 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Bidisha Roy | -- | 5037 | 2024-01-04 11:25:23 | | | |
| 2 | Peter Tang | + 1 word(s) | 5038 | 2024-01-05 01:51:23 | | |
Video Upload Options
Many of the ASDs exhibiting varying degrees of autism-like phenotypes have chromosomal anomalies in the Chr15q11–q13 region. Numerous potential candidate genes linked with ASD reside in this chromosomal segment. However, several clinical, in vivo, and in vitro studies selected one gene more frequently than others randomly and unbiasedly. This gene codes for UBE3A or Ubiquitin protein ligase E3A [also known as E6AP ubiquitin-protein ligase (E6AP)], an enzyme involved in the cellular degradation of proteins. This gene has been listed as one of the several genes with a high potential of causing ASD in the Autism Database. The gain of function mutations, triplication, or duplication in the UBE3A gene is also associated with ASDs like Angelman Syndrome (AS) and Dup15q Syndrome. The genetic imprinting of UBE3A in the brain and a preference for neuronal maternal-specific expression are the key features of various ASDs. Since the UBE3A gene is involved in two main important diseases associated with autism-like symptoms, there has been widespread research going on in understanding the link between this gene and autism.
1. Introduction
2. UBE3A—Structure and Function
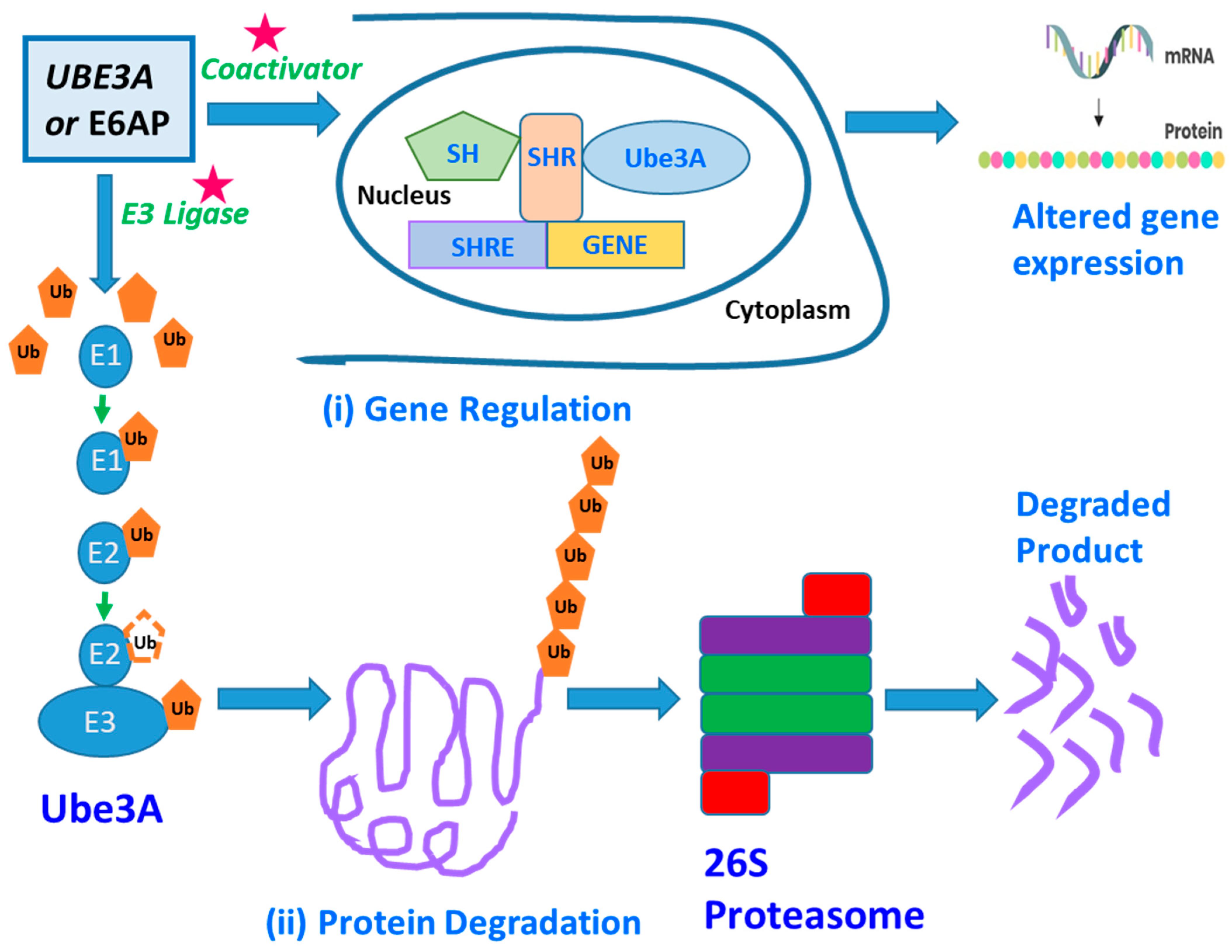
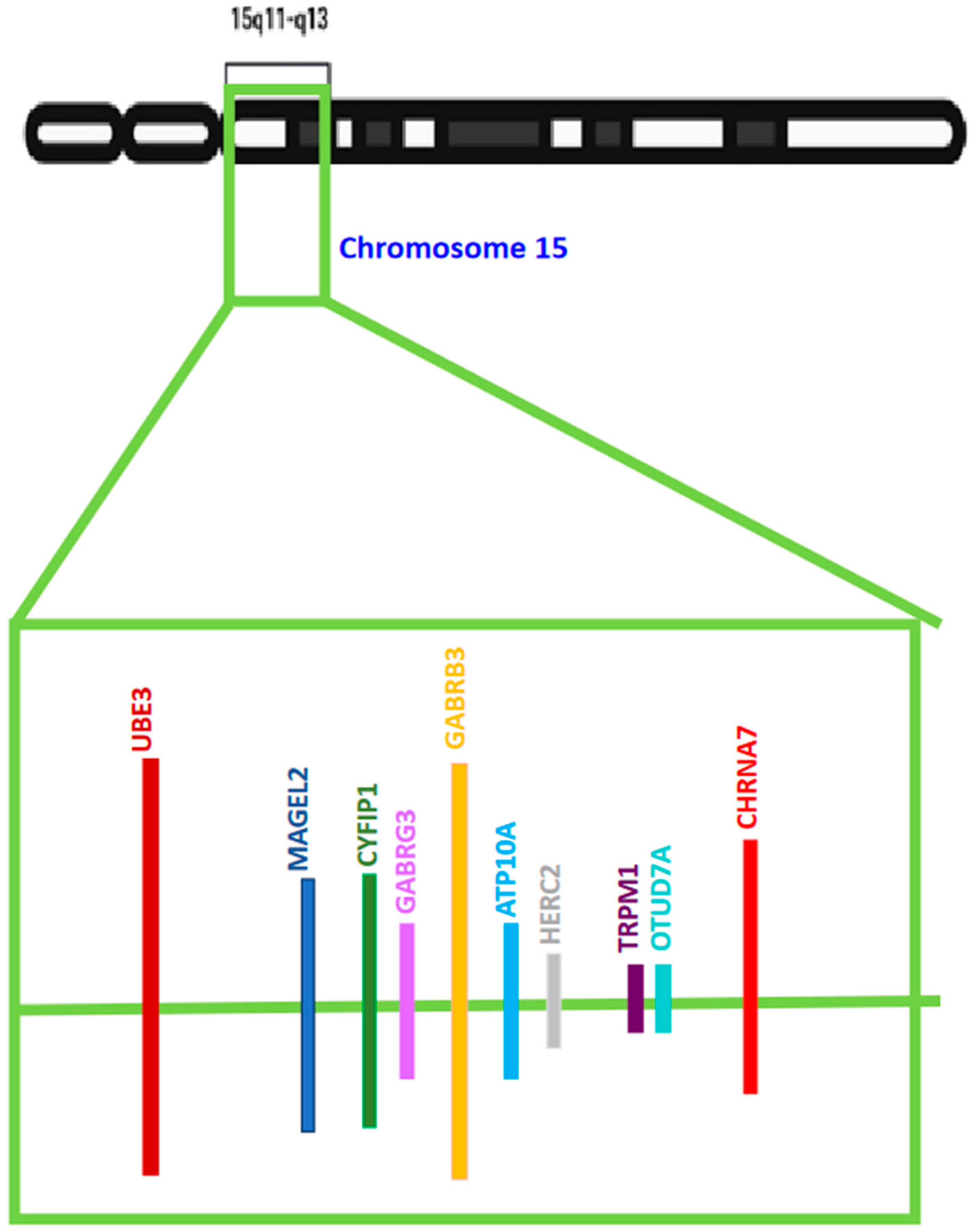
3. Potential Biomarkers Related to UBE3A
|
Biomarker |
Methodology |
Reference |
|---|---|---|
|
Beta EEG |
Spontaneous EEG recordings. |
[82] |
|
Awake electroencephalography (EEG) |
Spindle density, beta power, and slow-wave sleep (SWS) percentage in sleep EEG recordings as quantitative parameters for measurements of clinical manifestation of ASD symptoms. |
[42] |
|
UBE3A and SNORD116 mRNA levels |
Semi-quantitative techniques like reverse transcription droplet digital polymerase chain reaction (PCR) were used for UBE3A and SNORD116mRNA analysis from peripheral blood mononuclear cells (PBMCs). The data were normalized to a panel of internal control genes using the geNorm approach. |
[43] |
|
UBE3A protein |
Micro-dialysis of CSF fluid located in the rat hippocampus. |
[83] |
References
- Zeidan, J.; Fombonne, E.; Scorah, J.; Ibrahim, A.; Durkin, M.S.; Saxena, S.; Yusuf, A.; Shih, A.; Elsabbagh, M. Global prevalence of autism: A systematic review update. Autism Res. 2022, 15, 778–790.
