Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Hong Seok Choi | -- | 2402 | 2024-01-04 01:35:12 | | | |
| 2 | Peter Tang | + 2 word(s) | 2404 | 2024-01-04 02:49:47 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Bhattarai, P.Y.; Kim, G.; Bhandari, D.; Shrestha, P.; Choi, H.S. Regulation of m6A Methylome in Cancer. Encyclopedia. Available online: https://encyclopedia.pub/entry/53400 (accessed on 04 July 2026).
Bhattarai PY, Kim G, Bhandari D, Shrestha P, Choi HS. Regulation of m6A Methylome in Cancer. Encyclopedia. Available at: https://encyclopedia.pub/entry/53400. Accessed July 04, 2026.
Bhattarai, Poshan Yugal, Garam Kim, Dibikshya Bhandari, Pratikshya Shrestha, Hong Seok Choi. "Regulation of m6A Methylome in Cancer" Encyclopedia, https://encyclopedia.pub/entry/53400 (accessed July 04, 2026).
Bhattarai, P.Y., Kim, G., Bhandari, D., Shrestha, P., & Choi, H.S. (2024, January 04). Regulation of m6A Methylome in Cancer. In Encyclopedia. https://encyclopedia.pub/entry/53400
Bhattarai, Poshan Yugal, et al. "Regulation of m6A Methylome in Cancer." Encyclopedia. Web. 04 January, 2024.
Copy Citation
Reversible N6-adenosine methylation of mRNA, referred to as m6A modification, has emerged as an important regulator of post-transcriptional RNA processing. Numerous studies have highlighted its crucial role in the pathogenesis of diverse diseases, particularly cancer. Post-translational modifications of m6A-related proteins play a fundamental role in regulating the m6A methylome, thereby influencing the fate of m6A-methylated RNA. A comprehensive understanding of the mechanisms that regulate m6A-related proteins and the factors contributing to the specificity of m6A deposition has the potential to unveil novel therapeutic strategies for cancer treatment.
N6-methyladenosine (m6A) modification
m6A-related proteins
post-translational modification
m6A specificity
novel therapeutic targets
1. Introduction
Ribonucleotides in RNA molecules undergo a diverse range of chemical modifications of nitrogenous bases and ribose sugars. The term “epitranscriptome” collectively refers to all the chemical modifications in RNA molecules. To date, more than 170 types of chemical modifications have been reported in RNAs, including messenger RNA (mRNA), ribosomal RNA (rRNA), transfer RNA (tRNA), long noncoding RNA (lncRNA), and small nucleolar RNA [1]. N6-methyladenosine (m6A) is the most common internal chemical modification of mRNA. The m6A modification is catalyzed by a nuclear methyltransferase complex comprising methyltransferase-like (METTL)3, METTL14, and Wilms’ tumor-associated protein (WTAP) [2]. Proteins with a YT521-B homology (YTH) domain, such as YTHDF1, F2, F3, C1, and C2, specifically recognize and bind to m6A nucleotides [3]. In contrast, RNA demethylases with an Alk domain, such as ALKBH5 and ALKBH9 (FTO), can remove methyl groups from m6A [3].
2. Post-Translational Modification of m6A-Related Proteins
A profound understanding of m6A-related proteins has prompted inquiries into their regulation in both physiology and diseases. Herein, the researchers summarize the key post-translational modifications (PTMs) of m6A-related proteins verified with biochemical experiments and their influence on the regulation of the m6A methylome, particularly in cancer cells. Additionally, the researchers discuss the significance of these PTMs from a therapeutic perspective. The major PTMs found in m6A proteins and the signaling pathways that govern them are illustrated in Figure 1.
