Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mohanakrishnan Sathyamoorthy | -- | 2700 | 2024-01-05 17:17:49 | | | |
| 2 | Sirius Huang | -3 word(s) | 2697 | 2024-01-08 02:50:11 | | | | |
| 3 | Sirius Huang | Meta information modification | 2697 | 2024-03-11 02:55:25 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Moore, P.; Murdock, P.; Ramanathan, A.; Sathyamoorthy, M. Genetic Mutations Associated Myocardial Bridges. Encyclopedia. Available online: https://encyclopedia.pub/entry/53493 (accessed on 24 July 2026).
Moore P, Murdock P, Ramanathan A, Sathyamoorthy M. Genetic Mutations Associated Myocardial Bridges. Encyclopedia. Available at: https://encyclopedia.pub/entry/53493. Accessed July 24, 2026.
Moore, Peyton, Paul Murdock, Akash Ramanathan, Mohanakrishnan Sathyamoorthy. "Genetic Mutations Associated Myocardial Bridges" Encyclopedia, https://encyclopedia.pub/entry/53493 (accessed July 24, 2026).
Moore, P., Murdock, P., Ramanathan, A., & Sathyamoorthy, M. (2024, January 05). Genetic Mutations Associated Myocardial Bridges. In Encyclopedia. https://encyclopedia.pub/entry/53493
Moore, Peyton, et al. "Genetic Mutations Associated Myocardial Bridges." Encyclopedia. Web. 05 January, 2024.
Copy Citation
Myocardial bridging (MB) is a congenital coronary artery anomaly that has limited molecular disease state characterization. Though a large portion of individuals may be asymptomatic, the myocardial ischemia caused by this anomaly can lead to angina, acute coronary syndrome, coronary artery disease, and sudden cardiac death in patients.
myocardial bridging
genomics
congenital coronary vascular anomalies
single nucleotide variants
1. Introduction
Normal coronary artery anatomy consists of the coronary arteries arising from the aortic sinuses, which follow a converging path toward the heart’s anatomical apex. The three main coronary arteries (the right coronary artery (RCA), left anterior descending (LAD), and left circumflex (LCX)) each have common placements and travel along the epicardium of the heart in patients who underwent typical coronary development [1]. Myocardial bridging (MB) is a congenital anomaly that consists of a portion of an epicardial coronary artery diving into the myocardium layer for a section of its journey (Figure 1). The muscle layer that lies above the artery is recognized as a myocardial bridge, while the portion of the artery is labeled as a tunneled artery. This anomaly is typically seen in the LAD, but may also be present in any of the coronary arteries [2]. Clinical experience has verified this phenotypic presentation.
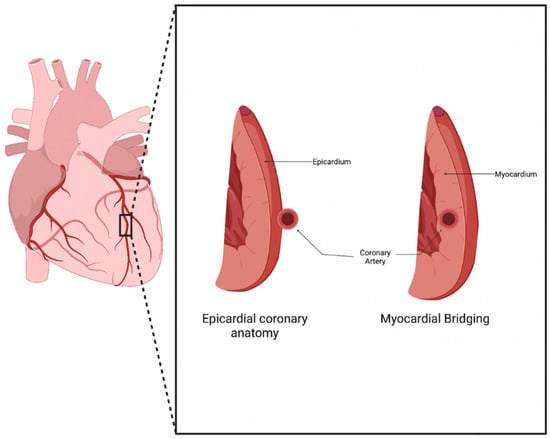
Figure 1. Myocardial Bridging anatomy. Normal coronary artery anatomy consists of the artery traveling along the epicardium of the heart, while a bridging artery travels within the myocardium [3].