- Masini, E.; Loi, E.; Vega-Benedetti, A.F.; Carta, M.; Doneddu, G.; Fadda, R.; Zavattari, P. An Overview of the Main Genetic, Epigenetic and Environmental Factors Involved in Autism Spectrum Disorder Focusing on Synaptic Activity. Int. J. Mol. Sci. 2020, 21, 8290.
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584.e523.
- Herrero, M.J.; Velmeshev, D.; Hernandez-Pineda, D.; Sethi, S.; Sorrells, S.; Banerjee, P.; Sullivan, C.; Gupta, A.R.; Kriegstein, A.R.; Corbin, J.G. Identification of amygdala-expressed genes associated with autism spectrum disorder. Mol. Autism 2020, 11, 39.
- Malzac, P.; Webber, H.; Moncla, A.; Graham, J.M.; Kukolich, M.; Williams, C.; Pagon, R.A.; Ramsdell, L.A.; Kishino, T.; Wagstaff, J. Mutation analysis of UBE3A in Angelman syndrome patients. Am. J. Hum. Genet. 1998, 62, 1353–1360.
- Lossie, A.C.; Whitney, M.M.; Amidon, D.; Dong, H.J.; Chen, P.; Theriaque, D.; Hutson, A.; Nicholls, R.D.; Zori, R.T.; Williams, C.A.; et al. Distinct phenotypes distinguish the molecular classes of Angelman syndrome. J. Med. Genet. 2001, 38, 834–845.
- Cook, E.H., Jr.; Lindgren, V.; Leventhal, B.L.; Courchesne, R.; Lincoln, A.; Shulman, C.; Lord, C.; Courchesne, E. Autism or atypical autism in maternally but not paternally derived proximal 15q duplication. Am. J. Hum. Genet. 1997, 60, 928–934.
- Schroer, R.J.; Phelan, M.C.; Michaelis, R.C.; Crawford, E.C.; Skinner, S.A.; Cuccaro, M.; Simensen, R.J.; Bishop, J.; Skinner, C.; Fender, D.; et al. Autism and maternally derived aberrations of chromosome 15q. Am. J. Med. Genet. 1998, 76, 327–336.
- Thomas, J.A.; Johnson, J.; Peterson Kraai, T.L.; Wilson, R.; Tartaglia, N.; LeRoux, J.; Beischel, L.; McGavran, L.; Hagerman, R.J. Genetic and clinical characterization of patients with an interstitial duplication 15q11–q13, emphasizing behavioral phenotype and response to treatment. Am. J. Med. Genet. A 2003, 119, 111–120.
- Hogart, A.; Wu, D.; LaSalle, J.M.; Schanen, N.C. The comorbidity of autism with the genomic disorders of chromosome 15q11.2–q13. Neurobiol. Dis. 2010, 38, 181–191.
- Borgatti, R.; Piccinelli, P.; Passoni, D.; Dalpra, L.; Miozzo, M.; Micheli, R.; Gagliardi, C.; Balottin, U. Relationship between clinical and genetic features in “inverted duplicated chromosome 15” patients. Pediatr. Neurol. 2001, 24, 111–116.
- Khatri, N.; Man, H.Y. The Autism and Angelman Syndrome Protein Ube3A/E6AP: The Gene, E3 Ligase Ubiquitination Targets and Neurobiological Functions. Front. Mol. Neurosci. 2019, 12, 109.
- Dindot, S.V.; Antalffy, B.A.; Bhattacharjee, M.B.; Beaudet, A.L. The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum. Mol. Genet. 2008, 17, 111–118.
- Rougeulle, C.; Glatt, H.; Lalande, M. The Angelman syndrome candidate gene, UBE3A/E6-AP, is imprinted in brain. Nat. Genet. 1997, 17, 14–15.
- Yamasaki, K.; Joh, K.; Ohta, T.; Masuzaki, H.; Ishimaru, T.; Mukai, T.; Niikawa, N.; Ogawa, M.; Wagstaff, J.; Kishino, T. Neurons but not glial cells show reciprocal imprinting of sense and antisense transcripts of Ube3a. Hum. Mol. Genet. 2003, 12, 837–847.
- Ortiz-Prado, E.; Iturralde, A.L.; Simbana-Rivera, K.; Gomez-Barreno, L.; Hidalgo, I.; Rubio-Neira, M.; Espinosa, N.; Izquierdo-Condoy, J.; Arteaga-Espinosa, M.E.; Lister, A.; et al. 15q Duplication Syndrome: Report on the First Patient from Ecuador with an Unusual Clinical Presentation. Case Rep. Med. 2021, 2021, 6662054.
