. Survival Comparison between Glioblastoma Multiforme and Other Incurable Cancers. J. Clin. Neurosci. 2010, 17, 417–421.
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110.
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477.
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20, S2–S8.
- Jakola, A.S.; Skjulsvik, A.J.; Myrmel, K.S.; Sjåvik, K.; Unsgård, G.; Torp, S.H.; Aaberg, K.; Berg, T.; Dai, H.Y.; Johnsen, K.; et al. Surgical Resection versus Watchful Waiting in Low-Grade Gliomas. Ann. Oncol. 2017, 28, 1942–1948.
- Zhang, P.; Xia, Q.; Liu, L.; Li, S.; Dong, L. Current Opinion on Molecular Characterization for GBM Classification in Guiding Clinical Diagnosis, Prognosis, and Therapy. Front. Mol. Biosci. 2020, 7, 562798.
- Guo, X.; Shi, Y.; Liu, D.; Li, Y.; Chen, W.; Wang, Y.; Wang, Y.; Xing, H.; Xia, Y.; Li, J.; et al. Clinical Updates on Gliomas and Implications of the 5th Edition of the WHO Classification of Central Nervous System Tumors. Front. Oncol. 2023, 13, 1131642.
- Shaw, E.G.; Berkey, B.; Coons, S.W.; Brachman, D.; Buckner, J.C.; Stelzer, K.J.; Barger, G.R.; Brown, P.D.; Gilbert, M.R.; Mehta, M. Initial Report of Radiation Therapy Oncology Group (RTOG) 9802: Prospective Studies in Adult Low-Grade Glioma (LGG). J. Clin. Oncol. 2006, 24, 1500.
- Lapointe, S.; Perry, A.; Butowski, N.A. Primary Brain Tumours in Adults. Lancet 2018, 392, 432–446.
- Fisher, J.P.; Adamson, D.C. Current FDA-Approved Therapies for High-Grade Malignant Gliomas. Biomedicines 2021, 9, 324.
- Hochberg, F.H.; Linggood, R.; Wolfson, L.; Baker, W.H.; Kornblith, P. Quality and Duration of Survival in Glioblastoma Multiforme: Combined Surgical, Radiation, and Lomustine Therapy. JAMA 1979, 241, 1016–1018.
- Walker, M.D.; Alexander, E.; Hunt, W.E.; MacCarty, C.S.; Mahaley, M.S.; Mealey, J.; Norrell, H.A.; Owens, G.; Ransohoff, J.; Wilson, C.B.; et al. Evaluation of BCNU and/or Radiotherapy in the Treatment of Anaplastic Gliomas: A Cooperative Clinical Trial. J. Neurosurg. 1978, 49, 333–343.
- Westphal, M.; Hilt, D.C.; Bortey, E.; Delavault, P.; Olivares, R.; Warnke, P.C.; Whittle, I.R.; Jääskeläinen, J.; Ram, Z. A Phase 3 Trial of Local Chemotherapy with Biodegradable Carmustine (BCNU) Wafers (Gliadel Wafers) in Patients with Primary Malignant Glioma. Neuro Oncol. 2003, 5, 79–88.
- Cohen, M.H.; Shen, Y.L.; Keegan, P.; Pazdur, R. FDA Drug Approval Summary: Bevacizumab (Avastin) as Treatment of Recurrent Glioblastoma Multiforme. Oncologist 2009, 14, 1131–1138.
- Stupp, R.; Taillibert, S.; Kanner, A.A.; Kesari, S.; Steinberg, D.M.; Toms, S.A.; Taylor, L.P.; Lieberman, F.; Silvani, A.; Fink, K.L.; et al. Maintenance Therapy with Tumor-Treating Fields Plus Temozolomide vs Temozolomide Alone for Glioblastoma: A Randomized Clinical Trial. JAMA 2015, 314, 2535–2543.
- Cheng, S.Y.; Huang, H.J.; Nagane, M.; Ji, X.D.; Wang, D.; Shih, C.C.; Arap, W.; Huang, C.M.; Cavenee, W.K. Suppression of Glioblastoma Angiogenicity and Tumorigenicity by Inhibition of Endogenous Expression of Vascular Endothelial Growth Factor. Proc. Natl. Acad. Sci. USA 1996, 93, 8502–8507.
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A Randomized Trial of Bevacizumab for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 699–708.
- Kim, M.M.; Umemura, Y.; Leung, D. Bevacizumab and Glioblastoma: Past, Present, and Future Directions. Cancer J. 2018, 24, 180–186.
- Wu, D.; Chen, Q.; Chen, X.; Han, F.; Chen, Z.; Wang, Y. The Blood–Brain Barrier: Structure, Regulation, and Drug Delivery. Signal Transduct. Target. Ther. 2023, 8, 217.
- Pardridge, W.M. CNS Drug Design Based on Principles of Blood-Brain Barrier Transport. J. Neurochem. 1998, 70, 1781–1792.
- Pulgar, V.M. Transcytosis to Cross the Blood Brain Barrier, New Advancements and Challenges. Front. Neurosci. 2019, 12, 1019.
- Hersh, D.S.; Wadajkar, A.S.; Roberts, N.B.; Perez, J.G.; Connolly, N.P.; Frenkel, V.; Winkles, J.A.; Woodworth, G.F.; Kim, A.J. Evolving Drug Delivery Strategies to Overcome the Blood Brain Barrier. Curr. Pharm. Des. 2016, 22, 1177–1193.
- Clark, D.E. In Silico Prediction of Blood–Brain Barrier Permeation. Drug Discov. Today 2003, 8, 927–933.
- Gleeson, M.P. Generation of a Set of Simple, Interpretable ADMET Rules of Thumb. J. Med. Chem. 2008, 51, 817–834.
- Hervé, F.; Ghinea, N.; Scherrmann, J.-M. CNS Delivery Via Adsorptive Transcytosis. AAPS J. 2008, 10, 455–472.
- Lalatsa, A.; Schätzlein, A.G.; Uchegbu, I.F. Drug Delivery across the Blood-Brain Barrier. In Comprehensive Biotechnology; Moo-Young, M., Ed.; Elsevier: Amsterdam, The Netherlands, 2011; Volume 5, pp. 657–667. ISBN 978-0-444-53352-4.
- Laquintana, V.; Trapani, A.; Denora, N.; Wang, F.; Gallo, J.M.; Trapani, G. New Strategies to Deliver Anticancer Drugs to Brain Tumors. Expert Opin. Drug Deliv. 2009, 6, 1017–1032.
- Ostermann, S.; Csajka, C.; Buclin, T.; Leyvraz, S.; Lejeune, F.; Decosterd, L.A.; Stupp, R. Plasma and Cerebrospinal Fluid Population Pharmacokinetics of Temozolomide in Malignant Glioma Patients. Clin. Cancer Res. 2004, 10, 3728–3736.
- Zhan, C.; Gu, B.; Xie, C.; Li, J.; Liu, Y.; Lu, W. Cyclic RGD Conjugated Poly(Ethylene Glycol)-Co-Poly(Lactic Acid) Micelle Enhances Paclitaxel Anti-Glioblastoma Effect. J. Control. Release 2010, 143, 136–142.
