Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Juergen Arnhold | -- | 3542 | 2024-01-08 11:04:41 | | | |
| 2 | Wendy Huang | Meta information modification | 3542 | 2024-01-09 12:32:30 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Arnhold, J. Chronic Inflammatory States and Altered Conditions in Tumors. Encyclopedia. Available online: https://encyclopedia.pub/entry/53545 (accessed on 05 July 2026).
Arnhold J. Chronic Inflammatory States and Altered Conditions in Tumors. Encyclopedia. Available at: https://encyclopedia.pub/entry/53545. Accessed July 05, 2026.
Arnhold, Jürgen. "Chronic Inflammatory States and Altered Conditions in Tumors" Encyclopedia, https://encyclopedia.pub/entry/53545 (accessed July 05, 2026).
Arnhold, J. (2024, January 08). Chronic Inflammatory States and Altered Conditions in Tumors. In Encyclopedia. https://encyclopedia.pub/entry/53545
Arnhold, Jürgen. "Chronic Inflammatory States and Altered Conditions in Tumors." Encyclopedia. Web. 08 January, 2024.
Copy Citation
In tumor cells, enhanced levels of cytotoxic agents are usually counteracted by an overexpression of protective mechanisms. In this manner, tumor cells can even survive therapeutically induced stress situations. Tumor cells also affect immune cells and other cells in their close neighborhood in such a way that cells in this tumor microenvironment change their properties and promote tumor progression. Numerous examples are given for how the disturbed balance between inflammation-associated cytotoxic agents and antagonizing principles is related to tumor growth, tumor cell invasion, and metastasis.
cytotoxic agents
antagonizing principles
chronic inflammation
hypoxia
tumor cells
tumor microenvironment
immunosuppression
matrix remodeling
1. Introduction
Inflammation is related to all stages of tumorigenesis, such as the initiation, promotion, progression, and metastasis [1][2][3]. Inflammatory processes are primarily directed to eliminate by means of the immune systems and the activation of components in the acute phase, complement, coagulation, and contact systems any harm from the host that can disturb normal functioning and tissue homeostasis [4][5].
In tumors, the associated inflammatory process is persistent [6]. During tumor development and the interaction between immune and tumor cells, three major phases can be distinguished, according to the concept of immunoediting [3][7]. These phases are schematically presented in Figure 1. The first phase, the elimination phase, corresponds to the original concept of immune surveillance, suggesting that immune cells are able to eliminate transformed cells [8][9]. Natural killer (NK) cells and cytotoxic T lymphocytes (CTLs) are the key players in the recognition and killing of transformed cells [10][11]. It is generally assumed that the formation and elimination of unwanted transformed cells occurs often in our organism.
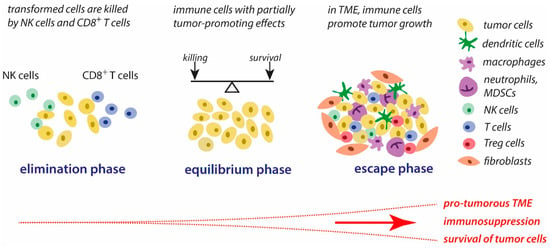
Figure 1. Concept of immunoediting in tumorigenesis. The three major phases of the interplay between tumor cells and immune cells are schematically presented. Further explanations are given in the text. Abbreviations: MDSCs—myeloid-derived suppressor cells, NK cells—natural killer cells, TME—tumor microenvironment, Tregs—regulatory T cells.
During the second phase, the equilibrium phase, the elimination of tumors cells and the development of tumor-mediated immune suppressing strategies co-exist. Thus, the elimination of unwanted cells is impeded, and part of these cells can survive. In this phase, tumor immunogenicity is reduced, and a pro-inflammatory tumor microenvironment (TME) is generated that promotes immune tolerance [3].
These alterations in the immune response are gradually strengthened to the immune escape that is characteristic of the third phase, the escape phase. As a result, the immune response is manipulated in multiple ways by tumor cells and the TME, ensuring that tumor growth and metastasis can occur and the processes of elimination of tumor cells by the host’s immune system are slowed down. The progressive development of tumor-mediated immune escape is closely associated with major changes in the tumor cell properties and conditions in the TME, such as increasing the deficiency of dioxygen, the accumulation of lactate, altered routes in the supply of energy substrates, the activation of stress-related genes and oncogenes, nutrient depletion, reversed pH gradient on tumor cells, and matrix remodeling. In general, these altered conditions promote the survival of tumor cells, favor the suppression of immune defense reactions against tumor components, elevate proliferative activities, and contribute to tumor progression [3][12].
During the gradual development of tumors, many immunological relevant factors are converted from an anti-tumor behavior in the early stages to tumor-promoting properties in advanced tumors. Concurrently, many therapeutic procedures are increasingly restricted in their efficiency during tumor progression and fail in the late stages. In cancer research, the focus is, therefore, directed to study and identify the immunosuppressive mechanisms in late-stage tumors with the hope of finding novel ways for therapeutical intervention [12][13][14][15].
In tumor-associated inflammations, host-derived cytotoxic agents originate from activated immune cells, damaged tissue cells in the TME, dysfunctional cellular processes in tumor cells, and can additionally result from therapeutic applications like radio- and chemotherapy. To prevent the disastrous action of these cytotoxic agents, numerous ready-to-use protective mechanisms exist that neutralize these cytotoxic agents. Disturbances in the control of host-derived cytotoxic agents by antagonizing principles are associated with the development of chronic inflammatory disease states [16].
2. Immunosuppression during Acute and Chronic Inflammation
Regarding the time elapsed, an acute inflammation can be subdivided into two major phases with distinct differences in the mediator profile and immune cell functions. First, in response to pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), pro-inflammatory cascades are induced via the activation of pattern recognition receptors (PRRs) [17][18]. As a result, immune cells are recruited and activated onto inflammatory sites and the maturation of dendritic cells is induced for the presentation of antigens to T cells [19]. Typical pro-inflammatory mediators are cytokines, such as interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor α (TNF-α), and the acute-phase proteins C-reactive protein (CRP) and serum amyloid A (SAA) [4][5][20][21][22]. These activities are directed to deactivate pathogens, eliminate virus-infected or transformed cells, remove any damaged cell material, and create better conditions for ongoing immune reactions. This very common concept of pattern recognition is valid for the initiation of novel inflammatory events in a broad range of diseases, including infections, rheumatoid arthritis, atherosclerosis, Alzheimer’s disease, and many others, as well as tumors.
During inflammatory response, different host-derived cytotoxic agents are released or generated, which are derived from activated immune and undergoing tissue cells, and in tumors also from tumor cells. These agents generally execute a dual role in living tissues. They are an essential part in many physiological functions, especially in immune cell-triggered processes. Cytotoxic agents are mandatory for combatting microbes, to restore tissue homeostasis after any threat, and to ensure normal functioning of the physiological processes in the organism [4]. Otherwise, they can damage unperturbed tissues and contribute to the initiation of novel inflammatory cascades. According to their mode of action, cytotoxic agents can be differentiated into oxidant-based agents (reactive species, oxidized heme proteins, free heme, transition metal ions) and protease-based agents (serine proteases, matrix metalloproteases, pro-inflammatory peptides) [16]. In healthy tissues, cytotoxic agents are inactivated by already existing antagonizing principles.
