Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Aleksandr Shikalov and Version 2 by Lindsay Dong.
Glioblastoma multiforme (GBM) is the most common type of glioma, with a median survival of 14.6 months post-diagnosis. Understanding the molecular profile of such tumors allowed the development of specific targeted therapies toward GBM, with a major role attributed to tyrosine kinase receptor inhibitors and immune checkpoint inhibitors. Targeted therapeutics are drugs that work by specific binding to GBM-specific or overexpressed markers on the tumor cellular surface and therefore contain a recognition moiety linked to a cytotoxic agent, which produces an antiproliferative effect.
- targeted therapy
- anticancer
- glioma
- brain tumors
1. Introduction
Glioma is the term referring to brain tumors that arise from glial or precursor cells. This group of oncological conditions includes astrocytoma, oligodendroglioma, ependymoma, oligoastrocytoma, and a few rare histologies. In medical practice, gliomas account for nearly 25% of all primary brain tumors [1]. According to the most recent WHO guideline for the classification of central nervous system (CNS) tumors, molecular, genetic, and epigenetic markers are used [2]; however, it is also possible to subgroup gliomas into a grading scale: low-grade gliomas (LGG, WHO grades 1–2) and high-grade gliomas (HGG, WHO grades 3–4).
The most common glioma is glioblastoma (GBM, WHO grade 4 astrocytoma), accounting for 14.3% of all primary brain and central nervous system neoplasms and 49.1% of all malignant brain tumors [1], with a poor prognosis for patients: the 5-year survival rate is at 6.8% [1] and it has a median survival of 14.6 months after diagnosis [3][4][3,4]. The patient’s prognosis remains weak despite conventional standard-of-care therapy that includes maximal tumor resection followed by radiotherapy and adjuvant temozolomide (TMZ) administration [3].
To better understand the molecular basis of GBM, extensive genomic [5] and transcriptomic [6] analyses were performed. In 2010, these allowed the classification of glioblastomas into subtypes according to their genetic expression profiles: classical (EGFR overexpression, CDKN2A deletion, and a lack of TP53 mutations), mesenchymal (altered NF1, PTEN mutations, a high activity of MET and CD44, and MERTK genes), proneural (altered PDGFRA, mutated TP53, point mutations in IDH1 and the increased expression of OLIG2), and neural (expression of neural markers such as NEFL, GABRA1, SYT1, and SLC12A5) [5]. Median survival rates of 11.5, 14.7 and 17.0 months for the mesenchymal, classical and proneural subtypes were determined.
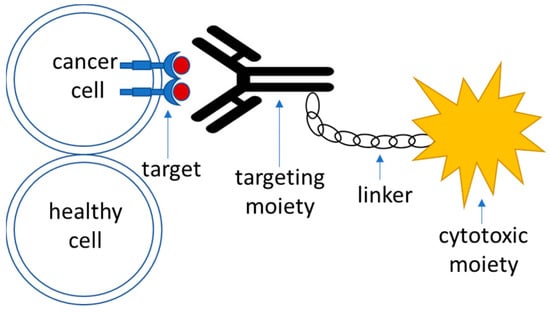
2. Conventional Standard of Care for Glioma Therapy
For glioblastoma patients, surgical resection of the tumor to the maximal feasible extent followed by focal tumor radiotherapy and concomitant temozolomide chemotherapy combined with additional irradiation therapy should be taken as the standard treatment (Stupp treatment) [3]. However, it is known that all glioblastomas will eventually relapse or progress [7][15]. For low-grade gliomas, the optimal treatment involves immediate surgical resection of the tumor to avoid further cancer progression and allow precise molecular characterization of the tumor [8][20], which is indeed crucial for the development of a treatment plan for any glioma [2][9][10][2,10,11]. For high-risk cohorts of low-grade-glioma-diagnosed patients (age > 40 years; patients who do not undergo gross total resection surgery [11][21]), postoperative care consists of 50–54 Gy radiotherapy followed by adjuvant therapy with DNA-alkylating or cytostatic agents. Lomustine, a DNA-alkylating agent, is usually preferred due to its tolerable toxicity and blood-brain barrier (BBB) permeation properties [12][22]. Taking the disease progression from low- to high-grades and the recurrent nature of gliomas [7][15], treatment options require constant attention and rapid development. There are several FDA-approved drugs and one medical device for glioma management aside from temozolomide (TMZ) [13][26]: lomustine [14][27], intravenous carmustine [15][28], carmustine wafer implants [16][29], bevacizumab [17][19], and tumor treatment fields [18][30]. These drugs and devices are mainly approved for the management of recurrent high-grade gliomas with only TMZ, carmustine wafer implants, and tumor treatment fields suitable for application in de novo diagnoses. Except for bevacizumab, FDA-approved drugs for gliomas represent the class of DNA-alkylating agents—their effect is not targeted on tumor cells; therefore, their application is linked with systemic adverse effects. Vascular endothelial growth factor (VEGF) was shown to be one of the key regulators of malignant angiogenesis in glioblastoma cells, while its inhibition