The genetic mechanisms driving the development of congenital coronary vascular anomalies (CCVAs), including those of myocardial bridging, lack sufficient research [4]. This finding is surprising as these anomalies are present and well characterized in patients throughout the world. The rates of myocardial bridging vary between the methods of evaluation, with autopsy and angiography being the most common techniques used to assess for the presence of bridging. Invasive angiography generally underreports the prevalence of myocardial bridging, as it typically detects the anomaly in 0.15–25% of patients [1]. This low prevalence is countered by autopsy reports, which illustrate the prevalence of MBs in 5–86% of patients [5]. Recent computed tomography (CT) studies have also shown bridging in up to 25% of patients, further showing the underestimation of its prevalence within angiography studies [1]. In a CCTA cohort from 2014–2023, the prevalence is 7% (70/1000) [6]. The discrepancy in these methods demonstrates the unreliability of the current diagnostic methods and suggests a need for improved techniques.
Though bridging was previously viewed as benign, this belief has been challenged in the recent literature. The vessel compression of the intramural artery has been noted during the contraction of the myocardium, often leading to chest pain and, sometimes, to acute coronary syndrome [7]. The overarching pathophysiology behind these symptoms stems from the myocardial ischemia secondary to the tunneling and compression of the artery, with the ischemia being exacerbated by the increased sympathetic tone [2]. Since the systolic phase of the myocardial cycle is only mildly involved in the perfusion of the myocardial tissue, other mechanisms contribute to the ischemia seen in bridging. These mechanisms include delayed early diastolic artery relaxation, atherosclerotic stenosis development proximal to the tunneled artery, functional disorders of coronary circulation including impaired endothelium-dependent vasodilatation and microvascular dysfunction, and the “branch steal” phenomenon (Figure 2). The branch steal effect describes the crossing of blood through the constricted segment during the end of systole and early diastole, leading to an increase in diastolic flow velocity in the artery. The ischemia due to these various mechanisms are important causes of the anginal symptoms that bridging patients experience [8]. Other cardiovascular events linked to myocardial bridging include cardiac arrhythmias and sudden cardiac death [9].
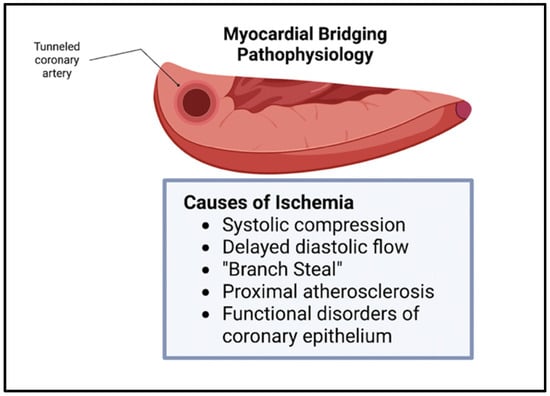
Figure 2. Myocardial Bridging physiology. Ischemia in myocardial bridging is secondary to systolic compression, delayed diastolic flow, branch steal, proximal atherosclerosis, and functional disorders of coronary epithelium [3].
2. Genetic Mutations Associated Myocardial Bridges: Potential Candidate Genes?
2.1. MYH7
ß-myosin heavy chain (MYH7) gene mutations have been associated with hypertrophic cardiomyopathy (HCM) and restrictive cardiomyopathy (RCM). The MYH7 mutations found in a 9-month-old female with RCM and associated myocardial hypertrophy and a 7-year-old female who died of sudden cardiac death (SCD) were classified as variants of unknown significance (VUS). Of note, both patients were found to have myocardial bridging of the left anterior descending (LAD) artery on autopsy [10][11]. Genetic testing of one of the patients revealed an A1157G mutation in exon 13 of the MYH7 gene. This mutation resulted in a nonsynonymous amino acid change at position 386 from tyrosine to cysteine and was classified as being likely pathogenic [10][11].
The MYH7 gene encodes for ß-myosin heavy chain, an essential component of the cardiac sarcomere. Though the researchers do not hypothesize a mechanism regarding bridge formation, there are other examples of associations between MYH7 mutations and bridge formation including the case of a 15-year-old with exertional dyspnea and chest pain [12]. The researchers view this as a potential candidate gene of interest for further studies, as mutations in the gene may have significant clinical manifestations outside of its already known associations with HCM and RCM.