- Lesniak, M.S.; Upadhyay, U.; Goodwin, R.; Tyler, B.; Brem, H. Local Delivery of Doxorubicin for the Treatment of Malignant Brain Tumors in Rats. Anticancer Res. 2005, 25, 3825–3831.
- Kondo, Y.; Kondo, S.; Tanaka, Y.; Haqqi, T.; Barna, B.P.; Cowell, J.K. Inhibition of Telomerase Increases the Susceptibility of Human Malignant Glioblastoma Cells to Cisplatin-Induced Apoptosis. Oncogene 1998, 16, 2243–2248.
- Torchilin, V. Tumor Delivery of Macromolecular Drugs Based on the EPR Effect. Adv. Drug Deliv. Rev. 2011, 63, 131–135.
- Maeda, H. The Enhanced Permeability and Retention (EPR) Effect in Tumor Vasculature: The Key Role of Tumor-Selective Macromolecular Drug Targeting. Adv. Enzyme Regul. 2001, 41, 189–207.
- Lammers, T.; Kiessling, F.; Hennink, W.E.; Storm, G. Drug Targeting to Tumors: Principles, Pitfalls and (Pre-) Clinical Progress. J. Control. Release 2012, 161, 175–187.
- Danhier, F. To Exploit the Tumor Microenvironment: Since the EPR Effect Fails in the Clinic, What Is the Future of Nanomedicine? J. Control. Release 2016, 244, 108–121.
- Xu, H.-L.; Yang, J.-J.; ZhuGe, D.-L.; Lin, M.-T.; Zhu, Q.-Y.; Jin, B.-H.; Tong, M.-Q.; Shen, B.-X.; Xiao, J.; Zhao, Y.-Z. Glioma-Targeted Delivery of a Theranostic Liposome Integrated with Quantum Dots, Superparamagnetic Iron Oxide, and Cilengitide for Dual-Imaging Guiding Cancer Surgery. Adv. Healthc. Mater. 2018, 7, 1701130.
- van den Bent, M.; Azaro, A.; De Vos, F.; Sepulveda, J.; Yung, W.K.A.; Wen, P.Y.; Lassman, A.B.; Joerger, M.; Tabatabai, G.; Rodon, J.; et al. A Phase Ib/II, Open-Label, Multicenter Study of INC280 (Capmatinib) Alone and in Combination with Buparlisib (BKM120) in Adult Patients with Recurrent Glioblastoma. J. Neurooncol. 2020, 146, 79–89.
- Brem, H.; Mahaley, M.S.; Vick, N.A.; Black, K.L.; Schold, S.C.; Burger, P.C.; Friedman, A.H.; Ciric, I.S.; Eller, T.W.; Cozzens, J.W.; et al. Interstitial Chemotherapy with Drug Polymer Implants for the Treatment of Recurrent Gliomas. J. Neurosurg. 1991, 74, 441–446.
- Brem, S.; Tyler, B.; Li, K.; Pradilla, G.; Legnani, F.; Caplan, J.; Brem, H. Local Delivery of Temozolomide by Biodegradable Polymers Is Superior to Oral Administration in a Rodent Glioma Model. Cancer Chemother. Pharmacol. 2007, 60, 643–650.
- Shi, M.; Sanche, L. Convection-Enhanced Delivery in Malignant Gliomas: A Review of Toxicity and Efficacy. J. Oncol. 2019, 2019, e9342796.
- Voulgaris, S.; Partheni, M.; Karamouzis, M.; Dimopoulos, P.; Papadakis, N.; Kalofonos, H.P. Intratumoral Doxorubicin in Patients with Malignant Brain Gliomas. Am. J. Clin. Oncol. 2002, 25, 60.
- Drappatz, J.; Brenner, A.; Wong, E.T.; Eichler, A.; Schiff, D.; Groves, M.D.; Mikkelsen, T.; Rosenfeld, S.; Sarantopoulos, J.; Meyers, C.A.; et al. Phase I Study of GRN1005 in Recurrent Malignant Glioma. Clin. Cancer Res. 2013, 19, 1567–1576.
- Li, Y.; He, H.; Jia, X.; Lu, W.-L.; Lou, J.; Wei, Y. A Dual-Targeting Nanocarrier Based on Poly(Amidoamine) Dendrimers Conjugated with Transferrin and Tamoxifen for Treating Brain Gliomas. Biomaterials 2012, 33, 3899–3908.
- Ruan, S.; Yuan, M.; Zhang, L.; Hu, G.; Chen, J.; Cun, X.; Zhang, Q.; Yang, Y.; He, Q.; Gao, H. Tumor Microenvironment Sensitive Doxorubicin Delivery and Release to Glioma Using Angiopep-2 Decorated Gold Nanoparticles. Biomaterials 2015, 37, 425–435.
- Gao, H.; Qian, J.; Cao, S.; Yang, Z.; Pang, Z.; Pan, S.; Fan, L.; Xi, Z.; Jiang, X.; Zhang, Q. Precise Glioma Targeting of and Penetration by Aptamer and Peptide Dual-Functioned Nanoparticles. Biomaterials 2012, 33, 5115–5123.
- Gu, G.; Xia, H.; Hu, Q.; Liu, Z.; Jiang, M.; Kang, T.; Miao, D.; Tu, Y.; Pang, Z.; Song, Q.; et al. PEG-Co-PCL Nanoparticles Modified with MMP-2/9 Activatable Low Molecular Weight Protamine for Enhanced Targeted Glioblastoma Therapy. Biomaterials 2013, 34, 196–208.
- Škrlj, N.; Drevenšek, G.; Hudoklin, S.; Romih, R.; Čurin Šerbec, V.; Dolinar, M. Recombinant Single-Chain Antibody with the Trojan Peptide Penetratin Positioned in the Linker Region Enables Cargo Transfer Across the Blood–Brain Barrier. Appl. Biochem. Biotechnol. 2013, 169, 159–169.
- Thomas, A.; Teicher, B.A.; Hassan, R. Antibody–Drug Conjugates for Cancer Therapy. Lancet Oncol. 2016, 17, e254–e262.
- Qin, A.; Musket, A.; Musich, P.R.; Schweitzer, J.B.; Xie, Q. Receptor Tyrosine Kinases as Druggable Targets in Glioblastoma: Do Signaling Pathways Matter? Neuro Oncol. Adv. 2021, 3, vdab133.
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134.
- McLendon, R.; Friedman, A.; Bigner, D.; Van Meir, E.G.; Brat, D.J.; Mastrogianakis, G.M.; Olson, J.J.; Mikkelsen, T.; Lehman, N.; Aldape, K.; et al. Comprehensive Genomic Characterization Defines Human Glioblastoma Genes and Core Pathways. Nature 2008, 455, 1061–1068.
- Snuderl, M.; Fazlollahi, L.; Le, L.P.; Nitta, M.; Zhelyazkova, B.H.; Davidson, C.J.; Akhavanfard, S.; Cahill, D.P.; Aldape, K.D.; Betensky, R.A.; et al. Mosaic Amplification of Multiple Receptor Tyrosine Kinase Genes in Glioblastoma. Cancer Cell 2011, 20, 810–817.
- Wang, W.; Wang, E.; Balthasar, J. Monoclonal Antibody Pharmacokinetics and Pharmacodynamics. Clin. Pharmacol. Ther. 2008, 84, 548–558.