In the second phase of an acute inflammation, the termination or resolution phase, destroyed cells and tissues are replaced by novel synthesized material via the induction of proliferative processes. The pro-inflammatory activities of immune cells are downregulated, and finally the former homeostasis is restored. The typical transiently acting cytokines of this phase are transforming growth factor β (TGF-β) and IL-10 [23][24][25]. In addition, growth factors [26][27] and lipid mediators, such as lipoxins and resolvins, are involved in the resolution of inflammation [28][29]. Cell signaling is characterized by the induction of the STAT3 pathway. Macrophages are polarized from the M1 type to M2 type [30][31]. The accumulation of myeloid-derived suppressor cells (MDSCs) contributes to the development of a transient immunosuppression in inflamed areas [32][33]. MDSCs are immature immune cells that promote immunosuppressive effects on lymphocytes, natural killer cells, macrophages, and dendritic cells [34]. In general, the aforementioned mechanisms impede the hyperactivation of pro-inflammatory cells and prevent any excessive tissue damage by host-derived cytotoxic agents during the resolution of inflammation. After the resolution, the number of immune cells and mediators is reduced to a level typical of healthy tissues [35]. Cytotoxic agents and their antagonizing counterparts are also involved in the resolution of inflammation, especially in the processes of matrix remodeling.
Under chronic inflammatory conditions, inflammation is only insufficiently terminated as pro-inflammatory cascades are activated again and again due to the incomplete inactivation of host-derived cytotoxic agents and the continuing release of DAMPs and antigens. At chronic inflammatory sites, pro-inflammatory mediators, the recruitment of immune cells, and tissue damage co-exist with a plethora of immunosuppressive mechanisms, such as the prevalence of MDSCs, the presence of inflammation-resolving and proliferative factors, as well as the depletion of essential metabolites for appropriate immune defense [16]. The resulting long-lasting immunosuppression can seriously affect the health status and enhance susceptibility to chronic infections and comorbidities [36][37]. For example, in elderly persons, persistent inflammation is closely associated with immune dysfunction. This condition is known as inflammaging [38], which is regarded as a risk factor for life-threatening diseases and adverse health outcomes [39][40]. In general, pro-inflammatory mechanisms and immunosuppressive conditions are closely associated with each other in many chronic diseases, including cancer.
In chronic inflammations, serious health problems can arise from the excessive release or generation of cytotoxic agents and from the decline or exhaustion of protective mechanisms. In consequence, the following release of novel DAMPs, antigens, and cytotoxic agents promotes ongoing inflammatory events and prevents the termination of inflammatory cascades. The worst case is a very low capacity of antagonizing principles or their exhaustion with the consequence of septic complications and organ failure [16]. This concept of the incomplete inactivation of cytotoxic agents by protective principles explains under which conditions an inflammation becomes persistent. It also provides the basis for a better understanding of the underlying molecular mechanisms for a wide range of chronic inflammatory diseases, including cancer.
In the description of the molecular processes during tumorigenesis, numerous cytotoxic agents derived from tumor cells, immune cells, and other tumor-associated cells are more expressed and exhibit higher activities compared to healthy tissue areas. In general, these agents are able to damage transformed cells with the subsequent elimination of these cells by the immune system. This is mostly observed during the early phases of tumorigenesis. However, in response to stress and the increasing accumulation of cytotoxic agents, numerous antagonizing principles are markedly upregulated in advanced tumors and promote the survival of tumor cells and protection against stress induced by chemo- and radiotherapy.
3. Key Elements of Immunosuppression in Tumors
Tumor progression and chronic inflammatory processes are highly linked to each other [1][2][3][6]. In the creation of an immunosuppressive milieu, both tumor cells and the TME contribute multiple mechanisms to the manipulation of the host’s immune answer and to tissue remodeling in tumors. An overview of the major mechanisms of tumor cell-mediated processes in the TME and immunosuppression is given in Figure 2.
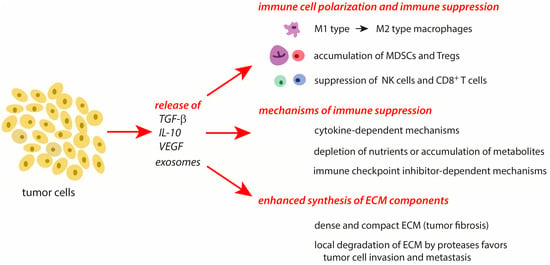
Figure 2. Major effects of tumor cells on the surrounding cells in the TME. The effects are mediated by the release of mediators from tumors cells and special conditions typical of tumors. Further explanations are given in the text. Abbreviations: IL-10—interleukin 10, MDSCs—myeloid-derived suppressor cells, NK cells—natural killer cells, TGF-β—transforming growth factor-β, Tregs—regulatory T cells, VEGF—vascular endothelial growth factor.
During tumor progression, the principal processes involved in immunosuppression and tissue remodeling during the resolution phase of an acute inflammation are also active. Whereas these mechanisms are only transiently expressed in a subsiding inflammation, they act permanently and more intensively in tumors. In cancer cells, several protective systems are highly upregulated in response to a stress-mediated increase of cytotoxic agents. Further, these cells actively secrete inflammation-resolving molecules (TGF-β, IL-10) and exosomes that affect the conditions and properties of immune cells in the TME [41]. Importantly, natural killer cells (NK) and cytotoxic T cells, which are able to deactivate tumor cells, are suppressed in their activity by the release of TGF-β [42][43][44]. In the TME, tumor-associated macrophages (TAMs) are triggered from the pro-inflammatory M1 type to the immune-resolving M2 type [45]. Regulatory T cells (Treg) and various types of myeloid-derived suppressor cells (MDSCs) accumulate in the TME during tumor progression. These cell types suppress the activation of other immune cells and contribute to the survival of tumors [3][13][32][35][41][46][47][48]. With their cargo, cancer cell-derived exosomes are able to modulate different processes of tumor progression such as angiogenesis, metastasis, and survival [41].
MDSCs are formed as all other myeloid cells from precursors in the bone marrow. Under inflammatory conditions, the need for novel immune cells increases and these cells undergo a forced formation process, known as emergency myelopoiesis, leading to immature properties. In most kinds of cancers, the accumulation of granulocytic MDSCs (G-MDSCs) dominates over monocytic MDSCs (M-MDSCs) [49]. In glioma, M-MDSCs are more expressed than G-MDSCs [49]. In humans, a third, less pronounced type of MDSCs, early-stage MDSCs (e-MDSCs), has been described [50][51]. The MDSC subtypes are usually identified by their morphological properties, density, and surface markers [49].
In the TME, immunosuppressive conditions can be differentiated by their underlying mechanisms as enzyme-dependent, cytokine-dependent, and immune checkpoint-dependent [14]. In addition, the expression of neoantigens and antigen-presenting molecules like MHC-1 are decreased in cancer cells [52][53].
Enzyme-dependent immunosuppressive pathways are characterized by a depletion of essential metabolites, such as ATP and tryptophan, or by an enhanced formation of metabolites like adenosine, citrulline, kynurenine, and prostaglandin E2 [14][54][55]. Several proteins (for example ectonucleotidases CD39 and CD73 [56] and indoleamine 2,3-dioxygenase [57]), which are involved in these pathways, are highly upregulated in tumors.