- Urraca, N.; Hope, K.; Victor, A.K.; Belgard, T.G.; Memon, R.; Goorha, S.; Valdez, C.; Tran, Q.T.; Sanchez, S.; Ramirez, J.; et al. Significant transcriptional changes in 15q duplication but not Angelman syndrome deletion stem cell-derived neurons. Mol. Autism 2018, 9, 6.
- Hershko, A.; Ciechanover, A. The ubiquitin system for protein degradation. Annu. Rev. Biochem. 1992, 61, 761–807.
- Pelzer, C.; Kassner, I.; Matentzoglu, K.; Singh, R.K.; Wollscheid, H.P.; Scheffner, M.; Schmidtke, G.; Groettrup, M. UBE1L2, a novel E1 enzyme specific for ubiquitin. J. Biol. Chem. 2007, 282, 23010–23014.
- Shang, F.; Deng, G.; Obin, M.; Wu, C.C.; Gong, X.; Smith, D.; Laursen, R.A.; Andley, U.P.; Reddan, J.R.; Taylor, A. Ubiquitin-activating enzyme (E1) isoforms in lens epithelial cells: Origin of translation, E2 specificity and cellular localization determined with novel site-specific antibodies. Exp. Eye Res. 2001, 73, 827–836.
- LaSalle, J.M.; Reiter, L.T.; Chamberlain, S.J. Epigenetic regulation of UBE3A and roles in human neurodevelopmental disorders. Epigenomics 2015, 7, 1213–1228.
- Vu, T.H.; Hoffman, A.R. Imprinting of the Angelman syndrome gene, UBE3A, is restricted to brain. Nat. Genet. 1997, 17, 12–13.
- Jones, K.A.; Han, J.E.; DeBruyne, J.P.; Philpot, B.D. Persistent neuronal Ube3a expression in the suprachiasmatic nucleus of Angelman syndrome model mice. Sci. Rep. 2016, 6, 28238.
- Grier, M.D.; Carson, R.P.; Lagrange, A.H. Toward a Broader View of Ube3a in a Mouse Model of Angelman Syndrome: Expression in Brain, Spinal Cord, Sciatic Nerve and Glial Cells. PLoS ONE 2015, 10, e0124649.
- Judson, M.C.; Sosa-Pagan, J.O.; Del Cid, W.A.; Han, J.E.; Philpot, B.D. Allelic specificity of Ube3a expression in the mouse brain during postnatal development. J. Comp. Neurol. 2014, 522, 1874–1896.
- Runte, M.; Huttenhofer, A.; Gross, S.; Kiefmann, M.; Horsthemke, B.; Buiting, K. The IC-SNURF-SNRPN transcript serves as a host for multiple small nucleolar RNA species and as an antisense RNA for UBE3A. Hum. Mol. Genet. 2001, 10, 2687–2700.
- Kalsner, L.; Chamberlain, S.J. Prader-Willi, Angelman, and 15q11–q13 Duplication Syndromes. Pediatr. Clin. N. Am. 2015, 62, 587–606.
- Whittington, J.E.; Holland, A.J.; Webb, T.; Butler, J.; Clarke, D.; Boer, H. Population prevalence and estimated birth incidence and mortality rate for people with Prader-Willi syndrome in one UK Health Region. J. Med. Genet. 2001, 38, 792–798.
- Cassidy, S.B.; Schwartz, S.; Miller, J.L.; Driscoll, D.J. Prader-Willi syndrome. Genet. Med. 2012, 14, 10–26.
- Dierssen, M.; Ramakers, G.J. Dendritic pathology in mental retardation: From molecular genetics to neurobiology. Genes Brain Behav. 2006, 5 (Suppl. S2), 48–60.
- Frohlich, J.; Reiter, L.T.; Saravanapandian, V.; DiStefano, C.; Huberty, S.; Hyde, C.; Chamberlain, S.; Bearden, C.E.; Golshani, P.; Irimia, A.; et al. Mechanisms underlying the EEG biomarker in Dup15q syndrome. Mol. Autism 2019, 10, 29.
- Greer, P.L.; Hanayama, R.; Bloodgood, B.L.; Mardinly, A.R.; Lipton, D.M.; Flavell, S.W.; Kim, T.K.; Griffith, E.C.; Waldon, Z.; Maehr, R.; et al. The Angelman Syndrome protein Ube3A regulates synapse development by ubiquitinating arc. Cell 2010, 140, 704–716.
- Margolis, S.S.; Salogiannis, J.; Lipton, D.M.; Mandel-Brehm, C.; Wills, Z.P.; Mardinly, A.R.; Hu, L.; Greer, P.L.; Bikoff, J.B.; Ho, H.Y.; et al. EphB-mediated degradation of the RhoA GEF Ephexin5 relieves a developmental brake on excitatory synapse formation. Cell 2010, 143, 442–455.