- Chacko, A.-M.; Li, C.; Pryma, D.A.; Brem, S.; Coukos, G.; Muzykantov, V.R. Targeted Delivery of Antibody-Based Therapeutic and Imaging Agents to CNS Tumors: Crossing the Blood-Brain-Barrier Divide. Expert Opin. Drug Deliv. 2013, 10, 907–926.
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and Challenges for the next Generation of Antibody–Drug Conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337.
- Ma, L.; Wang, C.; He, Z.; Cheng, B.; Zheng, L.; Huang, K. Peptide-Drug Conjugate: A Novel Drug Design Approach. Curr. Med. Chem. 2017, 24, 3373–3396.
- Dine, J.; Gordon, R.; Shames, Y.; Kasler, M.K.; Barton-Burke, M. Immune Checkpoint Inhibitors: An Innovation in Immunotherapy for the Treatment and Management of Patients with Cancer. Asia Pac. J. Oncol. Nurs. 2017, 4, 127–135.
- Ghouzlani, A.; Kandoussi, S.; Tall, M.; Reddy, K.P.; Rafii, S.; Badou, A. Immune Checkpoint Inhibitors in Human Glioma Microenvironment. Front. Immunol. 2021, 12, 679425.
- Huang, W.; Hao, Z.; Mao, F.; Guo, D. Small Molecule Inhibitors in Adult High-Grade Glioma: From the Past to the Future. Front. Oncol. 2022, 12, 911876.
- Liu, H.; Qiu, W.; Sun, T.; Wang, L.; Du, C.; Hu, Y.; Liu, W.; Feng, F.; Chen, Y.; Sun, H. Therapeutic Strategies of Glioblastoma (GBM): The Current Advances in the Molecular Targets and Bioactive Small Molecule Compounds. Acta Pharm. Sin. B 2022, 12, 1781–1804.
- Cesarini, V.; Scopa, C.; Silvestris, D.A.; Scafidi, A.; Petrera, V.; Del Baldo, G.; Gallo, A. Aptamer-Based In Vivo Therapeutic Targeting of Glioblastoma. Molecules 2020, 25, 4267.
- Qiu, M.; Wang, X.; Sun, H.; Zhang, J.; Deng, C.; Zhong, Z. Cyclic RGD-Peptide-Functionalized Polylipopeptide Micelles for Enhanced Loading and Targeted Delivery of Monomethyl Auristatin E. Mol. Pharm. 2018, 15, 4854–4861.
- Sievers, E.L.; Senter, P.D. Antibody-Drug Conjugates in Cancer Therapy. Annu. Rev. Med. 2013, 64, 15–29.
- Chen, B.; Dai, W.; He, B.; Zhang, H.; Wang, X.; Wang, Y.; Zhang, Q. Current Multistage Drug Delivery Systems Based on the Tumor Microenvironment. Theranostics 2017, 7, 538–558.
- Pillay, C.S.; Elliott, E.; Dennison, C. Endolysosomal Proteolysis and Its Regulation. Biochem. J. 2002, 363, 417–429.
- Casey, J.R.; Grinstein, S.; Orlowski, J. Sensors and Regulators of Intracellular pH. Nat. Rev. Mol. Cell Biol. 2010, 11, 50–61.
- Dubowchik, G.M.; Firestone, R.A. Cathepsin B-Sensitive Dipeptide Prodrugs. 1. A Model Study of Structural Requirements for Efficient Release of Doxorubicin. Bioorg. Med. Chem. Lett. 1998, 8, 3341–3346.
- Jeffrey, S.C.; Andreyka, J.B.; Bernhardt, S.X.; Kissler, K.M.; Kline, T.; Lenox, J.S.; Moser, R.F.; Nguyen, M.T.; Okeley, N.M.; Stone, I.J.; et al. Development and Properties of β-Glucuronide Linkers for Monoclonal Antibody−Drug Conjugates. Bioconjug. Chem. 2006, 17, 831–840.
- Tranoy-Opalinski, I.; Legigan, T.; Barat, R.; Clarhaut, J.; Thomas, M.; Renoux, B.; Papot, S. β-Glucuronidase-Responsive Prodrugs for Selective Cancer Chemotherapy: An Update. Eur. J. Med. Chem. 2014, 74, 302–313.
- Van Heeswijk, W.A.R.; Hoes, C.J.T.; Stoffer, T.; Eenink, M.J.D.; Potman, W.; Feijen, J. The Synthesis and Characterization of Polypeptide-Adriamycin Conjugates and Its Complexes with Adriamycin. Part I. J. Control. Release 1985, 1, 301–315.
- Wu, G.; Lupton, J.R.; Turner, N.D.; Fang, Y.-Z.; Yang, S. Glutathione Metabolism and Its Implications for Health. J. Nutr. 2004, 134, 489–492.
- Alas, M.; Saghaeidehkordi, A.; Kaur, K. Peptide–Drug Conjugates with Different Linkers for Cancer Therapy. J. Med. Chem. 2021, 64, 216–232.
- Ashman, N.; Bargh, J.D.; Spring, D.R. Non-Internalising Antibody–Drug Conjugates. Chem. Soc. Rev. 2022, 51, 9182–9202.
- Bargh, J.D.; Isidro-Llobet, A.; Parker, J.S.; Spring, D.R. Cleavable Linkers in Antibody–Drug Conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374.
- Ducry, L.; Stump, B. Antibody−Drug Conjugates: Linking Cytotoxic Payloads to Monoclonal Antibodies. Bioconjug. Chem. 2010, 21, 5–13.
- Zhang, P.; Cheetham, A.G.; Lock, L.L.; Cui, H. Cellular Uptake and Cytotoxicity of Drug–Peptide Conjugates Regulated by Conjugation Site. Bioconjug. Chem. 2013, 24, 604–613.
- Li, Y.; Zheng, X.; Gong, M.; Zhang, J. Delivery of a Peptide-Drug Conjugate Targeting the Blood Brain Barrier Improved the Efficacy of Paclitaxel against Glioma. Oncotarget 2016, 7, 79401–79407.
- Luo, Z.; Yan, Z.; Jin, K.; Pang, Q.; Jiang, T.; Lu, H.; Liu, X.; Pang, Z.; Yu, L.; Jiang, X. Precise Glioblastoma Targeting by AS1411 Aptamer-Functionalized Poly (l-γ-Glutamylglutamine)–Paclitaxel Nanoconjugates. J. Colloid Interface Sci. 2017, 490, 783–796.
- Sapra, P.; Shor, B. Monoclonal Antibody-Based Therapies in Cancer: Advances and Challenges. Pharmacol. Ther. 2013, 138, 452–469.
- Cooper, B.M.; Iegre, J.; Donovan, D.H.O.; Halvarsson, M.Ö.; Spring, D.R. Peptides as a Platform for Targeted Therapeutics for Cancer: Peptide–Drug Conjugates (PDCs). Chem. Soc. Rev. 2021, 50, 1480–1494.
- Doronina, S.O.; Mendelsohn, B.A.; Bovee, T.D.; Cerveny, C.G.; Alley, S.C.; Meyer, D.L.; Oflazoglu, E.; Toki, B.E.; Sanderson, R.J.; Zabinski, R.F.; et al. Enhanced Activity of Monomethylauristatin F through Monoclonal Antibody Delivery: Effects of Linker Technology on Efficacy and Toxicity. Bioconjug. Chem. 2006, 17, 114–124.