Cytokine-dependent immunosuppression can be induced by the activation of the IL-6/STAT-3 pathway that inhibits DC maturation and activates effector T cells [58][59]. Vascular endothelial growth factor A (VEGF-A) is immunosuppressive by the inhibition of the DC functions and maturation, by the infiltration of immunosuppressive cells (Treg, MDSC, and TAMs) into tumors, by reducing cytotoxic CD8+ T cell infiltration into tumors, and by the expression of the factors involved in the exhaustion of these cells [60][61][62]. Cytokines of the IL-10 and TGF-β pathways activate Treg and promote immunosuppression [63].
Several immune checkpoints are known to be involved in immunosuppressive activities. The inactivation of T cells is possible via the binding of a specific ligand to the inhibitory T cell receptor programmed cell death protein 1 (PD-1) [14]. This ligand known as PD-L1 or CD274 is expressed on the surface of a tumor and various other cells. The overexpression of PD-L1 on tumor cells is associated with a poor prognosis and the evasion of T cell recognition of cancers [64]. The PD-1/PD-L1 axis represents an important element in the generation of an immunosuppressive TME [14]. Other inhibitory immune checkpoint molecules on T cells are cytotoxic T lymphocyte-associated protein 4 (also known as CD 152), LAG3, TIM-3, and the tyrosine-based inhibitory domain (TIGIT) [65][66][67]. Cancer cells can express TIGIT ligands, such as CD155, and overcome cancer immunity.
In growing and metastasizing tumors, the balance between tumor-promoting and tumor-destroying factors is shifted towards the first types of factors. Although, many features of tumor-induced immunosuppression are known, this knowledge is incomplete concerning the interrelations between them and the consequences for further tumor processing. Numerous anti-tumor therapies, such as targeted therapies and chemotherapies, attempt to overcome immunosuppression in tumors. The application of immune checkpoint inhibitors provides a novel tool for the development of combined therapy approaches [14].
4. Poor Quality of the Tumor Vasculature
Tumorigenesis starts with the formation and uncontrolled growth of transformed cells. In developing tumors, the supply with dioxygen and nutrients is diminished due to the poor quality of the tumor vasculature, longer diffusion paths, insufficient lymphatic drainage, fluctuations in interstitial pressure, and intermittent vascular collapse in contrast to healthy tissues [68][69].
In solid tumors, the growth factor VEGF is overexpressed, whereby hypoxia promotes this expression [70][71]. This growth factor is responsible for the poor quality of the tumor vasculature with irregular, leaky, and immature vessels [72][73]. In addition, the VEGF is a key angiogenic factor in tumors and is involved in tumor progression and metastasis [73][74].
5. Direct Effects of Hypoxia in Tumor Cells
In tumors, hypoxia [75][76][77] provokes significant metabolic alterations in tumor cells, such as the activation and stabilization of hypoxia-inducible factor 1α (HIF-1α) [78], upregulation of glycolysis [79], downregulation of oxidative phosphorylation in the mitochondria [79], and enhanced formation of reactive species by dysfunctional mitochondria [80]. In addition, lactate accumulates [81][82] and induces numerous lactate-driven effects [81][83][84].
Cytosolic HIF-1α is a master regulator of glycolysis in many cells. This usually short-lived factor is controlled by prolyl hydroxylase that tags HIF-1α for proteasome degradation [85][86]. Hypoxia, stress-induced oxidation of Fe2+ in propyl hydroxylase, and an ascorbate deficiency prevent HIF-1α degradation [78]. As a result, HIF-1α upregulates several enzymes promoting glycolysis and downregulates pyruvate dehydrogenase that supplies acetyl-coenzyme A (Ac-CoA) for the citrate cycle [79]. Unlike HIF-1α, which is a ubiquitous protein, HIF-2α is predominantly expressed in highly vascularized tissues [87]. HIF-2α is active during prolonged hypoxia, replaces HIF-1α in a spatiotemporal manner [75][88], and is involved in controlling oxidative stress, the cell cycle, blood vessel remodeling, and RNA transport [89].
In healthy cells, lipogenesis is mostly induced by pyruvate-derived Ac-CoA. To ensure lipogenesis in hypoxic cancer cells, decreased pyruvate is replaced in the TCA cycle by glutamine, supporting the conversion of α-ketoglutarate into citrate and Ac-CoA [90]. In cancer cell growth, glutamine is an important carbon and nitrogen source for lipid, amino acid, and nucleotide synthesis [91].
In tumorigenesis, increased lactate production by tumors cells reprograms macrophages, T cells, and other immune cells in a way that they are immunosuppressive and anti-inflammatory [81][82][83][84]. Lactate also promotes angiogenesis and tumor progression [83][92], supporting the hyaluronan release from the adjacent fibroblasts. This hyaluronan cover protects additional tumor tissue from immune attacks [93].
6. Stress-Related Responses in Tumor Cells
In tumor cells, enhanced intracellular levels of reactive species, which result mainly from dysfunctional mitochondria, activate the transcription factor nuclear factor-erythroid 2-related factor 2 (Nrf2) that is low expressed under normal physiological conditions [94][95]. In cancer cells, Nrf2 promotes the syntheses of numerous enzymes involved in antioxidative defense and contributes to a resistance against chemo- and radiotherapy.
Prolonged hypoxia also mediates the processes of autophagy [96][97]. In the early stages of tumorigenesis, autophagy is directed to inhibit tumor growth [98][99][100]. Otherwise, in late-stage cancers, autophagy is known to stabilize cancer cells by maintaining the integrity of the mitochondria, reducing DNA damage, and increasing the resistance against stress. Under hypoxic conditions and a low supply of nutrients, autophagy can provide energy resources for the survival of cancer cells and resistance against chemotherapy [101]. With these mechanisms, autophagy contributes to tumor development [102][103][104] and facilitates metastasis [105][106][107][108].
Under stress situations, the percentage of misfolded proteins increases in the endoplasmic reticulum (ER) as a result of the action of reactive species and electrophiles. Misfolded proteins are subjected to ubiquitinylation with subsequent proteasomal degradation or autophagy. Three major stress sensors of an unfolded protein response (UPR) are activated by misfolded proteins in a wide range of tumor cells [109]. The overexpression of chaperones or mutations in the UPR pathways are used by tumor cells to antagonize ER stress [110][111][112][113]. A hyperactive UPR promotes tumorigenesis in advanced cancers [114].
To maintain a reducing environment in the cytosol of tumor cells, the enhanced uptake of cystine/cysteine by tumors and the conversion into glutathione are mandatory to protect cells from stress-mediated cell death [115]. In tumors, highly increased extracellular cysteine levels are observed, whereas cysteine is slightly enhanced within tumor cells [116]. Moreover, cysteine is mainly transported into tumor cells by the cystine/glutamate antiporter solute carrier family 7 member 11 (SLC7A11), which is widely expressed in human cancers [117][118]. In tumor cells, the conversion of cysteine into hydrogen sulfide activates the Nrf2-mediated gene expression of antioxidative proteins [119][120]. Hydrogen sulfide also accelerates the cell cycle in tumor cells [121].
In addition, a close link exists between the upregulation of reductive glutamine metabolism, also known as reductive carboxylation, and the transport of NADPH from cytosol into the mitochondria to upregulate mitochondrial GSH and protect against reactive species [122]. These pathways are important for anchorage-independent growth in cancer cells.
7. Intratumoral Hemorrhages
The rupture of blood vessels results in intratumoral hemorrhages, which are typical of many tumors [123][124]. The conditions in hemorrhages promote the formation of cytotoxic free heme and in turn the transcriptional processes in tumor cells via the binding of free heme to guanine-rich DNA and RNA structures (known as G4 elements) [125][126]. Free heme controls the expression of key target genes, including telomeres and oncogenes (such as c-Myc), and favors malignant transformation [127][128].