- Sun, J.; Zhu, G.; Liu, Y.; Standley, S.; Ji, A.; Tunuguntla, R.; Wang, Y.; Claus, C.; Luo, Y.; Baudry, M.; et al. UBE3A Regulates Synaptic Plasticity and Learning and Memory by Controlling SK2 Channel Endocytosis. Cell Rep. 2015, 12, 449–461.
- Sadikovic, B.; Fernandes, P.; Zhang, V.W.; Ward, P.A.; Miloslavskaya, I.; Rhead, W.; Rosenbaum, R.; Gin, R.; Roa, B.; Fang, P. Mutation Update for UBE3A variants in Angelman syndrome. Hum. Mutat. 2014, 35, 1407–1417.
- Mishra, A.; Prabha, P.K.; Singla, R.; Kaur, G.; Sharma, A.R.; Joshi, R.; Suroy, B.; Medhi, B. Epigenetic Interface of Autism Spectrum Disorders (ASDs): Implications of Chromosome 15q11–q13 Segment. ACS Chem. Neurosci. 2022, 13, 1684–1696.
- Burette, A.C.; Judson, M.C.; Li, A.N.; Chang, E.F.; Seeley, W.W.; Philpot, B.D.; Weinberg, R.J. Subcellular organization of UBE3A in human cerebral cortex. Mol. Autism 2018, 9, 54.
- Beer-Romero, P.; Glass, S.; Rolfe, M. Antisense targeting of E6AP elevates p53 in HPV-infected cells but not in normal cells. Oncogene 1997, 14, 595–602.
- Dodge, A.; Willman, J.; Willman, M.; Nenninger, A.W.; Morrill, N.K.; Lamens, K.; Greene, H.; Weeber, E.J.; Nash, K.R. Identification of UBE3A Protein in CSF and Extracellular Space of the Hippocampus Suggest a Potential Novel Function in Synaptic Plasticity. Autism Res. 2021, 14, 645–655.
- Hegde, A.N.; Haynes, K.A.; Bach, S.V.; Beckelman, B.C. Local ubiquitin-proteasome-mediated proteolysis and long-term synaptic plasticity. Front. Mol. Neurosci. 2014, 7, 96.
- Hegde, A.N. The ubiquitin-proteasome pathway and synaptic plasticity. Learn Mem. 2010, 17, 314–327.
- Saitoh, S.; Buiting, K.; Cassidy, S.B.; Conroy, J.M.; Driscoll, D.J.; Gabriel, J.M.; Gillessen-Kaesbach, G.; Glenn, C.C.; Greenswag, L.R.; Horsthemke, B.; et al. Clinical spectrum and molecular diagnosis of Angelman and Prader-Willi syndrome patients with an imprinting mutation. Am. J. Med. Genet. 1997, 68, 195–206.
- Sebat, J.; Lakshmi, B.; Malhotra, D.; Troge, J.; Lese-Martin, C.; Walsh, T.; Yamrom, B.; Yoon, S.; Krasnitz, A.; Kendall, J.; et al. Strong association of de novo copy number mutations with autism. Science 2007, 316, 445–449.
- Morrow, E.M.; Yoo, S.Y.; Flavell, S.W.; Kim, T.K.; Lin, Y.; Hill, R.S.; Mukaddes, N.M.; Balkhy, S.; Gascon, G.; Hashmi, A.; et al. Identifying autism loci and genes by tracing recent shared ancestry. Science 2008, 321, 218–223.
- Glessner, J.T.; Wang, K.; Cai, G.; Korvatska, O.; Kim, C.E.; Wood, S.; Zhang, H.; Estes, A.; Brune, C.W.; Bradfield, J.P.; et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 2009, 459, 569–573.
- Weiss, L.A.; Shen, Y.; Korn, J.M.; Arking, D.E.; Miller, D.T.; Fossdal, R.; Saemundsen, E.; Stefansson, H.; Ferreira, M.A.; Green, T.; et al. Association between microdeletion and microduplication at 16p11.2 and autism. N. Engl. J. Med. 2008, 358, 667–675.
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221.
- Cheon, S.; Dean, M.; Chahrour, M. The ubiquitin proteasome pathway in neuropsychiatric disorders. Neurobiol. Learn. Mem. 2019, 165, 106791.
- Buiting, K.; Williams, C.; Horsthemke, B. Angelman syndrome—insights into a rare neurogenetic disorder. Nat. Rev. Neurol. 2016, 12, 584–593.
- Kishino, T.; Wagstaff, J. Genomic organization of the UBE3A/E6-AP gene and related pseudogenes. Genomics 1998, 47, 101–107.
- Condon, K.H.; Ho, J.; Robinson, C.G.; Hanus, C.; Ehlers, M.D. The Angelman syndrome protein Ube3a/E6AP is required for Golgi acidification and surface protein sialylation. J. Neurosci. 2013, 33, 3799–3814.
- Krishnan, V.; Stoppel, D.C.; Nong, Y.; Johnson, M.A.; Nadler, M.J.; Ozkaynak, E.; Teng, B.L.; Nagakura, I.; Mohammad, F.; Silva, M.A.; et al. Autism gene Ube3a and seizures impair sociability by repressing VTA Cbln1. Nature 2017, 543, 507–512.