with the help of antisense oligonucleotides correlated positively with reduced tumor growth and, indeed, diminished vasculature formation [19][31]. This discovery led to the FDA approval of bevacizumab, a monoclonal antibody that targets VEGF-A on the surface of endothelial cells and blocks its interaction with its receptors VEGFR1 and VEGFR2, becoming the first FDA-approved targeting therapy for use in recurrent GBM. Although bevacizumab demonstrated enhanced progression-free survival (PFS), it did not provide a benefit to overall survival (OS). This suggests that solely targeting the VEGFR axis is inadequate for suppressing tumor progression [20][21][32,33].3. Delivery of Therapeutic Drugs to the Brain and Associated Difficulties
3.1. Blood–Brain Barrier as an Obstacle for Drug Delivery
The blood–brain barrier poses a significant challenge in treating conditions within the central nervous system (CNS) and brain tumors are not an exception. The components of BBB include endothelial cells with tight junctions (specific for CNS), smooth muscle cells, microglial cells, astrocytes and pericytes [22][34]. Tight junctions in endothelial cells create a nearly impenetrable barrier, restricting the passage of small and lipid-soluble molecules through the paracellular route. Relevant to drug delivery, the BBB allows the free passage of hydrophilic molecules with a molecular weight of up to 150 Da and hydrophobic molecules with a molecular weight of 400–600 Da [23][37], while the penetration of large or hydrophilic drugs is limited. In order to travel through the BBB, the drug may utilize receptor-mediated transcytosis mechanism (e.g., with transferrin and insulin receptors [24][38]). P-glycoprotein-1 (P-gp), which can pump out more than 60% of the marketed drugs, makes it more difficult to penetrate the BBB [25][39]. The transcellular route of drug transport through the BBB is favorable for molecules with a low molecular weight (<500 Da) and high lipophilicity [26][49]. The relationship between the polar surface area and brain permeability is proved to be inverse—drug candidates that have >80 Å of polar surface and a tendency to form multiple H-bonds require higher free energy to penetrate lipophilic membranes of the BBB cells [26][27][28][49,50,51]. However, the predominant usage of less polar or highly lipophilic compounds also has its drawbacks as lipophilic molecules would aggregate bound to plasma proteins, effectively decreasing the amount of available active compound [29][52]. For the treatment of high-grade gliomas [30][54], only a small number of cytotoxic drugs are being used, such as TMZ, which allows about 20% penetration through the BBB at the administered systemic dose [31][55]. Other well-studied drugs such as paclitaxel (PTX), doxorubicin, and cisplatin, are not included in the standard-of-care treatment for GBM due to their poor BBB permeability, even though preclinical in vitro studies have shown an inspiring decrease in the viability of cancer cells using these drugs [32][33][34][56,57,58].3.2. Targeted Drug Delivery to the Brain
3.2.1. Passive Targeting
Passive targeting is a major strategy in drug development to achieve therapeutic effects while using systemic administration, particularly for drugs that need to reach the brain. This strategy takes advantage of the abnormal vessel formation in tumors, resulting in enlarged pore sizes in the vasculature endothelium at the tumor site. As a result, compounds that usually cannot cross the blood–brain barrier can be delivered to the tumor site with a higher probability [35][36][77,78]. This condition is known as the Enhanced Permeation and Retention (EPR) effect. Pore cutoff sizes differ drastically in healthy (4–25 nm) and brain tumor blood vessels (380–780 nm). Taking that into account, as well as the fact that lymphatic drainage is also impaired in brain tumors, drug delivery nanosystems that are designed to utilize the EPR effect for reaching the tumor site may be up to several hundred nanometers in diameter and could stay in the targeted area for longer periods of time [37][79]. The size of the nanostructures is a variable parameter in drug design and could be changed to increase the tumor permeation rate and decrease systemic toxicity [38][80]. Dual-modality nanosystems, designed for both the diagnostics and therapeutics of gliomas, such as QSC-LP liposomes loaded with quantum dots, supramagnetic iron oxide and cytotoxic peptide cliengitide, have shown promising results in vivo by significantly prolonging overall survival and diminishing tumor size in a xenograft mouse model [39][81].3.2.2. Mechanical Targeting (Local Delivery)