8. Reversed pH Gradient in Cancer Cells
In cancer cells, the cytosolic pH is enhanced to 7.3–7.6 (versus about 7.2 in normal cells), whereas the extracellular pH is decreased to 6.8–7.0 (versus about 7.4 in normal cells) [129][130]. In the TME, the milieu is more acidic by about 0.3–0.7 pH units in comparison to the cytosolic pH of tumors cells [131]. Thus, the pH gradient across the plasma membrane of cancer cells is reversed in contrast to healthy cells. These deviations are mediated by hypoxia and lactate effects and accompanied by an increased expression and activity of ion transporters in the plasma membrane and intracellular pH regulators [132].
In many cell types, enhanced intracellular pH values by ~0.3–0.4 units are found in proliferating cells at the end of the S phase [133][134] and in migrating cells [135][136]. The reverse pH gradient in cancer cells promotes cell proliferation, survival, migration, and metastasis [129][130][137][138].
9. Matrix Remodeling in the TME
In the TME, the extracellular matrix (ECM) acts as a barrier around tumor cells against cytotoxic immune cells. Under hypoxic conditions, the stiffness of the matrix components, especially of collagen fibrils, increases due to more intense cross-linking reactions between matrix polymers [139][140]. These alterations are mainly caused by cancer-associated fibroblasts [141]. Several enzymes involved in matrix remodeling and cross-linking reactions are upregulated in tumors, namely lysyl oxidase, lysyl oxidase-like proteins, collagen prolyl 4-hydroxylase, and WINT1-inducible signaling pathway protein 1 (WISP1) [142][143][144][145].
Tumor fibrosis is closely associated with an overexpression of these enzymes [146]. As a result, ECM fibers, most of all collagens I and III, are converted into dense, linearized, and cross-linked fiber bundles with altered mechanical properties [147][148][149][150]. The enhanced stiffness of the ECM components favors the migration of single tumor cells, promotes angiogenesis, and dampens anti-tumor activities [151]. The elevated degree of tumor fibrosis is associated with a poor prognosis in many types of cancer [151][152][153].
In the TME, the deposition of the matrix components is important for angiogenesis, proliferation, tumor cell invasion, and metastasis. Several matrix metalloproteases such as MMP-2, -3, -9, and -14 are upregulated in malignant tumors [154][155]. An enhanced expression of heparinase also favors angiogenesis [156][157][158].
The enhanced proliferation of tumor cells is also supported by increased hyaluronan production [159][160][161]. Under hypoxic conditions, the production of hyaluronan is markedly increased by tumor cells [162]. M2-type macrophages accumulate preferentially in hyaluronan-rich areas of the TME [163]. In addition, hyaluronan favors the phenotype change in monocytes and macrophages to the M2-type [164].
References
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545.
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444.
- Nakamura, K.; Smyth, M.J. Targeting cancer-related inflammation in the era of immunotherapy. Immunol. Cell Biol. 2017, 95, 325–332.
- Arnhold, J. Immune response and tissue damage. In Cell and Tissue Destruction. Mechanisms, Protection, Disorders; Academic Press: London, UK; San Diego, CA, USA; Cambridge, MA, USA; Oxford, UK, 2020; pp. 155–204.
- Arnhold, J. Acute-phase proteins and additional protective systems. In Cell and Tissue Destruction. Mechanisms, Protection, Disorders; Academic Press: London, UK; San Diego, CA, USA; Cambridge, MA, USA; Oxford, UK, 2020; pp. 205–228.
- Wang, D.; DuBois, R.N. Immunosuppression associated with chronic inflammation in the tumor microenvironment. Carcinogenesis 2015, 36, 1085–1093.
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998.
- Burnet, M. Cancer; a biological approach. I. The process of control. II. The significance of somatic mutation. Br. Med. J. 1957, 1, 779–786.
- Thomas, L. On immunosurveillance in human cancer. Yale J. Biol. Med. 1982, 55, 329–333.
- Shimasaki, M.; Jain, A.; Campana, D. NK cells for cancer immunotherapy. Nat. Rev. Drug Discov. 2020, 19, 200–218.
- Raskov, H.; Orhan, A.; Christensen, J.P.; Gögenur, I. Cytotoxic CD8+ T cells in cancer and cancer immunotherapy. Br. J. Cancer 2021, 124, 359–367.
- Nakamura, K.; Smyth, M.J. Myeloid immunosuppression and immune checkpoints in the tumor microenvironment. Cell Mol. Immunol. 2020, 17, 1–12.
- Labani-Motlagh, A.; Ashja-Mahdavi, M.; Loskog, A. The tumor microenvironment: A milieu hindering and obstructing antitumor immune response. Front. Immunol. 2020, 11, 940.
- Barnestein, R.; Galland, L.; Kalfeist, L.; Ghiringhelli, F.; Ladoire, S.; Limagne, E. Immunosuppressive tumor microenvironment modulation by chemotherapies and targeted therapies to enhance immunotherapy effectiveness. OncoImmunology 2022, 11, 2120676.
- Tie, Y.; Tang, F.; Wei, Y.-Q.; Wie, X.-W. Immunosuppressive cells in cancer: Mechanisms and potential therapeutic targets. J. Hematol. Oncol. 2022, 15, 61.
- Arnhold, J. Host-derived cytotoxic agents in chronic inflammation and disease progression. Int. J. Mol. Sci. 2023, 24, 3016.
- Matzinger, P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 1994, 12, 991–1045.
- Janeway, C.A.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216.
- Suresh, R.; Moser, D.M. Pattern recognition in innate immunity, host defense, and immunopathology. Adv. Physiol. Educ. 2013, 37, 284–291.
- Pepys, M.B.; Baltz, M.I. Acute phase proteins with special reference to C-reactive protein and related proteins (pentraxins) and serum amyloid A protein. Adv. Immunol. 1983, 34, 141–212.
- Vandivier, R.W.; Henson, P.M.; Douglas, I.S. Burying the death: The impact of failed apoptotic cell removal (efferocytosis) on chronic inflammatory lung disease. Chest 2006, 129, 1673–1682.
- Pober, J.S.; Sessa, W.C. Inflammation and the blood microvascular system. Cold Spring Harbor Perspect. Biol. 2015, 7, 016345.
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.K.; Flavell, R.A. Transforming growth factor-beta regulation of immune response. Annu. Rev. Immunol. 2006, 24, 99–146.
- Li, M.O.; Flavell, R.A. Contextual regulation of inflammation: A duet of transforming growth factor-beta and interleukin-10. Immunity 2008, 28, 468–476.
- Couper, K.N.; Blount, D.G.; Riley, E.M. IL-10: The master regulator of immunity to infection. J. Immunol. 2008, 180, 5771–5777.
- Landén, N.X.; Li, D.; Ståhle, M. Transition from inflammation to proliferation: A critical step during wound healing. Cell Mol. Life Sci. 2016, 73, 3861–3885.
- Marega, M.; Chen, C.; Bellusci, S. Cross-talk between inflammation and fibroblast growth factor 10 during organogenesis and pathogenesis: Lessons learnt from the lung and other organs. Front. Cell Dev. Biol. 2021, 9, 656883.
- Serhan, C.N.; Savill, J. Resolution of inflammation: The beginning programs the end. Nat. Immunol. 2005, 6, 1191–1197.