2.2. DPP6
The DPP6 gene encodes a membrane protein that is a member of the peptidase S9B family of serine proteases and is known to bind specific voltage-gated potassium channels [13]. Mutations in the DPP6 gene have been previously associated with the development of idiopathic ventricular fibrillation (IVF), which is known to cause SCD [14]. Mutations in the DPP6 (Ala714Thr) gene were discovered in the father and sister of a child who died from SCD and was found to have a 1.1 × 0.5 cm myocardial bridge on autopsy. Further analysis of DPP6 and the other mutated genes in the family of the proband found that they were variants of unknown significance (VUS). Importantly, the DPP6 mutation was not the only mutation in the child. The VUS in MYH7, SCN2B, and NOTCH1 were also found. The researchers hypothesize that the SCD seen in the child was likely secondary to the HCM and its subsequent role in arrhythmic death, however, they are unsure of the consequences secondary to the isolated genetic variants [11]. Researchers suspect that the DPP6 mutation may have contributed to arrhythmias seen in the patient, but have low suspicion that the mutation played a role in bridge formation.
2.3. SCN2B
The SCN2B gene encodes voltage-gated sodium channel ß2-subunits, and is involved in cell–cell adhesion and cell migration [15]. Mutations in the SCN2B gene have been associated with cardiac arrhythmias in humans including atrial fibrillation and Brugada syndrome [16]. Mutations in the SCN2B gene (Glu31Asp) were discovered in a child that died from sudden cardiac death who was found to have a myocardial bridge on autopsy [11]. No mechanism regarding the gene mutation and the development of myocardial bridging was discussed in the report. Despite this, researchers hypothesize that the disruption in SCN2B’s known role in cell migration via genetic mutation may play a role in the abnormal migration of the coronary arteries during embryogenesis, which could subsequently result in the formation of myocardial bridging. Due to its known role in this vital process, further investigation into the potential relationship between mutations in SCN2B and bridge development may be promising.
2.4. NOTCH1
NOTCH1 encodes a cell-surface receptor that plays a role in the development of various cell and tissue types [17]. The importance of the NOTCH pathway during the development of the cardiovascular system has been extensively discussed. Notably, NOTCH has several roles during ventricular development including that of trabecular development via differentiation and the proliferation of cardiomyocytes [18]. A mutation in NOTCH1 (Arg2313Gln) was discovered in a young female that died from SCD who was found to have a myocardial bridge on autopsy [11]. Though the researchers made no direct hypothesis regarding a link between NOTCH1 and bridge development, it is plausible that mutations in a member of the NOTCH family of genes, known players in cardiovascular development, may play a role in the development of a coronary artery anomaly such as myocardial bridging.
Studies have demonstrated that NOTCH1 plays a pivotal role in cardiomyocyte differentiation and proliferation. The gene has been implicated in various congenital heart defects including hypoplastic left heart syndrome [19]. The core of NOTCH1’s role in angiogenesis is its impact on endothelial cells. The gene influences the delicate balance between tip cells and stalk cells during sprouting angiogenesis. Tip cells, located at the leading edge of growing vessels, steer vessel extension, while stalk cells support vessel elongation. NOTCH1 activity in tip cells curtails excessive sprouting by inducing the expression of Dll4, a NOTCH ligand. This creates a feedback loop where Dll4 interacts with the neighboring endothelial cells’ NOTCH receptors, repressing their tip cell potential and promoting stalk cell characteristics, thereby maintaining vessel integrity [20]. A disruption in this balance may result in a disruption of angiogenesis.
NOTCH1 is activated in proliferating embryonic and immature cardiomyocytes and is downregulated in the myocardium during postnatal development. NOTCH signaling in adults is activated transiently in response to myocardial injury, further suggesting that the gene is contributory to cardiac repair via angiogenesis [21][22]. Due to the many roles that the NOTCH1 gene plays in the angiogenesis and development of the cardiovascular system, further investigation assessing its potential association in bridge development is warranted as researchers view NOTCH1 to be a candidate gene of interest.