- Lopus, M.; Oroudjev, E.; Wilson, L.; Wilhelm, S.; Widdison, W.; Chari, R.; Jordan, M.A. Maytansine and Cellular Metabolites of Antibody-Maytansinoid Conjugates Strongly Suppress Microtubule Dynamics by Binding to Microtubules. Mol. Cancer Ther. 2010, 9, 2689–2699.
- Hamblett, K.J.; Kozlosky, C.J.; Siu, S.; Chang, W.S.; Liu, H.; Foltz, I.N.; Trueblood, E.S.; Meininger, D.; Arora, T.; Twomey, B.; et al. AMG 595, an Anti-EGFRvIII Antibody–Drug Conjugate, Induces Potent Antitumor Activity against EGFRvIII-Expressing Glioblastoma. Mol. Cancer Ther. 2015, 14, 1614–1624.
- Wang, Y.; Li, Y.; Cao, J.; Meng, Q.; Li, X.; Zhang, Y.; Lam, K.S.; Hong, A.; Liu, R.; Chen, X. Development and Characterization of a Novel Peptide—Drug Conjugate with DM1 for Treatment of FGFR2-Positive Tumors. Biomedicines 2021, 9, 849.
- Zhang, Q.-Y.; Yu, Q.-L.; Luan, W.-J.; Li, T.-F.; Xiao, Y.-N.; Zhang, L.; Li, Y.; Rong, R.; Ren, C.-G. LWJ-M30, a Conjugate of DM1 and B6, for the Targeted Therapy of Colorectal Cancer with Improved Therapeutic Effects. RSC Adv. 2023, 13, 10840–10846.
- Gazzah, A.; Bedard, P.L.; Hierro, C.; Kang, Y.-K.; Razak, A.A.; Ryu, M.-H.; Demers, B.; Fagniez, N.; Henry, C.; Hospitel, M.; et al. Safety, Pharmacokinetics, and Antitumor Activity of the Anti-CEACAM5-DM4 Antibody–Drug Conjugate Tusamitamab Ravtansine (SAR408701) in Patients with Advanced Solid Tumors: First-in-Human Dose-Escalation Study. Ann. Oncol. 2022, 33, 416–425.
- Pouzin, C.; Gibiansky, L.; Fagniez, N.; Chadjaa, M.; Tod, M.; Nguyen, L. Integrated Multiple Analytes and Semi-Mechanistic Population Pharmacokinetic Model of Tusamitamab Ravtansine, a DM4 Anti-CEACAM5 Antibody-Drug Conjugate. J. Pharmacokinet. Pharmacodyn. 2022, 49, 381–394.
- Zahaf, N.-I.; Schmidt, G. Bacterial Toxins for Cancer Therapy. Toxins 2017, 9, 236.
- Gill, D.M.; Pappenheimer, A.M., Jr.; Brown, R.; Kurnick, J.T. Studies on the mode of action of diphtheria toxin: VII: Toxin-stimulated hydrolysis of nicotinamide adenine dinucleotide in mammalian cell extracts. J. Exp. Med. 1969, 129, 1–21.
- Siegall, C.B.; Chaudhary, V.K.; FitzGerald, D.J.; Pastan, I. Functional Analysis of Domains II, Ib, and III of Pseudomonas Exotoxin. J. Biol. Chem. 1989, 264, 14256–14261.
- Loya, J.; Zhang, C.; Cox, E.; Achrol, A.S.; Kesari, S. Biological Intratumoral Therapy for the High-Grade Glioma Part I: Intratumoral Delivery and Immunotoxins. CNS Oncol. 2019, 8, CNS38.
- Loya, J.; Zhang, C.; Cox, E.; Achrol, A.S.; Kesari, S. Biological Intratumoral Therapy for the High-Grade Glioma Part II: Vector- and Cell-Based Therapies and Radioimmunotherapy. CNS Oncol. 2019, 8, CNS40.
- Cordier, D.; Krolicki, L.; Morgenstern, A.; Merlo, A. Targeted Radiolabeled Compounds in Glioma Therapy. Semin. Nucl. Med. 2016, 46, 243–249.
- Steiner, M.; Neri, D. Antibody-Radionuclide Conjugates for Cancer Therapy: Historical Considerations and New Trends. Clin. Cancer Res. 2011, 17, 6406–6416.
- Hirschberg, H.; Berg, K.; Peng, Q. Photodynamic Therapy Mediated Immune Therapy of Brain Tumors. Neuroimmunol. Neuroinflamm. 2018, 5, 27.
- Beck, T.J.; Kreth, F.W.; Beyer, W.; Mehrkens, J.H.; Obermeier, A.; Stepp, H.; Stummer, W.; Baumgartner, R. Interstitial Photodynamic Therapy of Nonresectable Malignant Glioma Recurrences Using 5-Aminolevulinic Acid Induced Protoporphyrin IX. Lasers Surg. Med. 2007, 39, 386–393.
- Madsen, S.J.; Angell-Petersen, E.; Spetalen, S.; Carper, S.W.; Ziegler, S.A.; Hirschberg, H. Photodynamic Therapy of Newly Implanted Glioma Cells in the Rat Brain. Lasers Surg. Med. 2006, 38, 540–548.
- Hamad, A.; Yusubalieva, G.M.; Baklaushev, V.P.; Chumakov, P.M.; Lipatova, A.V. Recent Developments in Glioblastoma Therapy: Oncolytic Viruses and Emerging Future Strategies. Viruses 2023, 15, 547.
- Li, J.; Meng, Q.; Zhou, X.; Zhao, H.; Wang, K.; Niu, H.; Wang, Y. Gospel of Malignant Glioma: Oncolytic Virus Therapy. Gene 2022, 818, 146217.
- Zhang, H.; Berezov, A.; Wang, Q.; Zhang, G.; Drebin, J.; Murali, R.; Greene, M.I. ErbB Receptors: From Oncogenes to Targeted Cancer Therapies. J. Clin. Investig. 2007, 117, 2051–2058.
- Hatanpaa, K.J.; Burma, S.; Zhao, D.; Habib, A.A. Epidermal Growth Factor Receptor in Glioma: Signal Transduction, Neuropathology, Imaging, and Radioresistance. Neoplasia 2010, 12, 675–684.
- Li, L.; Dutra, A.; Pak, E.; Labrie, J.E., III; Gerstein, R.M.; Pandolfi, P.P.; Recht, L.D.; Ross, A.H. EGFRvIII Expression and PTEN Loss Synergistically Induce Chromosomal Instability and Glial Tumors. Neuro Oncol. 2009, 11, 9–21.
- Mazzoleni, S.; Politi, L.S.; Pala, M.; Cominelli, M.; Franzin, A.; Sergi Sergi, L.; Falini, A.; De Palma, M.; Bulfone, A.; Poliani, P.L.; et al. Epidermal Growth Factor Receptor Expression Identifies Functionally and Molecularly Distinct Tumor-Initiating Cells in Human Glioblastoma Multiforme and Is Required for Gliomagenesis. Cancer Res. 2010, 70, 7500–7513.