- Chandrasekharan, J.A.; Sharma-Walia, N. Lipoxins: Nature’s way to resolve inflammation. J. Inflamm. Res. 2015, 8, 181–192.
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737.
- Martinez, F.O.; Gordon, S.; Locati, M.; Montavani, A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: New molecules and patterns of gene expression. J. Immunol. 2006, 177, 7303–7311.
- Sanchez-Pino, M.D.; Dean, M.J.; Ochoa, A.C. Myeloid-derived suppressor cells (MDSC): When good intentions go awry. Cell Immunol. 2021, 362, 104302.
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 485–498.
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumor progression. Br. J. Cancer 2019, 120, 16–25.
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174.
- Sepkowitz, K.A. Opportunistic infections in patients with and patients without acquired immunodeficiency syndrome. Clin. Infect. Dis. 2002, 34, 1098–1107.
- Kampitak, T.; Suwanpimolkul, G.; Browne, S.; Suankratay, C. Anti-interferon-autoantibody and opportunistic infections: Case series and review of the literature. Infection 2011, 39, 65–71.
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Beneticits, G. Inflammaging—An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254.
- Newman, A.B.; Sanders, J.L.; Kizer, J.R.; Boudrou, R.M.; Odden, M.C.; Zeki Al Hazzouri, A.; Arnold, A.M. Trajectories of function and biomarkers with age: The CHS All stars study. Int. J. Epidemiol. 2016, 45, 1135–1145.
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 1135–1145.
- Othman, N.; Jamal, R.; Abu, N. Cancer-derived exosomes as effectors of key inflammation-related players. Front. Immunol. 2019, 10, 2103.
- Wrzesinski, S.H.; Wan, Y.Y.; Flavell, R.A. Transforming growth factor-β and immune response: Implications for anticancer therapy. Clin. Cancer Res. 2007, 13, 5262–5270.
- Bierie, B.; Moses, H.L. Transforming growth factor β (TGF-β) and inflammation in cancer. Cytokine Growth Factor Rev. 2010, 21, 49–59.
- Szczepanski, M.J.; Szajnik, M.; Welsh, A.; Whiteside, T.L.; Boyiadzis, M. Blast-derived microvesicles in sera from patients with acute myeloid leukemia suppress natural killer cell function via membrane-associated transforming growth factor-beta 1. Haematologia 2011, 96, 1302–1309.
- He, Z.; Zhang, S. Tumor-associated macrophages and their functional transformation in the hypoxic tumor microenvironment. Front. Immunol. 2021, 12, 741305.
- Ostrand-Rosenberg, S.; Sinha, P. Myeloid-derived suppressor cells: Linking inflammation and cancer. J. Immunol. 2009, 182, 4499–4506.
- Umansky, V.; Blattner, C.; Gebhardt, C.; Utikal, J. The role of myeloid-derived suppressor cells (MDSC) in cancer progression. Vaccines 2016, 4, 36.
- Hedge, S.; Leader, A.M.; Merad, M. MDSC: Marker, development, states, and unaddressed complexity. Immunity 2021, 54, 875–884.
- Cassetta, L.; Bruderek, K.; Skrzeczynska-Moncznik, J.; Osiecka, O.; Hu, X.; Rundgren, I.M.; Lin, A.; Santegoets, K.; Horzum, U.; Godinhi-Santos, A.; et al. Differential expansion of circulating human MDSC subsets in patients with cancer, infection and inflammation. J. Immunother. Cancer 2020, 8, e001223.
- Casetta, L.; Baekkevold, E.S.; Brandau, S.; Bujko, A.; Casatella, M.A.; Dorhoi, A.; Krieg, C.; Lin, A.; Loré, K.; Marini, O.; et al. Deciphering myeloid-derived suppressor cells: Isolation and markers in humans, mice and non-human primates. Cancer Immunol. Immunother. 2019, 68, 687–697.
- Bronte, V.; Brandau, S.; Chen, S.-H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150.
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017, 8, 1136.
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.M.C.S.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198.
- Molinier-Frenkel, V.; Castellano, F. Immunosuppressive enzymes in the tumor microenvironment. FEBS Lett. 2017, 591, 3135–3157.
- Bader, J.E.; Voss, K.; Rathmell, J.C. Targeting metabolism to improve the tumor microenvironment for cancer immunotherapy. Mol. Cell 2020, 78, 1019–1033.
- Allard, B.; Longhi, M.S.; Robson, S.C.; Stagg, J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol. Rev. 2017, 276, 121–144.
- Moon, Y.W.; Hajjar, J.; Hwu, P.; Naing, A. Targeting the indoleamine 2,3-dioxygenase pathway in cancer. J. Immunother. Cancer 2015, 3, 51.
- Kitamura, H.; Ohno, Y.; Toyoshima, Y.; Ohtake, J.; Homma, S.; Kawamura, H.; Takahashi, N.; Taketomi, A. Interleukin-6/STAT3 signaling as a promising target to improve the efficacy of cancer immunotherapy. Cancer Sci. 2017, 108, 1947–1952.
- Park, S.-J.; Nakagawa, T.; Kitamura, H.; Atsumi, T.; Kamon, H.; Sawa, S.-I.; Kamimura, D.; Ueda, N.; Iwakura, Y.; Ishihara, K. IL-6 Regulates in vivo dendritic cell differentiation through STAT3 activation. J. Immunol. 2004, 1950, 3844–3854.
- Voron, T.; Colussi, O.; Marcheteau, E.; Pernot, S.; Nizard, M.; Pointet, A.-L.; Latreche, S.; Bergaya, S.; Benhamouda, N.; Tanchot, C. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J. Exp. Med. 2015, 212, 139–148.
- Ohm, J.E.; Carbone, D.P. VEGF as a mediator of tumor-associated immunodeficiency. Immunol. Res. 2001, 23, 263–272.
- Huang, H.; Langenkamp, E.; Georganaki, M.; Loskog, A.; Fuchs, P.F.; Dieterich, L.C.; Kreuger, J.; Dimberg, A. VEGF suppresses T-lymphocyte infiltration in the tumor microenvironment through Inhibition of NF-κB-induced endothelial activation. FASEB J. 2015, 29, 227–238.
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089.
- Patel, S.P.; Kurzrock, R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856.
- Walunas, T.L.; Lenschow, D.J.; Bakker, C.Y.; Linsley, P.S.; Freeman, G.J.; Green, J.M.; Thompson, C.B.; Bluestone, J.A. CTLA-4 can function as a negative regulator of T cell activation. Immunity 1994, 1, 405–413.
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355.
- Kawashima, S.; Inozume, T.; Kawazu, M.; Ueno, T.; Nagasaki, J.; Tanji, E.; Honobe, A.; Ohnuma, T.; Kawamura, T.; Umeda, Y. TIGIT/CD155 axis mediates resistance to immunotherapy in patients with melanoma with the inflamed tumor microenvironment. J. Immunother. Cancer 2021, 9, e003134.
- Konerding, M.A.; Fait, E.; Gaumann, A. 3D microvascular architecture of pre-cancerous lesion and invasive carcinomas in the colon. Br. J. Cancer 2001, 84, 1354–1362.
- Siemann, D.W. The unique characteristics of tumor vasculature and preclinical evidence for its selective disruption by tumor-vascular disrupting agents. Cancer Treat. Rev. 2011, 37, 63–74.