Local application of the therapeutic agents to the tumor site, which scholars will refer to as ‘mechanical’ targeting, could be achieved via intracerebroventricular injection, intratumoral injection, convection-enhanced delivery, and adding anti-cancer compounds intraoperatively into the post-lesion cavity, such as carmustine wafers [17][40][19,70]. In this case, as there is no need to pass the BBB and no application of drugs occurs in the circulation, therefore, the systemic toxic effects are significantly lower than in the case with intravenous application, as well as there being increased therapeutic efficacy of the active compounds as they are delivered locally to the site of the tumor [41][42][43][44][82,83,84,85].3.2.3. Active Targeting
Active targeting is a method of drug delivery that utilizes ligands (monoclonal antibodies and their derivatives, peptides, etc.) for tumor-specific recognition. Upon recognition of tumor-specific biomarkers which are represented by overexpressed or mutated surface proteins, the drug promotes its cytotoxic effect either directly or indirectly based on a type of drug system and its corresponding specifications. As an example, contrary to passive drug delivery through the EPR effect described above, targeting BBB-overexpressed glioma markers with corresponding ligands showed successful delivery to the tumor site in a phase I trial of the peptide–drug conjugate GRN1005 [45][67], as well as nanoparticles in preclinical in vitro [46][61] and in vivo studies [47][48][49][50][62,65,88,89].4. Drug Delivery Systems for Active Targeting of Brain Tumors
4.1. General Scheme for Drug Delivery Systems That Use Active Targeting
Targeted drug delivery systems that utilize active targeting strategy are generally composed of the following parts (Figure 1):-
Target—this is a moiety that is specific to the cell of interest—in this case, a tumor cell. It is required to be expressed or significantly overrepresented in the tumor over healthy cells; high-level expression is advantageous, although not necessary [51][90]. It could also represent a molecule within an altered biochemical pathway that is specific to a tumor cell.
-
Recognition moiety—a part of the drug delivery system that recognizes the target specifically and allows address delivery to a specified tumor site with minimal toxicity and off-target effects.
-
Linker—an engineered connective unit that ties the recognition part of the drug delivery system to the payload.
-
Payload—a cytotoxic agent that acts upon delivery to the tumor cells either directly or indirectly through the mediation of the host’s immune response.
Figure 1.
A schematic outline of the components of an active targeting drug delivery system.
4.2. Targets
Targeted glioma therapies commonly aim for the recognition/inhibition of the tumor-overexpressed variants of the receptor tyrosine kinase (RTK) family of transmembrane proteins that are responsible for cell growth and proliferation signaling. RTKs represent the most abundant and clinically relevant group of surface targets for GBM drug development [6]. In glioma initiation, several subclasses of RTKs are prone to alterations such as amplification and point mutations [6][52][6,91]. From a structural point of view, RTKs contain an extracellular domain required for ligand binding, a transmembrane domain, and an intracellular domain with tyrosine residues required for receptor activation upon phosphorylation [53][93]. The binding of receptor-specific ligands triggers dimerization of the RTK and subsequent autophosphorylation at intracellular tyrosine residues (activation sites). Activated residues act as landing pads for small G-proteins whose recruitment is necessary for downstream signaling through the RAF/MEK/ERK and PI3K/AKT pathways, which control tumor formation, cell survival, proliferation, and invasion [52][91]. In the context of GBM, alteration in the RTK/PI3K/AKT pathway, which is responsible for constitutive RTK signaling, is considered to be the most elevated core signaling pathway [54][92]. Alterations (mutations/deletions) in oncosuppressor genes such as TP53, NF1, and PTEN additionally promote RTK activity, which accelerates malignant progression and plays an important role in tumor initiation [54][55][92,94].4.3. Recognition Moiety
Recognition moieties in the development of targeted drug delivery systems are represented by several classes of molecules. As mentioned before, the recognition particle of the drug should be tumor-specific, with minimal off-target effects as well as no reactivity with healthy tissues [56][98]. Recognition moieties utilized in drug delivery systems include-
Monoclonal antibodies and their derivatives (such as antibody fragments (Fabs), single chain variable fragments (scFvs), and bispecific antibodies) [57][99]. The most frequently used format for the development of antibody–drug conjugates is IgG1 immunoglobulin, as it is both easy to manufacture as well as the fact that it shows a strong cytotoxic effect [58][100].