2.5. SLMAP
A variant of unknown significance in the SLMAP gene (c.599c>T) was found in a 20-year-old male that passed away from sudden cardiac death. Further autopsy revealed a 2-cm myocardial bridge that began in the left anterior descending artery. The missense mutation was found to have a very low frequency in the general population, and the mutation was found to be likely pathogenic using three in silico predictors [23]. SLMAP is a known cardiac membrane protein that plays a role in excitation–contraction coupling in cardiac myocytes. SLMAP mutations have been previously linked to cardiac conditions including the development of heart failure [24]. SLMAP has also been associated with the development of Brugada syndrome, a cardiac channelopathy, through the silencing of SLMAP by small-interfering RNA (siRNA) [25]. No relationship between SLMAP and angiogenesis, cell migration, or coronary artery embryogenesis were discussed, limiting our suspicion for its involvement in myocardial bridge development. Based on SLMAP’s role in cardiac ion channel physiology rather than processes vital to coronary artery development and maturation, the researchers do not view this as a potential candidate gene for further studies.
2.6. COMMD10
Deletions in COMMD10 were identified in a 19-year-old athlete who was experiencing syncopal episodes. The coronary CTA of the patient discovered a 20 mm bridge in the LAD. An in-depth genetic analysis was performed using array-comparative genomic hybridization (array-CGH) and whole exome sequencing. The array-CGH demonstrated a copy number variation (CNV) alteration affecting intron 5 of the COMMD10 gene, which is responsible for modulating the activity of the cullin-RING E3 ubiquitin ligase complexes (CRL) and reducing NFK-ß activation. COMMD10 has also been found to be expressed in the endothelial cells and smooth muscle cells of various tissues, which demonstrates a potential involvement in angiogenesis during stages of embryogenesis. The gene has also been found to be expressed in cardiac tissue, which suggests it may have a role in cardiac development. The researchers hypothesize that COMMD10 plays an important role during cardiac development, which may include the development of congenital heart disorders and anomalies such as myocardial bridging [26]. Due to these findings, researchers agree that COMMD10 is a candidate gene of interest and believe that further research regarding the association of the gene with myocardial bridge development is warranted.
2.7. MACROD2
MACROD2 is a deacetylase involved in removing ADP-ribose from mono-ADP-ribosylated proteins and is frequently involved in patients with complex syndromes [27]. A macrodeletion of MACROD2 was discovered in an athlete suffering from syncope who was found to have a 20 mm myocardial bridge. An array-CGH demonstrated CNVs, which may be associated with the presence of congenital heart disorders (CHD). A recent genome-wide association study that included over 4000 patients affected by CHD and 8000 controls revealed a statistically significant association between MACROD2 polymorphisms and the development of the transposition of the great vessels. Further data has shown the expression of MACROD2 is present in human embryonic cardiac cells, further strengthening the possibility of the gene’s involvement as a transcriptional regulator in cardiomyocytes [26]. Due to its presence in human embryonic cardiac cells and its potential role as a transcriptional regulator in cardiac myocytes, the researchers hypothesized its contributing role in the development of CHDs. With these findings, researchers agree that this may be a candidate gene of interest and support further investigation into this genotype–phenotype relationship.
2.8. FBN1
A heterozygous mutation in the FBN1 gene was found on whole exome sequencing in a 12-year-old girl that was experiencing recurrent syncopal episodes after exercise. The identified change was a missense mutation of c.3535A>G resulting in an amino acid change of p.I1175M in exon 29. Further bioinformatic analysis suggested that this variant may affect the structure and function of the protein product [28]. The FBN1 gene encodes the fibrillin-1 protein, an extracellular matrix component that regulates growth factor signaling pathways. Pathologic mutations in FBN1 are well-established causes of Marfan syndrome and other congenital heart and vascular defects [29]. There are no established associations between FBN1 and coronary artery development, however, with the gene’s extensive role in other cardiovascular pathologies, genes in this cascade (FBN1 and FBN2) should be further characterized in myocardial bridge development as candidate genes of interest.