- Jubb, A.M.; Pham, T.Q.; Hanby, A.M.; Frantz, G.D.; Peale, F.V.; Wu, T.D.; Koeppen, H.W.; Hilan, K.J. Expression of vascular endothelial growth factor, hypoxia inducible factor 1 alpha, and carbonic anhydrase IX in human tumours. J. Clin. Pathol. 2004, 57, 504–512.
- Shweiki, D.; Itin, A.; Soffer, D.; Keshet, E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 1992, 359, 843–845.
- Roskoski, R., Jr. Vascular endothelial growth factor(VEGF) signaling in tumor progression. Crit. Rev. Oncol./Hematol. 2007, 62, 179–213.
- Saharinen, P.; Eklund, L.; Pulkki, K.; Bono, P.; Alitalo, K. VEGF and angiopoietin signaling in tumor angiogenesis and metastasis. Trends Mol. Med. 2011, 17, 347–362.
- Yang, Y.; Cao, Y. The impact of VEGF on cancer metastasis and systemic disease. Semin. Cancer Biol. 2022, 86, 251–261.
- Carreau, A.; El Hafny-Rahbi, B.; Matejuk, A.; Grillon, C.; Kieda, C. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J. Cell Mol. Med. 2011, 15, 1239–1253.
- McKeown, S.R. Defined normoxia, physoxia and hypoxia in tumors—Implications for treatment response. Br. J. Radiol. 2014, 87, 20130676.
- Vaupel, P.; Mayer, A.; Höckel, M. Impact of hemoglobin levels on tumor oxygenation: The higher, the better? Strahlenther. Onkol. 2006, 182, 63–71.
- Kozlov, A.M.; Lone, A.; Betts, D.H.; Cumming, R.C. Lactate preconditioning promotes a HIF-1α-mediated metabolic shift from OXPHOS to glycolysis in normal human diploid fibroblasts. Sci. Rep. 2020, 10, 8388.
- Nishimura, K.; Fukuda, A.; Hisatake, K. Mechanisms of the metabolic shift during somatic cell reprogramming. Int. J. Mol. Sci. 2019, 20, 2254.
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793.
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819.
- Choi, J.; Gyamfi, J.; Jang, H.; Koo, J.S. The role of tumor-associated macrophage in breast cancer biology. Histol. Histopathol. 2018, 33, 133–145.
- Mu, X.; Shi, W.; Xu, Y.; Xu, C.; Zhao, T.; Geng, B.; Yang, J.; Pan, J.; Hu, S.; Zhang, C.; et al. Tumor-derived lactate induces M2 macrophage polarization via the activation of the ERK/STAT3 signaling pathway in breast cancer. Cell Cycle 2018, 17, 428–438.
- Ratter, J.M.; Rooljackers, H.M.M.; Hoolveld, G.J.; Hijmans, A.G.M.; de Galan, B.E.; Tack, C.J.; Stienstra, R. In vitro and in vivo effects of lactate on metabolism and cytokine production of human primary PBMCs and monocytes. Front. Immunol. 2018, 9, 2564.
- De Saedeleer, C.J.; Copetti, T.; Porporato, P.E.; Verrax, J.; Feron, O.; Sonveaux, P. Lactate activates HIF-1 in oxidative but not in Warburg-phenotype human tumor cells. PLoS ONE 2012, 7, e46571.
- Pires, B.R.B.; Mencalha, A.L.; Ferreira, G.M.; Panis, C.; Silva, R.C.M.C.; Abdelhay, E. The hypoxia-inducible factor-1 alpha signaling pathway and its relation to cancer and immunology. Am. J. Immunol. 2014, 10, 215–224.
- Davis, L.; Recktenwald, M.; Hutt, E.; Fuller, S.; Briggs, M.; Goel, A.; Daringer, N. Targeting HIF-2α in the tumor microenvironment: Redefining the role of HIF-2α for solid cancer therapy. Cancers 2022, 14, 1259.
- Löfstedt, T.; Fredlund, E.; Holmquist-Mengelbier, L.; Pietras, A.; Overnberger, M.; Pollinger, L.; Påhlman, S. Hypoxia inducible factor-2α in cancer. Cell Cycle 2007, 6, 919–926.
- Schödel, J.; Oikonomopoulos, S.; Ragoussis, J.; Pugh, C.W.; Ratcliffe, P.J.; Mole, D.R. High-resolution genome-wide mapping of HIF-binding sites by CHIP-seq. Blood 2011, 117, e207.
- Stalnecker, C.A.; Cluntun, A.A.; Cerione, R.A. Balancing redox stress: Anchorage-independent growth requires reductive carboxylation. Transl. Cancer Res. 2016, 5, S433–S437.
- Jin, J.; Byun, J.-K.; Choi, Y.-K.; Park, K.-G. Targeting glutamine metabolism as a therapeutic strategy for cancer. Exp. Mol. Med. 2023, 55, 706–715.
- Végran, F.; Boidot, R.; Michiels, C.; Sonveaux, P.; Feron, O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports on NFκB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011, 71, 2550–2560.
- Chamnee, T.; Ontong, P.; Itano, N. Hyaluronan: A modulator of the tumor microenvironment. Cancer Lett. 2016, 375, 20–30.
- Lennicke, C.; Rahn, J.; Lichtenfels, R.; Wessjohann, L.A.; Seliger, B. Hydrogen peroxide—Production, fate and role in redox signaling of tumor cells. Cell Commun. Signal. 2015, 13, 39.
- Cyran, A.M.; Zhitkovich, A. HIF1, HSF1, and NRF2: Oxidant-responsive trio raising cellular defenses and engaging immune system. Chem. Res. Toxicol. 2022, 35, 1690–1700.
- Wang, S.; Song, P.; Zou, M.-H. AMP-activated protein kinase, stress responses and cardiovascular diseases. Clin. Sci. 2012, 122, 555–573.
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226.
- Barnard, R.A.; Regan, D.P.; Hansen, R.J.; Maycotte, P.; Thorborn, A.; Gustafson, D.L. Autophagy inhibition delays early but not late-stage metastatic disease. J. Pharmacol. Exp. Ther. 2016, 358, 282–293.
- Guo, J.Y.; Xia, B.; White, E. Autophagy-mediated tumor promotion. Cell 2013, 155, 1216–1219.
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410.
- Jiang, X.; Overholtzer, M.; Thompson, C.B. Autophagy in cellular metabolism and cancer. J. Clin. Investig. 2015, 125, 47–54.
- Jin, S.; DiPaola, R.S.; Mathew, R.; White, E. Metabolic catastrophe as a means to cancer cell death. J. Cell Sci. 2007, 120, 379–383.
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64.
- Lum, J.J.; Bauer, D.E.; Kong, M.; Harris, M.H.; Li, C.; Lindsten, Z.; Thompson, C.B. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 2005, 120, 237–248.
- Fung, C.; Lock, R.; Gao, S.; Salas, E.; Debnath, J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol. Biol. Cell 2008, 19, 797–806.
- Macintosh, R.L.; Timpson, P.; Thorborn, J.; Anderson, K.I.; Thorborn, A.; Ryan, K.M. Inhibition of autophagy impairs tumor cell invasion in an organotypic model. Cell Cycle 2012, 11, 2022–2029.
- Peng, Y.-F.; Shi, Y.-H.; Ding, Z.-B.; Ke, A.-W.; Gu, C.-Y.; Zhou, J.; Qiu, S.-J.; Dai, Z.; Fan, J. Autophagy inhibition suppresses pulmonary metastasis of HCC in mice via impairing anoikis resistance and colonization of HCC cells. Autophagy 2013, 9, 2056–2068.