- Pang, L.Y.; Saunders, L.; Argyle, D.J. Epidermal Growth Factor Receptor Activity Is Elevated in Glioma Cancer Stem Cells and Is Required to Maintain Chemotherapy and Radiation Resistance. Oncotarget 2017, 8, 72494–72512.
- Wen, P.Y.; Kesari, S. Malignant Gliomas in Adults. N. Engl. J. Med. 2008, 359, 492–507.
- Lassman, A.B.; van den Bent, M.J.; Gan, H.K.; Reardon, D.A.; Kumthekar, P.; Butowski, N.; Lwin, Z.; Mikkelsen, T.; Nabors, L.B.; Papadopoulos, K.P.; et al. Safety and Efficacy of Depatuxizumab Mafodotin + Temozolomide in Patients with EGFR-Amplified, Recurrent Glioblastoma: Results from an International Phase I Multicenter Trial. Neuro Oncol. 2019, 21, 106–114.
- Narita, Y.; Muragaki, Y.; Kagawa, N.; Asai, K.; Nagane, M.; Matsuda, M.; Ueki, K.; Kuroda, J.; Date, I.; Kobayashi, H.; et al. Safety and Efficacy of Depatuxizumab Mafodotin in Japanese Patients with Malignant Glioma: A Nonrandomized, Phase 1/2 Trial. Cancer Sci. 2021, 112, 5020–5033.
- Van Den Bent, M.; Eoli, M.; Sepulveda, J.M.; Smits, M.; Walenkamp, A.; Frenel, J.-S.; Franceschi, E.; Clement, P.M.; Chinot, O.; De Vos, F.; et al. INTELLANCE 2/EORTC 1410 Randomized Phase II Study of Depatux-M Alone and with Temozolomide vs Temozolomide or Lomustine in Recurrent EGFR Amplified Glioblastoma. Neuro Oncol. 2020, 22, 684–693.
- Gan, H.K.; Reardon, D.A.; Lassman, A.B.; Merrell, R.; van den Bent, M.; Butowski, N.; Lwin, Z.; Wheeler, H.; Fichtel, L.; Scott, A.M.; et al. Safety, Pharmacokinetics, and Antitumor Response of Depatuxizumab Mafodotin as Monotherapy or in Combination with Temozolomide in Patients with Glioblastoma. Neuro Oncol. 2018, 20, 838–847.
- Phillips, A.C.; Boghaert, E.R.; Vaidya, K.S.; Mitten, M.J.; Norvell, S.; Falls, H.D.; DeVries, P.J.; Cheng, D.; Meulbroek, J.A.; Buchanan, F.G.; et al. ABT-414, an Antibody–Drug Conjugate Targeting a Tumor-Selective EGFR Epitope. Mol. Cancer Ther. 2016, 15, 661–669.
- Rosenthal, M.; Curry, R.; Reardon, D.A.; Rasmussen, E.; Upreti, V.V.; Damore, M.A.; Henary, H.A.; Hill, J.S.; Cloughesy, T. Safety, Tolerability, and Pharmacokinetics of Anti-EGFRvIII Antibody–Drug Conjugate AMG 595 in Patients with Recurrent Malignant Glioma Expressing EGFRvIII. Cancer Chemother. Pharmacol. 2019, 84, 327–336.
- Desjardins, A.; Chandramohan, V.; Landi, D.B.; Johnson, M.O.; Khasraw, M.; Peters, K.B.; Low, J.; Herndon, J.E.; Threatt, S.; Bullock, C.A.; et al. A Phase 1 Trial of D2C7-It in Combination with an Fc-Engineered Anti-CD40 Monoclonal Antibody (2141-V11) Administered Intratumorally via Convection-Enhanced Delivery for Adult Patients with Recurrent Malignant Glioma (MG). J. Clin. Oncol. 2022, 40, e14015.
- Neyns, B.; Sadones, J.; Joosens, E.; Bouttens, F.; Verbeke, L.; Baurain, J.-F.; D’Hondt, L.; Strauven, T.; Chaskis, C.; Veld, P.I.; et al. Stratified Phase II Trial of Cetuximab in Patients with Recurrent High-Grade Glioma. Ann. Oncol. 2009, 20, 1596–1603.
- Hasselbalch, B.; Lassen, U.; Hansen, S.; Holmberg, M.; Sørensen, M.; Kosteljanetz, M.; Broholm, H.; Stockhausen, M.-T.; Poulsen, H.S. Cetuximab, Bevacizumab, and Irinotecan for Patients with Primary Glioblastoma and Progression after Radiation Therapy and Temozolomide: A Phase II Trial. Neuro Oncol. 2010, 12, 508–516.
- Westphal, M.; Heese, O.; Steinbach, J.P.; Schnell, O.; Schackert, G.; Mehdorn, M.; Schulz, D.; Simon, M.; Schlegel, U.; Senft, C.; et al. A Randomised, Open Label Phase III Trial with Nimotuzumab, an Anti-Epidermal Growth Factor Receptor Monoclonal Antibody in the Treatment of Newly Diagnosed Adult Glioblastoma. Eur. J. Cancer 2015, 51, 522–532.
- Cantanhede, I.G.; de Oliveira, J.R.M. PDGF Family Expression in Glioblastoma Multiforme: Data Compilation from Ivy Glioblastoma Atlas Project Database. Sci. Rep. 2017, 7, 15271.
- Cao, Y. Multifarious Functions of PDGFs and PDGFRs in Tumor Growth and Metastasis. Trends Mol. Med. 2013, 19, 460–473.
- Nazarenko, I.; Hede, S.-M.; He, X.; Hedrén, A.; Thompson, J.; Lindström, M.S.; Nistér, M. PDGF and PDGF Receptors in Glioma. Upsala J. Med. Sci. 2012, 117, 99–112.
- Camorani, S.; Esposito, C.L.; Rienzo, A.; Catuogno, S.; Iaboni, M.; Condorelli, G.; de Franciscis, V.; Cerchia, L. Inhibition of Receptor Signaling and of Glioblastoma-Derived Tumor Growth by a Novel PDGFRβ Aptamer. Mol. Ther. 2014, 22, 828–841.
- Plate, K.H.; Breier, G.; Farrell, C.L.; Risau, W. Platelet-Derived Growth Factor Receptor-Beta Is Induced during Tumor Development and Upregulated during Tumor Progression in Endothelial Cells in Human Gliomas. Lab. Investig. 1992, 67, 529–534.
- Lokker, N.A.; Sullivan, C.M.; Hollenbach, S.J.; Israel, M.A.; Giese, N.A. Platelet-Derived Growth Factor (PDGF) Autocrine Signaling Regulates Survival and Mitogenic Pathways in Glioblastoma Cells: Evidence That the Novel PDGF-C and PDGF-D Ligands May Play a Role in the Development of Brain Tumors. Cancer Res. 2002, 62, 3729–3735.
- Vassbotn, F.S.; Östman, A.; Langeland, N.; Holmsen, H.; Westermark, B.; Heldin, C.-H.; Nistér, M. Activated Platelet-Derived Growth Factor Autocrine Pathway Drives the Transformed Phenotype of a Human Glioblastoma Cell Line. J. Cell. Physiol. 1994, 158, 381–389.