- Su, H.; Fan, W.; Coskun, P.E.; Vesa, J.; Gold, J.A.; Jiang, Y.H.; Potluri, P.; Procaccio, V.; Acab, A.; Weiss, J.H.; et al. Mitochondrial dysfunction in CA1 hippocampal neurons of the UBE3A deficient mouse model for Angelman syndrome. Neurosci. Lett. 2011, 487, 129–133.
- Llewellyn, K.J.; Nalbandian, A.; Gomez, A.; Wei, D.; Walker, N.; Kimonis, V.E. Administration of CoQ10 analogue ameliorates dysfunction of the mitochondrial respiratory chain in a mouse model of Angelman syndrome. Neurobiol. Dis. 2015, 76, 77–86.
- Santini, E.; Turner, K.L.; Ramaraj, A.B.; Murphy, M.P.; Klann, E.; Kaphzan, H. Mitochondrial Superoxide Contributes to Hippocampal Synaptic Dysfunction and Memory Deficits in Angelman Syndrome Model Mice. J. Neurosci. 2015, 35, 16213–16220.
- Nawaz, Z.; Lonard, D.M.; Smith, C.L.; Lev-Lehman, E.; Tsai, S.Y.; Tsai, M.J.; O’Malley, B.W. The Angelman syndrome-associated protein, E6-AP, is a coactivator for the nuclear hormone receptor superfamily. Mol. Cell. Biol. 1999, 19, 1182–1189.
- Smith, C.L.; DeVera, D.G.; Lamb, D.J.; Nawaz, Z.; Jiang, Y.H.; Beaudet, A.L.; O’Malley, B.W. Genetic ablation of the steroid receptor coactivator-ubiquitin ligase, E6-AP, results in tissue-selective steroid hormone resistance and defects in reproduction. Mol. Cell. Biol. 2002, 22, 525–535.
- Khan, O.Y.; Fu, G.; Ismail, A.; Srinivasan, S.; Cao, X.; Tu, Y.; Lu, S.; Nawaz, Z. Multifunction steroid receptor coactivator, E6-associated protein, is involved in development of the prostate gland. Mol. Endocrinol. 2006, 20, 544–559.
- Oda, H.; Kumar, S.; Howley, P.M. Regulation of the Src family tyrosine kinase Blk through E6AP-mediated ubiquitination. Proc. Natl. Acad. Sci. USA 1999, 96, 9557–9562.
- Mishra, A.; Jana, N.R. Regulation of turnover of tumor suppressor p53 and cell growth by E6-AP, a ubiquitin protein ligase mutated in Angelman mental retardation syndrome. Cell. Mol. Life Sci. 2008, 65, 656–666.
- Mishra, A.; Godavarthi, S.K.; Jana, N.R. UBE3A/E6-AP regulates cell proliferation by promoting proteasomal degradation of p27. Neurobiol. Dis. 2009, 36, 26–34.
- Reiter, L.T.; Seagroves, T.N.; Bowers, M.; Bier, E. Expression of the Rho-GEF Pbl/ECT2 is regulated by the UBE3A E3 ubiquitin ligase. Hum. Mol. Genet. 2006, 15, 2825–2835.
- Kaphzan, H.; Buffington, S.A.; Jung, J.I.; Rasband, M.N.; Klann, E. Alterations in intrinsic membrane properties and the axon initial segment in a mouse model of Angelman syndrome. J. Neurosci. 2011, 31, 17637–17648.
- Kaphzan, H.; Hernandez, P.; Jung, J.I.; Cowansage, K.K.; Deinhardt, K.; Chao, M.V.; Abel, T.; Klann, E. Reversal of impaired hippocampal long-term potentiation and contextual fear memory deficits in Angelman syndrome model mice by ErbB inhibitors. Biol. Psychiatry 2012, 72, 182–190.
- Mabb, A.M.; Judson, M.C.; Zylka, M.J.; Philpot, B.D. Angelman syndrome: Insights into genomic imprinting and neurodevelopmental phenotypes. Trends Neurosci. 2011, 34, 293–303.
- Mishra, A.; Dikshit, P.; Purkayastha, S.; Sharma, J.; Nukina, N.; Jana, N.R. E6-AP promotes misfolded polyglutamine proteins for proteasomal degradation and suppresses polyglutamine protein aggregation and toxicity. J. Biol. Chem. 2008, 283, 7648–7656.
- Mishra, A.; Godavarthi, S.K.; Maheshwari, M.; Goswami, A.; Jana, N.R. The ubiquitin ligase E6-AP is induced and recruited to aggresomes in response to proteasome inhibition and may be involved in the ubiquitination of Hsp70-bound misfolded proteins. J. Biol. Chem. 2009, 284, 10537–10545.