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 12.
- Moenner, M.; Pluquet, O.; Bouchecareilh, M.; Chevet, E. Integrated endoplasmic reticulum stress responses in cancer. Cancer Res. 2007, 67, 10361–10364.
- Fernandez, P.M.; Tabbara, S.O.; Jacobs, L.K.; Manning, F.C.; Tsangaris, T.N.; Schwartz, A.M.; Kennedy, K.A.; Patierno, S.R. Overexpression of the glucose-regulated stress gene GRP78 in malignant but not benign human breast lesions. Breast Cancer Res. Treat. 2000, 59, 15–26.
- Xue, Z.; He, Y.; Ye, K.; Gu, Z.; Mao, Y.; Qi, L. A conserved structural determinant located at the interdomain region of mammalian inositol-requiring enzyme 1alpha. J. Biol. Chem. 2011, 286, 30859–30866.
- Hong, S.Y.; Hagen, T. Multiple myeloma Leu167Ile (c.499C>A) mutation prevents XBP1 mRNA splicing. Br. J. Haematol. 2013, 161, 898–901.
- Ghosh, R.; Wang, L.; Wang, E.S.; Perera, B.G.; Igbaria, A.; Morita, S.; Prado, K.; Thamsen, M.; Caswell, D.; Macias, H.; et al. Allosteric inhibition of the IRE1α RNase preserves cell viability and function during endoplasmic reticulum stress. Cell 2014, 158, 534–548.
- Oakes, S.A. Endoplasmic reticulum stress signaling in cancer cells. Am. J. Pathol. 2020, 190, 934–946.
- Min, J.-Y.; Chun, K.-S.; Kim, D.-H. The versatile utility of cysteine as a target for cancer treatment. Front. Oncol. 2023, 12, 997919.
- Banjac, A.; Perisic, T.; Sato, H.; Seiler, A.; Bannai, S.; Weiss, N.; Kölle, P.; Tschoep, K.; Issels, R.D.; Daniel, P.T.; et al. The cystine/cysteine cycle: A redox cycle regulating susceptibility versus resistance to cell death. Oncogene 2008, 27, 1618–1628.
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system xc− in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal. 2013, 18, 522–555.
- Lin, W.; Wang, C.; Liu, G.; Bi, C.; Wang, X.; Zhou, Q.; Jin, H. SLC7A11/xCT in cancer: Biological functions and therapeutic implications. Am. J. Cancer Res. 2020, 10, 3106–3126.
- Sen, N.; Paul, B.D.; Gadalla, M.M.; Mustafa, A.K.; Sen, T.; Xu, R.; Kim, S.; Snyder, S.H. Hydrogen sulfide-linked sulfhydration of NF-κB mediates its antiapoptotic actions. Mol. Cell 2012, 45, 13–24.
- Yang, G.; Zhao, K.; Ju, Y.; Mani, S.; Cao, Q.; Puukila, S.; Khaper, N.; Wu, L.; Wang, R. Hydrogen sulfide protects against cellular senescence via S-sulfhydration of Keap1 and activation of Nrf2. Antioxid. Redox Signal. 2013, 18, 1906–1919.
- Ma, Z.; Bi, Q.; Wang, Y. Hydrogen sulfide accelerates cell cycle progression in oral squamous cell carcinoma cell lines. Oral Dis. 2015, 21, 156–162.
- Jiang, L.; Shestov, A.A.; Swain, P.; Yang, C.; Parker, S.J.; Wang, Q.A.; Terada, L.S.; Adams, N.D.; McCabe, M.T.; Pietrak, B.; et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 2016, 532, 255–258.
- Kaya, B.; Çiçek, O.; Erdi, F.; Findig, S.; Karatas, Y.; Esen, H.; Keskin, F.; Kalkan, E. Intratumoral hemorrhage-related differences in the expression of vascular endothelial growth factor, basic fibroblast growth factor and thioredoxin reductase 1 in human glioblastoma. Mol. Clin. Oncol. 2006, 5, 343–346.
- Yin, T.; He, S.; Liu, X.; Jiang, W.; Ye, T.; Lin, Z.; Sang, Y.; Su, C.; Wan, Y.; Shen, G.; et al. Extravascular red blood cells and hemoglobin promote tumor growth and therapeutic resistance as endogenous danger signals. J. Immunol. 2015, 194, 429–437.
- Poon, L.C.; Methot, S.P.; Morabi-Pazocki, W.; Pio, F.; Bennet, A.J.; Sen, D. Guanine-rich RNAs and DNAs that bind heme robustly catalyze oxygen transfer reactions. J. Am. Chem. Soc. 2011, 133, 1877–1884.
- Gray, L.T.; Lombardi, E.P.; Verga, D.; Nicolas, A.; Teulade-Fichou, M.-P.; Londoño-Vallejo, A.; Maizels, N. G-Quadruplexes sequester free heme in living cells. Cell Chem. Biol. 2019, 26, 1681–1689.
- Brooks, T.A.; Hurley, L.H. Targeting MYC expression through G-quadruplexes. Genes Cancer 2010, 1, 641–649.
- Brooks, T.A.; Kendrick, S.; Hurley, L.H. Making sense of G-quadruplex and i-motif functions in oncogene promoters. FEBS J. 2010, 277, 3459–3469.
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated pH: A perfect storm for cancer progression. Nat. Rev. Cancer 2011, 11, 671–677.
- Liu, Y.; White, K.A.; Barber, D.L. Intracellular pH regulates cancer and stem cell behaviors: A protein dynamics perspective. Front. Oncol. 2020, 10, 1401.
- Hao, G.; Xu, Z.P.; Li, L. Manipulating extracellular tumour pH: An effective target for cancer therapy. RSC Adv. 2018, 8, 22182–22192.
- Casey, J.R.; Grinstein, S.; Orlowski, J. Sensors and regulators of intracellular pH. Nat. Rev. Mol. Cell Biol. 2010, 11, 50–61.
- Flinck, M.; Kramer, S.H.; Schnipper, J.; Andersen, A.P.; Pedersen, S.F. The acid-base transport proteins NHE1 and NBCn1 regulate cell cycle progression in human breast cancer cells. Cell Cycle 2018, 17, 1056–1067.
- Putney, L.K.; Barber, D.L. Na-H exchange-dependent increase in intracellular pH times G2/M entry and transition. J. Biol. Chem. 2003, 278, 44645–44649.
- Parks, S.K.; Pouyssegur, J. The Na(+)/HCO3(-) co-transporter SLC4A4 plays a role in growth and migration of colon and breast cancer cells. J. Cell Physiol. 2015, 230, 1954–1963.
- Schwab, A.; Stock, C. Ion channels and transporters in tumour cell migration and invasion. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130102.
- Schulze, A.; Harris, A.L. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature 2012, 491, 364–373.
- Lee, S.; Shanti, A. Effect of exogenous pH on cell growth of breast cancer cells. Int. J. Mol. Sci. 2021, 22, 9910.
- Makris, E.A.; Responte, D.J.; Paschos, N.K.; Hu, J.C.; Athanasiou, K.A. Developing functional musculoskeletal tissues through hypoxia and lysyl oxidase-induced collagen cross-linking. Proc. Natl. Acad. Sci. USA 2014, 111, E4832–E4841.
- Makris, E.A.; Hu, J.C.; Athanasiou, K.A. Hypoxia-induced collagen crosslinking as a mechanism for enhancing mechanical properties of engineered articular cartilage. Osteoarthr. Cartil. 2013, 21, 634–641.