2.9. DES
A heterozygous missense variant (c.1300G>A; p.E434K) of the DES gene was identified using whole-exome sequencing in a Chinese family with cardiomyopathy and sudden cardiac death. A myocardial bridge was discovered in the proband; however, none was present in the other four patients. Further genetic analysis predicted the mutation to be pathogenic, which was strengthened with its absence in 200 controls. The DES gene encodes for desmin, an intermediate filament protein that stabilizes sarcomeres and cell contacts in the cardiac intercalated disc [30]. The researchers have little suspicion to believe that the isolated DES mutation was the causative agent for bridge development. This belief is secondary to the lack of evidence of bridging found in the proband’s siblings or parents, or in other reports.
2.10. PRKAG2
The PRKAG2 gene is best known for its role in Wolff-Parkinson-White (WPW) and PRKAG2 syndrome. PRKAG2 syndrome is glycogen accumulation within cardiac tissue, which clinically presents as HCM. A mutation in the PRKAG2 gene was found in a 23-year-old female with a known history of WPW and HCM, who presented after a non-ST elevation myocardial infarction. The patient underwent genetic testing which revealed a heterozygous missense Arg302Gln mutation in the PRKAG2 gene. The angiography revealed severe bridging of the LAD in the patient [31]. Though this mutation may have contributed to myocardial hypertrophy secondary to the glycogen accumulation, researchers do not view this as a likely candidate gene for bridging development.
2.11. Summary of Genes
Literature review revealed a total of 10 genes in patients that were also found to have a myocardial bridging phenotype (Figure 3). Each of these genes have different roles in various processes including angiogenesis, embryogenesis, and cell differentiation, as discussed above. Patients in the cases varied in their presentation, with many being symptomatic and others having incidental findings of myocardial bridging on imaging or autopsy. Regardless of the presentation, the background of each gene was reviewed to determine its potential role in myocardial bridge development.
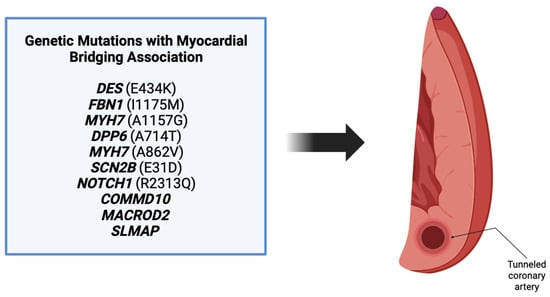
Figure 3. Ten genes were found associated with myocardial bridge development. These genes include DES, FBN1, MYH7, DPP6, MYH7, SCN2B, NOTCH1, COMMD10, MACROD2, and SLMAP [3].
Of the mutations discovered, researchers only view six to be candidate genes for further research: MYH7, SCN2B, NOTCH1, COMMD10, MACROD2, and FBN1/FBN2 (Table 3). These genes have been found to be key in various processes that are vital for coronary artery development and/or migration, and it is plausible that disruptions in these genes may serve as a genesis for the development of myocardial bridging. These findings warrant further investigation into the potential mechanistic basis behind mutations in the candidate genes and the development of myocardial bridging.
Table 3. Genetic variants (single nucleotide variants) associated with myocardial bridging and candidate gene likelihood.
| Genes | Mutations | Candidate Gene |
|---|---|---|
| MYH7 | A1157G | Yes |
| DPP6 | A714T | No |
| SCN2B | E31D | Yes |
| NOTCH1 | R2313Q | Yes |
| SLMAP | S200L | No |
| COMMD10 | Intron 5 CNV | Yes |
| MACROD2 | CNVs | Yes |
| FBN1/FBN2 | I1175M | Yes |
| DES | E434K | No |
| PRKAG2 | R302Q | No |
References
- Villa, A.D.M.; Sammut, E.; Nair, A.; Rajani, R.; Bonamini, R.; Chiribiri, A. Coronary artery anomalies overview: The normal and the abnormal. World J. Radiol. 2016, 8, 537–555.