- Phuphanich, S.; Raizer, J.; Chamberlain, M.; Canelos, P.; Narwal, R.; Hong, S.; Miday, R.; Nade, M.; Laubscher, K. Phase II Study of MEDI-575, an Anti-Platelet-Derived Growth Factor-α Antibody, in Patients with Recurrent Glioblastoma. J. Neurooncol. 2017, 131, 185–191.
- Jain, R.K.; di Tomaso, E.; Duda, D.G.; Loeffler, J.S.; Sorensen, A.G.; Batchelor, T.T. Angiogenesis in Brain Tumours. Nat. Rev. Neurosci. 2007, 8, 610–622.
- Clara, C.A.; Marie, S.K.N.; de Almeida, J.R.W.; Wakamatsu, A.; Oba-Shinjo, S.M.; Uno, M.; Neville, M.; Rosemberg, S. Angiogenesis and Expression of PDGF-C, VEGF, CD105 and HIF-1α in Human Glioblastoma. Neuropathology 2014, 34, 343–352.
- Puputti, M.; Tynninen, O.; Sihto, H.; Blom, T.; Mäenpää, H.; Isola, J.; Paetau, A.; Joensuu, H.; Nupponen, N.N. Amplification of KIT, PDGFRA, VEGFR2, and EGFR in Gliomas. Mol. Cancer Res. 2006, 4, 927–934.
- Chaudhry, I.H.; O’Donovan, D.G.; Brenchley, P.E.C.; Reid, H.; Roberts, I.S.D. Vascular Endothelial Growth Factor Expression Correlates with Tumour Grade and Vascularity in Gliomas. Histopathology 2001, 39, 409–415.
- Tamura, R.; Ohara, K.; Sasaki, H.; Morimoto, Y.; Yoshida, K.; Toda, M. Histopathological Vascular Investigation of the Peritumoral Brain Zone of Glioblastomas. J. Neurooncol. 2018, 136, 233–241.
- Roskoski, R. Vascular Endothelial Growth Factor (VEGF) Signaling in Tumor Progression. Crit. Rev. Oncol. Hematol. 2007, 62, 179–213.
- Hamerlik, P.; Lathia, J.D.; Rasmussen, R.; Wu, Q.; Bartkova, J.; Lee, M.; Moudry, P.; Bartek, J.; Fischer, W.; Lukas, J.; et al. Autocrine VEGF-VEGFR2-Neuropilin-1 Signaling Promotes Glioma Stem-like Cell Viability and Tumor Growth. J. Exp. Med. 2012, 209, 507–520.
- Tamura, R.; Tanaka, T.; Akasaki, Y.; Murayama, Y.; Yoshida, K.; Sasaki, H. The Role of Vascular Endothelial Growth Factor in the Hypoxic and Immunosuppressive Tumor Microenvironment: Perspectives for Therapeutic Implications. Med. Oncol. 2019, 37, 2.
- Friedman, H.S.; Prados, M.D.; Wen, P.Y.; Mikkelsen, T.; Schiff, D.; Abrey, L.E.; Yung, W.K.A.; Paleologos, N.; Nicholas, M.K.; Jensen, R.; et al. Bevacizumab Alone and in Combination with Irinotecan in Recurrent Glioblastoma. J. Clin. Oncol. 2009, 27, 4733–4740.
- Vredenburgh, J.J.; Desjardins, A.; Herndon, J.E.; Marcello, J.; Reardon, D.A.; Quinn, J.A.; Rich, J.N.; Sathornsumetee, S.; Gururangan, S.; Sampson, J.; et al. Bevacizumab Plus Irinotecan in Recurrent Glioblastoma Multiforme. J. Clin. Oncol. 2007, 25, 4722–4729.
- Brandes, A.A.; Gil-Gil, M.; Saran, F.; Carpentier, A.F.; Nowak, A.K.; Mason, W.; Zagonel, V.; Dubois, F.; Finocchiaro, G.; Fountzilas, G.; et al. A Randomized Phase II Trial (TAMIGA) Evaluating the Efficacy and Safety of Continuous Bevacizumab Through Multiple Lines of Treatment for Recurrent Glioblastoma. Oncology 2019, 24, 521–528.
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus Radiotherapy–Temozolomide for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 709–722.
- Herrlinger, U.; Schäfer, N.; Steinbach, J.P.; Weyerbrock, A.; Hau, P.; Goldbrunner, R.; Friedrich, F.; Rohde, V.; Ringel, F.; Schlegel, U.; et al. Bevacizumab Plus Irinotecan Versus Temozolomide in Newly Diagnosed O6-Methylguanine–DNA Methyltransferase Nonmethylated Glioblastoma: The Randomized GLARIUS Trial. J. Clin. Oncol. 2016, 34, 1611–1619.
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and Bevacizumab in Progressive Glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963.
- Mamluk, R.; Carvajal, I.M.; Morse, B.A.; Wong, H.; Abramowitz, J.; Aslanian, S.; Lim, A.-C.; Gokemeijer, J.; Storek, M.J.; Lee, J.; et al. Anti-Tumor Effect of CT-322 as an Adnectin Inhibitor of Vascular Endothelial Growth Factor Receptor-2. MAbs 2010, 2, 199–208.
- Schiff, D.; Kesari, S.; de Groot, J.; Mikkelsen, T.; Drappatz, J.; Coyle, T.; Fichtel, L.; Silver, B.; Walters, I.; Reardon, D. Phase 2 Study of CT-322, a Targeted Biologic Inhibitor of VEGFR-2 Based on a Domain of Human Fibronectin, in Recurrent Glioblastoma. Investig. New Drugs 2015, 33, 247–253.
- Kwak, Y.; Kim, S.-I.; Park, C.-K.; Paek, S.H.; Lee, S.-T.; Park, S.-H. C-MET Overexpression and Amplification in Gliomas. Int. J. Clin. Exp. Pathol. 2015, 8, 14932–14938.
- Xie, Q.; Bradley, R.; Kang, L.; Koeman, J.; Ascierto, M.L.; Worschech, A.; De Giorgi, V.; Wang, E.; Kefene, L.; Su, Y.; et al. Hepatocyte Growth Factor (HGF) Autocrine Activation Predicts Sensitivity to MET Inhibition in Glioblastoma. Proc. Natl. Acad. Sci. USA 2012, 109, 570–575.
- Lu, K.V.; Chang, J.P.; Parachoniak, C.A.; Pandika, M.M.; Aghi, M.K.; Meyronet, D.; Isachenko, N.; Fouse, S.D.; Phillips, J.J.; Cheresh, D.A.; et al. VEGF Inhibits Tumor Cell Invasion and Mesenchymal Transition through a MET/VEGFR2 Complex. Cancer Cell 2012, 22, 21–35.
- El Atat, O.; Naser, R.; Abdelkhalek, M.; Habib, R.A.; El Sibai, M. Molecular Targeted Therapy: A New Avenue in Glioblastoma Treatment (Review). Oncol. Lett. 2023, 25, 46.
- Cruz Da Silva, E.; Mercier, M.-C.; Etienne-Selloum, N.; Dontenwill, M.; Choulier, L. A Systematic Review of Glioblastoma-Targeted Therapies in Phases II, III, IV Clinical Trials. Cancers 2021, 13, 1795.