- Mulherkar, S.A.; Sharma, J.; Jana, N.R. The ubiquitin ligase E6-AP promotes degradation of alpha-synuclein. J. Neurochem. 2009, 110, 1955–1964.
- Valluy, J.; Bicker, S.; Aksoy-Aksel, A.; Lackinger, M.; Sumer, S.; Fiore, R.; Wust, T.; Seffer, D.; Metge, F.; Dieterich, C.; et al. A coding-independent function of an alternative Ube3a transcript during neuronal development. Nat. Neurosci. 2015, 18, 666–673.
- Jessen, K.R. Glial cells. Int. J. Biochem. Cell Biol. 2004, 36, 1861–1867.
- Nakatani, J.; Tamada, K.; Hatanaka, F.; Ise, S.; Ohta, H.; Inoue, K.; Tomonaga, S.; Watanabe, Y.; Chung, Y.J.; Banerjee, R.; et al. Abnormal behavior in a chromosome-engineered mouse model for human 15q11–q13 duplication seen in autism. Cell 2009, 137, 1235–1246.
- Smith, S.E.; Zhou, Y.D.; Zhang, G.; Jin, Z.; Stoppel, D.C.; Anderson, M.P. Increased gene dosage of Ube3a results in autism traits and decreased glutamate synaptic transmission in mice. Sci. Transl. Med. 2011, 3, 103ra197.
- Kuhnle, S.; Mothes, B.; Matentzoglu, K.; Scheffner, M. Role of the ubiquitin ligase E6AP/UBE3A in controlling levels of the synaptic protein Arc. Proc. Natl. Acad. Sci. USA 2013, 110, 8888–8893.
- Jiang, Y.H.; Armstrong, D.; Albrecht, U.; Atkins, C.M.; Noebels, J.L.; Eichele, G.; Sweatt, J.D.; Beaudet, A.L. Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron 1998, 21, 799–811.
- Heck, D.H.; Zhao, Y.; Roy, S.; LeDoux, M.S.; Reiter, L.T. Analysis of cerebellar function in Ube3a-deficient mice reveals novel genotype-specific behaviors. Hum. Mol. Genet. 2008, 17, 2181–2189.
- Godavarthi, S.K.; Dey, P.; Maheshwari, M.; Jana, N.R. Defective glucocorticoid hormone receptor signaling leads to increased stress and anxiety in a mouse model of Angelman syndrome. Hum. Mol. Genet. 2012, 21, 1824–1834.
- Shi, S.Q.; Bichell, T.J.; Ihrie, R.A.; Johnson, C.H. Ube3a imprinting impairs circadian robustness in Angelman syndrome models. Curr. Biol. 2015, 25, 537–545.
- Weeber, E.J.; Jiang, Y.H.; Elgersma, Y.; Varga, A.W.; Carrasquillo, Y.; Brown, S.E.; Christian, J.M.; Mirnikjoo, B.; Silva, A.; Beaudet, A.L.; et al. Derangements of hippocampal calcium/calmodulin-dependent protein kinase II in a mouse model for Angelman mental retardation syndrome. J. Neurosci. 2003, 23, 2634–2644.
- Yashiro, K.; Riday, T.T.; Condon, K.H.; Roberts, A.C.; Bernardo, D.R.; Prakash, R.; Weinberg, R.J.; Ehlers, M.D.; Philpot, B.D. Ube3a is required for experience-dependent maturation of the neocortex. Nat. Neurosci. 2009, 12, 777–783.
- Sato, M.; Stryker, M.P. Genomic imprinting of experience-dependent cortical plasticity by the ubiquitin ligase gene Ube3a. Proc. Natl. Acad. Sci. USA 2010, 107, 5611–5616.
- Wallace, M.L.; Burette, A.C.; Weinberg, R.J.; Philpot, B.D. Maternal loss of Ube3a produces an excitatory/inhibitory imbalance through neuron type-specific synaptic defects. Neuron 2012, 74, 793–800.
- Gregory, S.G.; Connelly, J.J.; Towers, A.J.; Johnson, J.; Biscocho, D.; Markunas, C.A.; Lintas, C.; Abramson, R.K.; Wright, H.H.; Ellis, P.; et al. Genomic and epigenetic evidence for oxytocin receptor deficiency in autism. BMC Med. 2009, 7, 62.
- Baker, E.K.; Butler, M.G.; Hartin, S.N.; Ling, L.; Bui, M.; Francis, D.; Rogers, C.; Field, M.J.; Slee, J.; Gamage, D.; et al. Relationships between UBE3A and SNORD116 expression and features of autism in chromosome 15 imprinting disorders. Transl. Psychiatry 2020, 10, 362.
- Urraca, N.; Cleary, J.; Brewer, V.; Pivnick, E.K.; McVicar, K.; Thibert, R.L.; Schanen, N.C.; Esmer, C.; Lamport, D.; Reiter, L.T. The interstitial duplication 15q11.2–q13 syndrome includes autism, mild facial anomalies and a characteristic EEG signature. Autism Res. 2013, 6, 268–279.