- Sternheim, D.; Power, D.A.; Samtani, R.; Kini, A.; Fuster, V.; Sharma, S. Myocardial Bridging: Diagnosis, Functional Assessment, and Management. J. Am. Coll. Cardiol. 2021, 78, 2196–2212.
- Image Created with biorender.com. Available online: Biorender.com (accessed on 13 August 2023).
- Picazo, B.; Perez-Pomares, J.M. Human Genetics of Coronary Artery Anomalies. In Congenital Heart Diseases: The Broken Heart; Springer: Vienna, Austria, 2016; pp. 535–539.
- Lee, M.S.; Chen, C.H. Myocardial Bridging: An Up-to-Date Review. J. Invasive Cardiol. 2015, 27, 521–528.
- With Permission, Research Database—Consultants in Cardiovascular Medicine and Science—Fort Worth—PLLC, Fort Worth, Texas.
- Happach, V.C.; Delk, G.T.; Ganti, L. Myocardial Bridging, the Hidden Risk Factor for Ischemia. Mil. Med. 2022, 187, E1230–E1232.
- Ciliberti, G.; Laborante, R.; Di Francesco, M.; Restivo, A.; Rizzo, G.; Galli, M.; Canonico, F.; Zito, A.; Princi, G.; Vergallo, R.; et al. Comprehensive functional and anatomic assessment of myocardial bridging: Unlocking the Gordian Knot. Front. Cardiovasc. Med. 2022, 9, 970422.
- Murtaza, G.; Mukherjee, D.; Gharacholou, S.M.; Nanjundappa, A.; Lavie, C.J.; Khan, A.A.; Shanmugasundaram, M.; Paul, T.K. An Updated Review on Myocardial Bridging. Cardiovasc. Revascularization Med. 2020, 21, 1169–1179.
- Greenway, S.C.; Wilson, G.J.; Wilson, J.; George, K.; Kantor, P.F. Sudden Death in an Infant with Angina, Restrictive Cardiomyopathy, and Coronary Artery Bridging An Unusual Phenotype for a beta-Myosin Heavy Chain (MYH7) Sarcomeric Protein Mutation. Circ.-Heart Fail. 2012, 5, E92–E93.
- Grassi, S.; Campuzano, O.; Coll, M.; Brion, M.; Arena, V.; Iglesias, A.; Carracedo, A.; Brugada, R.; Oliva, A. Genetic variants of uncertain significance: How to match scientific rigour and standard of proof in sudden cardiac death? Legal. Med. 2020, 45, 101712.
- Joseph, A.; Hernandez, N.B.; Davies, R.; Tan, W. Managing Myocardial Bridge and Right Ventricular Outflow Tract Obstruction in an Adolescent with Hypertrophic Cardiomyopathy. World J. Pediatr. Congenit. Heart Surg. 2023, 14, 530–532.
- DPP6 Dipeptidyl Peptidase Like 6 —Gene—NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene/1804 (accessed on 15 August 2023).
- Chopra, N.; Knollmann, B.C. Genetics of sudden cardiac death syndromes. Curr. Opin. Cardiol. 2011, 26, 196–203.
- SCN2B Sodium Voltage-Gated Channel Beta Subunit 2 —Gene—NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene/6327 (accessed on 15 August 2023).
- Bao, Y.; Willis, B.C.; Frasier, C.R.; Lopez-Santiago, L.F.; Lin, X.; Ramos-Mondragon, R.; Auerbach, D.S.; Chen, C.; Wang, Z.; Anumonwo, J.; et al. Scn2b Deletion in Mice Results in Ventricular and Atrial Arrhythmias. Circ. Arrhythm. Electrophysiol. 2016, 9, e003923.
- NOTCH1 Notch Receptor 1 —Gene—NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene/4851 (accessed on 20 August 2023).
- Niessen, K.; Karsan, A. Notch signaling in cardiac development. Circ. Res. 2008, 102, 1169–1181.
- Ye, S.; Wang, C.; Xu, Z.; Lin, H.; Wan, X.; Yu, Y.; Adhicary, S.; Zhang, J.Z.; Zhou, Y.; Liu, C.; et al. Impaired Human Cardiac Cell Development due to NOTCH1 Deficiency. Circ. Res. 2023, 132, 187–204.