- Najafi, M.; Farhood, B.; Mortezaee, K. Extracellular matrix (ECM) stiffness and degradation as cancer drivers. J. Cell Biochem. 2019, 120, 2782–2790.
- Aro, E.; Khatri, R.; Gerard-O’Riley, R.; Mangiavini, L.; Myllyharju, J.; Schipani, E. Hypoxia-inducible factor-1 (HIF-1) but not HIF-2 is essential for hypoxic induction of collagen prolyl 4-hydroxylases in primary newborn mouse epiphyseal growth plate chondrocytes. J. Biol. Chem. 2012, 287, 37134–37144.
- Xiong, G.; Stewart, R.L.; Chen, J.; Gao, T.; Scott, T.L.; Samayoa, L.M.; O’Connor, K.; Lane, A.N.; Xu, R. Collagen prolyl 4-hydroxylase 1 is essential for HIF-1alpha stabilization and TNBC chemoresistance. Nat. Commun. 2018, 9, 4456.
- Saatci, O.; Kaymak, A.; Raza, U.; Ersan, P.G.; Akbulut, O.; Banister, C.E.; Sikirzhytski, V.; Tokat, U.M.; Aykut, G.; Ansari, S.A.; et al. Targeting lysyl oxidase (LOX) overcomes chemotherapy resistance in triple negative breast cancer. Nat. Commun. 2020, 11, 2416.
- Janjanam, J.; Pano, G.; Wang, R.; Minden-Birkenmaier, B.A.; Breeze-Jones, H.; Baker, E.; Garcin, C.; Clayton, G.; Shirinifard, A.; Zaske, A.M.; et al. Matricellular protein WISP2 is an endogenous inhibitor of collagen linearization and cancer metastasis. Cancer Res. 2021, 81, 5666–5677.
- Murdocca, M.; De Masi, C.; Pucci, S.; Mango, R.; Novelli, G.; Di Natale, C.; Sangiuolo, F. LOX-1 and cancer: An indissoluble liaison. Cancer Gene Ther. 2021, 28, 1088–1098.
- Bodelon, C.; Mullooly, M.; Pfeiffer, R.M.; Fan, S.; Abubakar, M.; Lenz, P.; Vacek, P.M.; Weaver, D.L.; Herschorn, S.D.; Johnson, J.M.; et al. Mammary collagen architecture and its association with mammographic density and lesion severity among women undergoing image-guided breast biopsy. Breast Cancer Res. 2021, 23, 105.
- Chen, D.; Liu, Z.; Liu, W.; Fu, M.; Jiang, W.; Xu, S.; Wang, G.; Chen, F.; Lu, J.; Chen, H.; et al. Predicting postoperative peritoneal metastasis in gastric cancer with serosal invasion using a collagen nomogram. Nat. Commun. 2021, 12, 179.
- Angenendt, L.; Mikesch, J.H.; Gorlich, D.; Busch, A.; Arnhold, I.; Rudack, C.; Hartmann, W.; Wardelmann, E.; Berdel, W.E.; Stenner, M.; et al. Stromal collagen type VI associates with features of malignancy and predicts poor prognosis in salivary gland cancer. Cell. Oncol. Dordr. 2018, 41, 517–525.
- Salmon, H.; Franciszkiewicz, K.; Damotte, D.; Dieu-Nosjean, M.C.; Validire, P.; Trautmann, A.; Mami-Chouaib, F.; Donnadieu, E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Investig. 2012, 122, 899–910.
- Piersma, B.; Hayward, M.K.; Weaver, V.M. Fibrosis and cancer: A strained relationship. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188356.
- Landoldt, L.; Spagnoli, G.C.; Hertig, A.; Brocheriou, I.; Marti, H.-P. Fibrosis and cancer: Shared features and mechanisms suggest common targeted therapeutic approaches. Nephrol. Dial. Transplant. 2022, 37, 1024–1032.
- Wu, B.; Sodji, Q.H.; Oyelere, A.K. inflammation, fibrosis and cancer: Mechanisms, therapeutic options and challenges. Cancers 2022, 14, 552.
- Luo, L.; Yang, J.-X.; Luo, T.; Liu, D.; Wu, G.-H.; He, J.-M. A study on the mechanism of PP2A in the recovery of SCI in rats through downregulation of MMP-9 via MAPK signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 7195–7203.
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine cathepsins: From structure, function and regulation to new frontiers. Biochim. Biophys. Acta 2012, 1824, 68–88.
- Piperigkou, Z.; Kyriakopoulou, K.; Koutsakis, C.; Mastronikolis, S.; Karamanos, N.K. Key Matrix Remodeling Enzymes: Functions and Targeting in Cancer. Cancers 2021, 13, 1441.
- Barash, U.; Cohen-Kaplan, V.; Dowek, I.; Sanderson, R.D.; Ilan, N.; Vlodavsky, I. Proteoglycans in health and disease: New concepts for heparanase function in tumor progression and metastasis. FEBS J. 2010, 277, 3890–3903.
- Wei, R.-R.; Sun, D.-N.; Yang, H.; Yan, J.; Zhang, X.; Zheng, X.-L.; Fu, X.-H.; Geng, M.-Y.; Huang, X.; Ding, J. CTC clusters induced by heparanase enhance breast cancer metastasis. Acta Pharmacol. Sin. 2018, 39, 1326–1337.
- Song, J.M.; Im, J.; Nho, R.S.; Han, Y.H.; Upadhyaya, P.; Kassie, F. Hyaluronan-CD44/RHAMM interaction-dependent cell proliferation and survival in lung cancer cells. Mol. Carcinog. 2019, 58, 321–333.
- Makkar, S.; Riehl, T.E.; Chen, B.; Yan, Y.; Alvarado, D.M.; Ciorba, M.A.; Stenson, W.F. Hyaluronic acid binding to TLR4 promotes proliferation and blocks apoptosis in colon cancer. Mol. Cancer Ther. 2019, 18, 2446–2456.
- Yang, Y.M.; Noureddin, M.; Liu, C.; Ohashi, K.; Kim, S.Y.; Ramnath, D.; Powell, E.E.; Sweet, M.J.; Roh, Y.S.; Hsin, I.-F.; et al. Hyaluronan synthase 2-mediated hyaluronan production mediates Notch1 activation and liver fibrosis. Sci. Transl. Med. 2019, 11, eaat9284.
- Chen, J.E.; Lumibao, J.; Blazek, A.; Gaskins, H.R.; Harley, B. Hypoxia activates enhanced invasive potential and endogenous hyaluronic acid production by glioblastoma cells. Biomater. Sci. 2018, 6, 854–862.
- Kobayashi, N.; Miyoshi, S.; Mikami, T.; Koyama, H.; Kitazawa, M.; Takeoka, M.; Sano, K.; Amano, J.; Isogai, Z.; Niida, S.; et al. Hyaluronan deficiency in tumor stroma impairs macrophage trafficking and tumor neovascularization. Cancer Res. 2010, 70, 7073–7083.
- Kuang, D.M.; Wu, Y.; Chen, N.; Cheng, J.; Zhuang, S.M.; Zheng, L. Tumor-derived hyaluronan induces formation of immunosuppressive macrophages through transient early activation of monocytes. Blood 2007, 110, 587–595.
More
Information
Subjects:
Oncology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
505
Revisions:
2 times
(View History)
Update Date:
09 Jan 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No