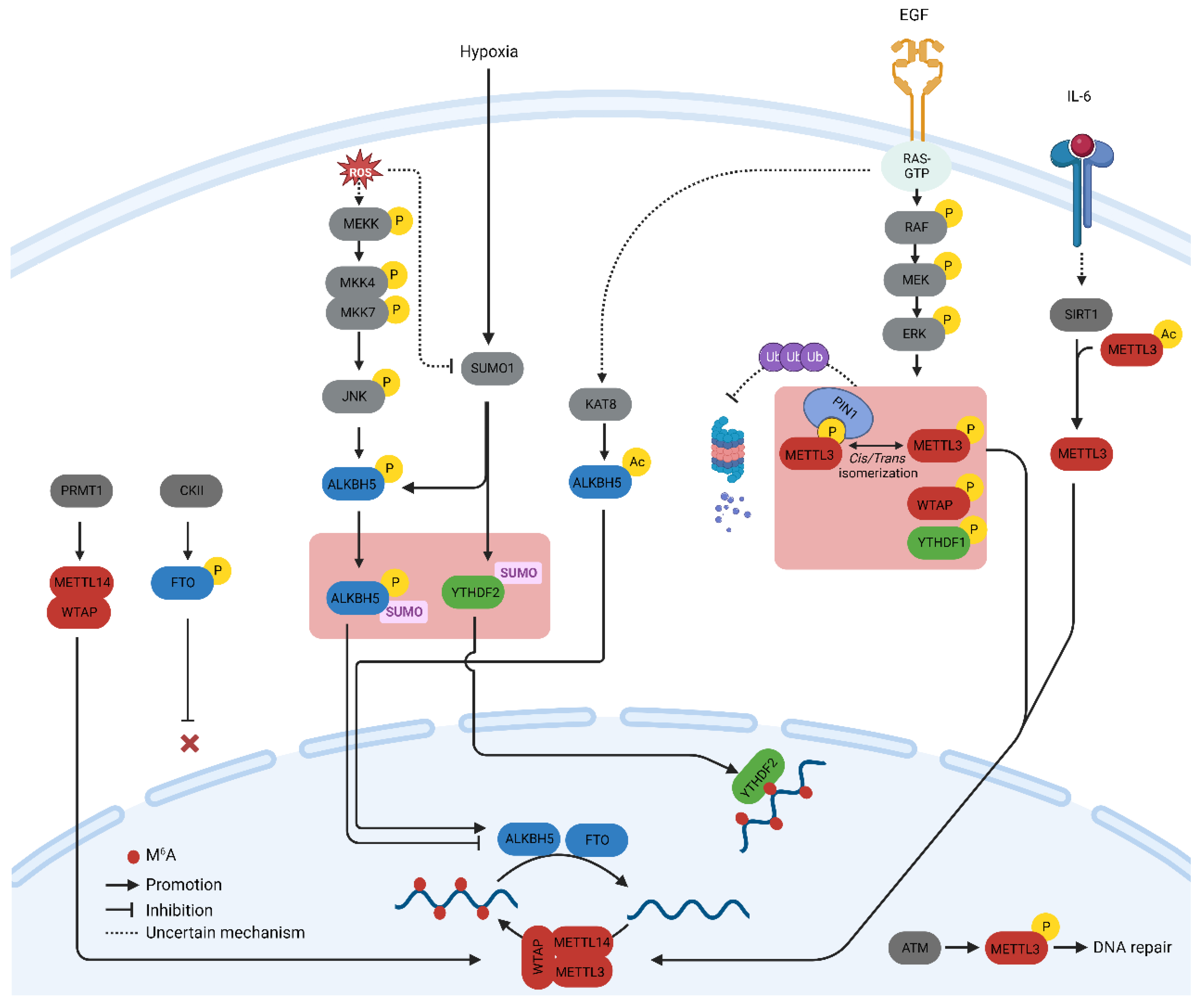
Figure 1. Schematic depiction of signaling pathways orchestrating the post-translational modification of m6A-related proteins.
2.1. Phosphorylation
Protein phosphorylation is the most common and arguably the most important PTM that regulates cellular processes such as signal transduction, protein synthesis, cell division, and apoptosis [4]. Phosphorylation is mediated by a cascade of protein kinases that are crucial regulators of m6A-related proteins. The phosphorylation of METTL3 and WTAP at specific serine sites is driven by the extracellular signal-regulated kinases (ERK) pathway. ERK phosphorylates METTL3 at S43, S50, and S525, while it phosphorylates WTAP at S306 and S341 [5]. This phosphorylation, along with the subsequent deubiquitination by USP5, stabilizes the m6A methyltransferase complex. Reduced METTL3/WTAP phosphorylation increases the stability of m6A-labeled pluripotent factor transcripts, such as Nanong, and maintains pluripotency in mouse embryonic stem cells [5]. The same phosphorylation pattern, which promotes tumorigenesis, has been observed in breast and melanoma cancer cells [5]. The ERK pathway is often activated by extracellular growth factors such as epidermal growth factor (EGF). Activation of the epidermal growth factor receptor (EGFR) by EGF triggers the phosphorylation of METTL3, which subsequently catalyzes the methylation of small nuclear 7SK mRNA. This methylation event enhances the affinity of 7SK for heterogeneous nuclear ribonucleoproteins, resulting in the dissociation of the HEXIM1/P-TEFb complex and the promotion of transcriptional elongation. This process underscores the intricate role of the MEK/ERK pathway in the regulation of METTL3 function [6].