- Cloughesy, T.; Finocchiaro, G.; Belda-Iniesta, C.; Recht, L.; Brandes, A.A.; Pineda, E.; Mikkelsen, T.; Chinot, O.L.; Balana, C.; Macdonald, D.R.; et al. Randomized, Double-Blind, Placebo-Controlled, Multicenter Phase II Study of Onartuzumab Plus Bevacizumab Versus Placebo Plus Bevacizumab in Patients with Recurrent Glioblastoma: Efficacy, Safety, and Hepatocyte Growth Factor and O6-Methylguanine–DNA Methyltransferase Biomarker Analyses. J. Clin. Oncol. 2017, 35, 343–351.
- Wen, P.Y.; Schiff, D.; Cloughesy, T.F.; Raizer, J.J.; Laterra, J.; Smitt, M.; Wolf, M.; Oliner, K.S.; Anderson, A.; Zhu, M.; et al. A Phase II Study Evaluating the Efficacy and Safety of AMG 102 (Rilotumumab) in Patients with Recurrent Glioblastoma. Neuro Oncol. 2011, 13, 437–446.
- Babina, I.S.; Turner, N.C. Advances and Challenges in Targeting FGFR Signalling in Cancer. Nat. Rev. Cancer 2017, 17, 318–332.
- Helsten, T.; Elkin, S.; Arthur, E.; Tomson, B.N.; Carter, J.; Kurzrock, R. The FGFR Landscape in Cancer: Analysis of 4853 Tumors by Next-Generation Sequencing. Clin. Cancer Res. 2016, 22, 259–267.
- Imamura, T. Physiological Functions and Underlying Mechanisms of Fibroblast Growth Factor (FGF) Family Members: Recent Findings and Implications for Their Pharmacological Application. Biol. Pharm. Bull. 2014, 37, 1081–1089.
- Qin, A.; Johnson, A.; Ross, J.S.; Miller, V.A.; Ali, S.M.; Schrock, A.B.; Gadgeel, S.M. Detection of Known and Novel FGFR Fusions in Non-Small Cell Lung Cancer by Comprehensive Genomic Profiling. J. Thorac. Oncol. 2019, 14, 54–62.
- Tiong, K.H.; Mah, L.Y.; Leong, C.-O. Functional Roles of Fibroblast Growth Factor Receptors (FGFRs) Signaling in Human Cancers. Apoptosis 2013, 18, 1447–1468.
- Day, E.K.; Sosale, N.G.; Xiao, A.; Zhong, Q.; Purow, B.; Lazzara, M.J. Glioblastoma Cell Resistance to EGFR and MET Inhibition Can Be Overcome via Blockade of FGFR-SPRY2 Bypass Signaling. Cell Rep. 2020, 30, 3383–3396.e7.
- Guagnano, V.; Kauffmann, A.; Wöhrle, S.; Stamm, C.; Ito, M.; Barys, L.; Pornon, A.; Yao, Y.; Li, F.; Zhang, Y.; et al. FGFR Genetic Alterations Predict for Sensitivity to NVP-BGJ398, a Selective Pan-FGFR Inhibitor. Cancer Discov. 2012, 2, 1118–1133.
- Reardon, D.A.; Dresemann, G.; Taillibert, S.; Campone, M.; van den Bent, M.; Clement, P.; Blomquist, E.; Gordower, L.; Schultz, H.; Raizer, J.; et al. Multicentre Phase II Studies Evaluating Imatinib plus Hydroxyurea in Patients with Progressive Glioblastoma. Br. J. Cancer 2009, 101, 1995–2004.
- Dresemann, G.; Weller, M.; Rosenthal, M.A.; Wedding, U.; Wagner, W.; Engel, E.; Heinrich, B.; Mayer-Steinacker, R.; Karup-Hansen, A.; Fluge, Ø.; et al. Imatinib in Combination with Hydroxyurea versus Hydroxyurea Alone as Oral Therapy in Patients with Progressive Pretreated Glioblastoma Resistant to Standard Dose Temozolomide. J. Neurooncol. 2010, 96, 393–402.
- Raymond, E.; Brandes, A.A.; Dittrich, C.; Fumoleau, P.; Coudert, B.; Clement, P.M.J.; Frenay, M.; Rampling, R.; Stupp, R.; Kros, J.M.; et al. Phase II Study of Imatinib in Patients with Recurrent Gliomas of Various Histologies: A European Organisation for Research and Treatment of Cancer Brain Tumor Group Study. J. Clin. Oncol. 2008, 26, 4659–4665.
- Lassman, A.B.; Pugh, S.L.; Gilbert, M.R.; Aldape, K.D.; Geinoz, S.; Beumer, J.H.; Christner, S.M.; Komaki, R.; DeAngelis, L.M.; Gaur, R.; et al. Phase 2 Trial of Dasatinib in Target-Selected Patients with Recurrent Glioblastoma (RTOG 0627). Neuro Oncol. 2015, 17, 992–998.
- Franceschi, E.; Stupp, R.; van den Bent, M.J.; van Herpen, C.; Laigle Donadey, F.; Gorlia, T.; Hegi, M.; Lhermitte, B.; Strauss, L.C.; Allgeier, A.; et al. EORTC 26083 Phase I/II Trial of Dasatinib in Combination with CCNU in Patients with Recurrent Glioblastoma. Neuro Oncol. 2012, 14, 1503–1510.
- Balaña, C.; Gil, M.J.; Perez, P.; Reynes, G.; Gallego, O.; Ribalta, T.; Capellades, J.; Gonzalez, S.; Verger, E. Sunitinib Administered Prior to Radiotherapy in Patients with Non-Resectable Glioblastoma: Results of a Phase II Study. Targ. Oncol. 2014, 9, 321–329.
- Pan, E.; Yu, D.; Yue, B.; Potthast, L.; Chowdhary, S.; Smith, P.; Chamberlain, M. A Prospective Phase II Single-Institution Trial of Sunitinib for Recurrent Malignant Glioma. J. Neurooncol. 2012, 110, 111–118.
- Hutterer, M.; Nowosielski, M.; Haybaeck, J.; Embacher, S.; Stockhammer, F.; Gotwald, T.; Holzner, B.; Capper, D.; Preusser, M.; Marosi, C.; et al. A Single-Arm Phase II Austrian/German Multicenter Trial on Continuous Daily Sunitinib in Primary Glioblastoma at First Recurrence (SURGE 01-07). Neuro Oncol. 2014, 16, 92–102.
- O’Hare, T.; Shakespeare, W.C.; Zhu, X.; Eide, C.A.; Rivera, V.M.; Wang, F.; Adrian, L.T.; Zhou, T.; Huang, W.-S.; Xu, Q.; et al. AP24534, a Pan-BCR-ABL Inhibitor for Chronic Myeloid Leukemia, Potently Inhibits the T315I Mutant and Overcomes Mutation-Based Resistance. Cancer Cell 2009, 16, 401–412.
- Lee, E.Q.; Muzikansky, A.; Reardon, D.A.; Dietrich, J.; Nayak, L.; Duda, D.G.; Chukwueke, U.N.; Beroukhim, R.; Doherty, L.M.; Kane, C.; et al. Phase II Trial of Ponatinib in Patients with Bevacizumab-Refractory Glioblastoma. J. Clin. Oncol. 2018, 36, 2032.