- Al Ageeli, E.; Drunat, S.; Delanoe, C.; Perrin, L.; Baumann, C.; Capri, Y.; Fabre-Teste, J.; Aboura, A.; Dupont, C.; Auvin, S.; et al. Duplication of the 15q11–q13 region: Clinical and genetic study of 30 new cases. Eur. J. Med. Genet. 2014, 57, 5–14.
- Patzold, L.M.; Richdale, A.L.; Tonge, B.J. An investigation into sleep characteristics of children with autism and Asperger’s Disorder. J. Paediatr. Child Health 1998, 34, 528–533.
- Wiggs, L.; Stores, G. Sleep patterns and sleep disorders in children with autistic spectrum disorders: Insights using parent report and actigraphy. Dev. Med. Child Neurol. 2004, 46, 372–380.
- Liu, X.; Hubbard, J.A.; Fabes, R.A.; Adam, J.B. Sleep disturbances and correlates of children with autism spectrum disorders. Child Psychiatry Hum. Dev. 2006, 37, 179–191.
- Souders, M.C.; Mason, T.B.; Valladares, O.; Bucan, M.; Levy, S.E.; Mandell, D.S.; Weaver, T.E.; Pinto-Martin, J. Sleep behaviors and sleep quality in children with autism spectrum disorders. Sleep 2009, 32, 1566–1578.
- Devnani, P.A.; Hegde, A.U. Autism and sleep disorders. J. Pediatr. Neurosci. 2015, 10, 304–307.
- Cohen, S.; Conduit, R.; Lockley, S.W.; Rajaratnam, S.M.; Cornish, K.M. The relationship between sleep and behavior in autism spectrum disorder (ASD): A review. J. Neurodev. Disord. 2014, 6, 44.
- Ferrarelli, F.; Tononi, G. Reduced sleep spindle activity point to a TRN-MD thalamus-PFC circuit dysfunction in schizophrenia. Schizophr. Res. 2017, 180, 36–43.
- Wamsley, E.J.; Tucker, M.A.; Shinn, A.K.; Ono, K.E.; McKinley, S.K.; Ely, A.V.; Goff, D.C.; Stickgold, R.; Manoach, D.S. Reduced sleep spindles and spindle coherence in schizophrenia: Mechanisms of impaired memory consolidation? Biol. Psychiatry 2012, 71, 154–161.
- Limoges, E.; Mottron, L.; Bolduc, C.; Berthiaume, C.; Godbout, R. Atypical sleep architecture and the autism phenotype. Brain 2005, 128 Pt 5, 1049–1061.
- Tessier, S.; Lambert, A.; Chicoine, M.; Scherzer, P.; Soulieres, I.; Godbout, R. Intelligence measures and stage 2 sleep in typically-developing and autistic children. Int. J. Psychophysiol. 2015, 97, 58–65.
- Christensen, J.A.; Kempfner, J.; Zoetmulder, M.; Leonthin, H.L.; Arvastson, L.; Christensen, S.R.; Sorensen, H.B.; Jennum, P. Decreased sleep spindle density in patients with idiopathic REM sleep behavior disorder and patients with Parkinson’s disease. Clin. Neurophysiol. 2014, 125, 512–519.
- Fernandez, L.M.J.; Luthi, A. Sleep Spindles: Mechanisms and Functions. Physiol. Rev. 2020, 100, 805–868.
- Johnston, M.V.; Ammanuel, S.; O’Driscoll, C.; Wozniak, A.; Naidu, S.; Kadam, S.D. Twenty-four hour quantitative-EEG and in-vivo glutamate biosensor detects activity and circadian rhythm dependent biomarkers of pathogenesis in Mecp2 null mice. Front. Syst. Neurosci. 2014, 8, 118.
- Ammanuel, S.; Chan, W.C.; Adler, D.A.; Lakshamanan, B.M.; Gupta, S.S.; Ewen, J.B.; Johnston, M.V.; Marcus, C.L.; Naidu, S.; Kadam, S.D. Heightened Delta Power during Slow-Wave-Sleep in Patients with Rett Syndrome Associated with Poor Sleep Efficiency. PLoS ONE 2015, 10, e0138113.
- Arazi, A.; Meiri, G.; Danan, D.; Michaelovski, A.; Flusser, H.; Menashe, I.; Tarasiuk, A.; Dinstein, I. Reduced sleep pressure in young children with autism. Sleep 2020, 43, zsz309.
- Saravanapandian, V.; Nadkarni, D.; Hsu, S.H.; Hussain, S.A.; Maski, K.; Golshani, P.; Colwell, C.S.; Balasubramanian, S.; Dixon, A.; Geschwind, D.H.; et al. Abnormal sleep physiology in children with 15q11.2–q13.1 duplication (Dup15q) syndrome. Mol. Autism 2021, 12, 54.