4.4. Linker
For antibody- and peptide-conjugated drugs, linkers could be classified into cleavable and non-cleavable subgroups. Cleavable linkers in conjugated drugs are developed for the cytotoxic drugs to be released upon cellular uptake of the drug conjugate, which reduces off-target drug release with associated unwanted toxicity in healthy tissues [65][66][114,115]. In general, there are three main triggers for linker cleavage and drug release for conjugated drugs: acid-cleavable linkers undergo degradation and drug release due to the usage of the difference between pH levels in plasma (approx. pH 7.4) and endosomal/lysosomal compartments of the tumor cells [67][68][116,117] that could be as low as pH 4.5–4.7 [69][118]; enzyme-cleavable linkers are being cleaved upon internalization in the endosomal or lysosomal compartment of the cell based on the linker structure, with help of intracellular cathepsin B and beta-glucuronidase enzymes [70][71][72][73][119,120,121,122], and redox-sensitive disulphide linkers undergo reductive cleavage based on the difference in the number of reducing equivalents in the form of glutathione between the plasma (2–20 μM/ L) and cytoplasm (0.5–10 mM) [74][123]. Non-cleavable linkers in the conjugated drugs require retention of the drug activity upon degradation of the recognition moiety within the endosomal compartment of the cells while the linker or its part stays intact. The advantage of using such types of drugs is associated with a decreased chance of off-target effects, as the drug is more likely to transform to its active form only upon internalization to the cells of interest [75][76][77][124,125,126].4.5. Payload (Cytotoxic Agent)
Cytotoxic payloads that are commonly used in the arsenal of targeted therapeutics include-
Microtubule assembly inhibitors: auristatins that inhibit tubulin polymerase and promote cell cycle arrest [82][137] such as monomethyl auristatin E (MMAE) [83][138] and monomethyl auristatin F (MMAF) [84][139], or maytansines and their derivatives that bind to tubulin and therefore interfere with the assembly of microtubules [85][140]. Common examples include mertansine (DM1) [86][87][88][141,142,143] or ravtansine (DM4) [89][90][144,145] as parts of the cytotoxic agents in targeted therapeutic drugs.
-
Bacterial toxins—toxic compounds produced by Pseudomonas aeruginosa (Pseudomonas Exotoxin A) and Corynebacterium diphtheria (Diphtheria toxin) are the most commonly used toxins of bacterial origin that are utilized for cancer therapies [91][146]. Both toxins act upon binding and irreversibly modify the eukaryotic EF2 elongation factor, which results in impaired protein synthesis and cell death [92][93][147,148].
-
Radioligands fused with peptides or antibodies with tumor specificity are used as a cytotoxic moiety for the targeted delivery of drugs to the tumor sites [94][95][96][97][71,72,150,151], which allows tumor-specific distribution of the radioactivity as compared to standard beam irradiation in the standard-of-care Stupp protocol [3].
-
Photodynamic therapy (PDT) is a therapeutic approach that requires the incorporation of non-toxic and inactive photosensitizer molecules into the cells of interest with subsequent light irradiation, in which the therapeutic molecules become activated. Upon irradiation, photosensitizer molecules transfer light energy and excitate molecular oxygen present in the surrounding tissues to a triplet or singlet state. In the triplet state, oxygen is capable of the generation of reactive oxygen species that react with molecules containing double bonds, leading to their damage and subsequent cell death [98][152]. In glioma studies and treatment, PDT is implemented via placing fiberoptics at the site of the tumor in order to irradiate cells within the brain that have obtained photosensitizer molecules [99][100][153,154].
-
Oncolytic virotherapy is a novel approach for the targeted therapy of neoplasms that utilizes viruses for the elimination of tumor cells. While the exact mechanisms and the nature of tropism for such viruses might still be unknown, the main theories involve either direct killing of the targeted cells or indirect modulation of the host’s immune system response in order to mediate immunogenic cytotoxicity [101][102][158,159].