- Reardon, D.A.; Kim, T.M.; Frenel, J.-S.; Simonelli, M.; Lopez, J.; Subramaniam, D.S.; Siu, L.L.; Wang, H.; Krishnan, S.; Stein, K.; et al. Treatment with Pembrolizumab in Programmed Death Ligand 1–Positive Recurrent Glioblastoma: Results from the Multicohort Phase 1 KEYNOTE-028 Trial. Cancer 2021, 127, 1620–1629.
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients with Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010.
- Lim, M.; Weller, M.; Idbaih, A.; Steinbach, J.; Finocchiaro, G.; Raval, R.R.; Ansstas, G.; Baehring, J.; Taylor, J.W.; Honnorat, J.; et al. Phase III Trial of Chemoradiotherapy with Temozolomide plus Nivolumab or Placebo for Newly Diagnosed Glioblastoma with Methylated MGMT Promoter. Neuro Oncol. 2022, 24, 1935–1949.
- Omuro, A.; Brandes, A.A.; Carpentier, A.F.; Idbaih, A.; Reardon, D.A.; Cloughesy, T.; Sumrall, A.; Baehring, J.; van den Bent, M.; Bähr, O.; et al. Radiotherapy Combined with Nivolumab or Temozolomide for Newly Diagnosed Glioblastoma with Unmethylated MGMT Promoter: An International Randomized Phase III Trial. Neuro Oncol. 2023, 25, 123–134.
- Fong, B.; Jin, R.; Wang, X.; Safaee, M.; Lisiero, D.N.; Yang, I.; Li, G.; Liau, L.M.; Prins, R.M. Monitoring of Regulatory T Cell Frequencies and Expression of CTLA-4 on T Cells, before and after DC Vaccination, Can Predict Survival in GBM Patients. PLoS ONE 2012, 7, e32614.
- Belcaid, Z.; Phallen, J.A.; Zeng, J.; See, A.P.; Mathios, D.; Gottschalk, C.; Nicholas, S.; Kellett, M.; Ruzevick, J.; Jackson, C.; et al. Focal Radiation Therapy Combined with 4-1BB Activation and CTLA-4 Blockade Yields Long-Term Survival and a Protective Antigen-Specific Memory Response in a Murine Glioma Model. PLoS ONE 2014, 9, e101764.
- Das, M.; Zhu, C.; Kuchroo, V.K. Tim-3 and Its Role in Regulating Anti-Tumor Immunity. Immunol. Rev. 2017, 276, 97–111.
- Li, G.; Wang, Z.; Zhang, C.; Liu, X.; Cai, J.; Wang, Z.; Hu, H.; Wu, F.; Bao, Z.; Liu, Y.; et al. Molecular and Clinical Characterization of TIM-3 in Glioma through 1024 Samples. OncoImmunology 2017, 6, e1328339.
- Triebel, F.; Jitsukawa, S.; Baixeras, E.; Roman-Roman, S.; Genevee, C.; Viegas-Pequignot, E.; Hercend, T. LAG-3, a Novel Lymphocyte Activation Gene Closely Related to CD4. J. Exp. Med. 1990, 171, 1393–1405.
- Workman, C.J.; Rice, D.S.; Dugger, K.J.; Kurschner, C.; Vignali, D.A.A. Phenotypic Analysis of the Murine CD4-Related Glycoprotein, CD223 (LAG-3). Eur. J. Immunol. 2002, 32, 2255–2263.
- Yang, K.; Wu, Z.; Zhang, H.; Zhang, N.; Wu, W.; Wang, Z.; Dai, Z.; Zhang, X.; Zhang, L.; Peng, Y.; et al. Glioma Targeted Therapy: Insight into Future of Molecular Approaches. Mol. Cancer 2022, 21, 39.
- Harris-Bookman, S.; Mathios, D.; Martin, A.M.; Xia, Y.; Kim, E.; Xu, H.; Belcaid, Z.; Polanczyk, M.; Barberi, T.; Theodros, D.; et al. Expression of LAG-3 and Efficacy of Combination Treatment with Anti-LAG-3 and Anti-PD-1 Monoclonal Antibodies in Glioblastoma. Int. J. Cancer 2018, 143, 3201–3208.
- Mair, M.J.; Kiesel, B.; Feldmann, K.; Widhalm, G.; Dieckmann, K.; Wöhrer, A.; Müllauer, L.; Preusser, M.; Berghoff, A.S. LAG-3 Expression in the Inflammatory Microenvironment of Glioma. J. Neurooncol. 2021, 152, 533–539.
- Panda, A.; Rosenfeld, J.A.; Singer, E.A.; Bhanot, G.; Ganesan, S. Genomic and Immunologic Correlates of LAG-3 Expression in Cancer. OncoImmunology 2020, 9, 1756116.
- Sood, D.; Tang-Schomer, M.; Pouli, D.; Mizzoni, C.; Raia, N.; Tai, A.; Arkun, K.; Wu, J.; Black, L.D.; Scheffler, B.; et al. 3D Extracellular Matrix Microenvironment in Bioengineered Tissue Models of Primary Pediatric and Adult Brain Tumors. Nat. Commun. 2019, 10, 4529.
- Mohiuddin, E.; Wakimoto, H. Extracellular Matrix in Glioblastoma: Opportunities for Emerging Therapeutic Approaches. Am. J. Cancer Res. 2021, 11, 3742–3754.
- Brack, S.S.; Silacci, M.; Birchler, M.; Neri, D. Tumor-Targeting Properties of Novel Antibodies Specific to the Large Isoform of Tenascin-C. Clin. Cancer Res. 2006, 12, 3200–3208.
- Czabanka, M.; Parmaksiz, G.; Bayerl, S.H.; Nieminen, M.; Trachsel, E.; Menssen, H.D.; Erber, R.; Neri, D.; Vajkoczy, P. Microvascular Biodistribution of L19-SIP in Angiogenesis Targeting Strategies. Eur. J. Cancer 2011, 47, 1276–1284.
- Spaeth, N.; Wyss, M.T.; Pahnke, J.; Biollaz, G.; Trachsel, E.; Drandarov, K.; Treyer, V.; Weber, B.; Neri, D.; Buck, A. Radioimmunotherapy Targeting the Extra Domain B of Fibronectin in C6 Rat Gliomas: A Preliminary Study about the Therapeutic Efficacy of Iodine-131-Labeled SIP(L19). Nucl. Med. Biol. 2006, 33, 661–666.
- Nam, L.; Coll, C.; Erthal, L.C.S.; De la Torre, C.; Serrano, D.; Martínez-Máñez, R.; Santos-Martínez, M.J.; Ruiz-Hernández, E. Drug Delivery Nanosystems for the Localized Treatment of Glioblastoma Multiforme. Materials 2018, 11, 779.
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21.
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-Cell RNA-Seq Highlights Intratumoral Heterogeneity in Primary Glioblastoma. Science 2014, 344, 1396–1401.
- Perrin, S.L.; Samuel, M.S.; Koszyca, B.; Brown, M.P.; Ebert, L.M.; Oksdath, M.; Gomez, G.A. Glioblastoma Heterogeneity and the Tumour Microenvironment: Implications for Preclinical Research and Development of New Treatments. Biochem. Soc. Trans. 2019, 47, 625–638.
More
Information
Subjects:
Medicine, Research & Experimental
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
361
Revisions:
2 times
(View History)
Update Date:
17 Jan 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No