- Blanco, R.; Gerhardt, H. VEGF and Notch in Tip and Stalk Cell Selection. Cold Spring Harb. Perspect. Med. 2013, 3, a006569.
- Kachanova, O.; Lobov, A.; Malashicheva, A. The Role of the Notch Signaling Pathway in Recovery of Cardiac Function after Myocardial Infarction. Int. J. Mol. Sci. 2022, 23, 12509.
- Li, Y.; Hiroi, Y.; Liao, J.K. Notch Signaling as an Important Mediator of Cardiac Repair and Regeneration after Myocardial Infarction. Trends Cardiovasc. Med. 2010, 20, 228–231.
- Grassi, S.; Vidal, M.C.; Campuzano, O.; Arena, V.; Alfonsetti, A.; Rossi, S.S.; Scarnicci, F.; Iglesias, A.; Brugada, R.; Oliva, A. Sudden Death without a Clear Cause after Comprehensive Investigation: An Example of Forensic Approach to Atypical/Uncertain Findings. Diagnostics 2021, 11, 886.
- Nader, M. The SLMAP/Striatin complex: An emerging regulator of normal and abnormal cardiac excitation-contraction coupling. Eur. J. Pharmacol. 2019, 858, 172491.
- Ishikawa, T.; Sato, A.; Marcou, C.A.; Tester, D.J.; Ackerman, M.J.; Crotti, L.; Schwartz, P.J.; On, Y.K.; Park, J.E.; Nakamura, K.; et al. A Novel Disease Gene for Brugada Syndrome Sarcolemmal Membrane-Associated Protein Gene Mutations Impair Intracellular Trafficking of hNav1.5. Circ.-Arrhythmia Electrophysiol. 2012, 5, 1098–1107.
- Brancaccio, M.; Mennitti, C.; Cesaro, A.; Monda, E.; D’Argenio, V.; Casaburi, G.; Mazzaccara, C.; Ranieri, A.; Fimiani, F.; Barretta, F.; et al. Multidisciplinary In-Depth Investigation in a Young Athlete Suffering from Syncope Caused by Myocardial Bridge. Diagnostics 2021, 11, 2144.
- Lombardo, B.; Esposito, D.; Iossa, S.; Vitale, A.; Verdesca, F.; Perrotta, C.; Di Leo, L.; Costa, V.; Pastore, L.; Franze, A. Intragenic Deletion in MACROD2: A Family with Complex Phenotypes Including Microcephaly, Intellectual Disability, Polydactyly, Renal and Pancreatic Malformations. Cytogenet. Genome Res. 2019, 158, 25–31.
- Sun, Y.X.; Hu, B.; Feng, L.; Dong, J.T.; Huang, X.S.; Cai, S.J.; Yuan, Y. A Case of Syncope in a Child due to the Large Segment of Myocardial Bridge. Int. Heart J. 2022, 63, 416–420.
- Ergoren, M.C.; Turkgenc, B.; Teralı, K.; Rodoplu, O.; Verstraeten, A.; Van Laer, L.; Mocan, G.; Loeys, B.; Tetik, O.; Temel, S.G. Identification and characterization of a novel FBN1 gene variant in an extended family with variable clinical phenotype of Marfan syndrome. Connect. Tissue Res. 2019, 60, 146–154.
- Liu, Y.X.; Yu, R.; Sheng, Y.; Fan, L.L.; Deng, Y. Case report: Whole-exome sequencing identifies a novel DES mutation (p. E434K) in a Chinese family with cardiomyopathy and sudden cardiac death. Front. Cardiovasc. Med. 2022, 9, 971501.
- Banankhah, P.; Fishbein, G.A.; Dota, A.; Ardehali, R. Cardiac manifestations of PRKAG2 mutation. BMC Med. Genet. 2018, 19, 1.
More
Information
Subjects:
Cardiac & Cardiovascular Systems
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
3 times
(View History)
Update Date:
11 Mar 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No