The S43 site in METTL3 is also phosphorylated by the ataxia telangiectasia mutation (ATM) in response to double-strand breaks (DSBs). The ATM kinase triggers the activation of METTL3, which is subsequently guided to DNA damage sites, where it introduces m6A modifications to adenosine within DNA damage-associated RNAs.
Reactive Oxygen Species (ROS) play a significant role in increasing global m6A levels by regulating ALKBH5 [7]. This rapid and effective upregulation of m6A affects thousands of genes, particularly those involved in DNA damage repair. Mechanistically, ROS stimulate ALKBH5 SUMOylation via ERK/JNK signaling-mediated phosphorylation at serine residues S87 and S325, which, in turn, inhibits the m6A demethylase activity of ALKBH5 by impeding substrate accessibility. The ROS-triggered ERK/JNK/ALKBH5 pathway is also active in hematopoietic stem/progenitor cells (HSPCs) in vivo in mice, highlighting its physiological significance in safeguarding genomic stability within HSPCs. This cited study revealed a molecular mechanism involving ALKBH5 phosphorylation and increased mRNA m6A levels that preserve cellular genomic integrity in response to ROS [7].
2.2. Methylation
The methylation of Lys and Arg amino acids on non-histone proteins is a common post-translational modification that regulates signal transduction via various pathways and influences cellular functions, such as chromatin remodeling, gene transcription, and DNA repair [8]. Notably, protein and RNA methylation share the same substrate as the source of the methyl group, that is, S-adenosine methionine (SAM), implying that a detailed understanding of the relationship between these two modifications may offer a more powerful therapeutic option.
Arginine methylation of METTL14 at arginine methyltransferases (PRMT) by arginine 255 (R255) enhances the stability of the interaction between the m6A methyltransferase complex and its RNA substrate [9]. This in turn boosts global m6A modifications and supports the differentiation of mouse embryonic stem cells (mESCs) into the endoderm. These findings highlight the intricate interplay between protein and RNA methylation in the regulation of gene expression [9]. Consequently, the suppression of PRMT with MS023 inhibits cancer cell proliferation induced by METTL14 overexpression [10]. In addition, PRMT1-induced methylation of WTAP promotes m6A methyltransferase function in multiple myeloma [11].
2.3. Acetylation
Protein acetylation uses acetyl-CoA, a component of the cellular metabolic pathway, as a source of an acetyl group [12]. The incorporation of metabolites into the protein structure allows cells to incorporate metabolic cues into intricate cellular decision-making processes such as protein acetylation. Therefore, studies on acetylation of m6A-related proteins offer valuable insights into the mechanisms by which cellular metabolism affects the global m6A methylome.
METTL3 acetylation regulates its localization and profoundly affects metastatic spread. IL-6, whose mRNA transcript undergoes METTL3-mediated m6A modification, promotes METTL3 deacetylation via the nicotinamide adenine dinucleotide (NAD)-dependent histone deacetylase silent information regulator (SIRT1) [13]. Deacetylation of METTL3 promotes its nuclear translocation and consequently elevates global m6A levels. This deacetylation-driven shift in METTL3 to the nucleus can be counteracted by inhibiting SIRT1, an effect further potentiated with aspirin treatment, which ultimately impairs lung metastasis. Similarly, acetyl-CoA acetyltransferase 1 (ACAT1) induces METTL3 acetylation and promotes its stability by inhibiting ubiquitin-mediated proteasomal degradation [14]. METTL3 upregulation mediated by ACAT1 is associated with increased migration and invasion of triple-negative breast cancer (TNBC) cells [14].
2.4. SUMOylation
SUMOylation is a process that entails the covalent bonding of a protein from the small ubiquitin-like modifier (SUMO) family to lysine residues within target proteins. This occurs through an enzymatic cascade that is similar to the ubiquitination pathway but with distinct characteristics [15]. SUMO1 predominantly modifies METTL3 at lysine residues K177, K211, K212, and K215, and these modifications can be diminished through the action of the SUMO1-specific protease, sentrin/SUMO-specific protease 1 (SENP1) [16]. The SUMOylation of METTL3 does not impact its stability, localization, or interaction with METTL14 and WTAP. However, it notably suppresses its m6A methyltransferase activity, leading to a reduction in m6A levels within mRNAs. The modification of m6A in mRNA molecules, induced by METTL3 SUMOylation, has direct implications for alterations in gene expression profiles. These changes, in turn, can influence processes such as soft-agar colony formation and xenograft tumor growth in H1299 cells [16].
2.5. O-GlcNAcylation
O-GlcNAcylation is a PTM that responds to nutrient availability and stress. This includes the addition of O-linked N-acetylglucosamine groups to serine and threonine residues of proteins. O-GlcNAc modifies the m6A mRNA reader YTHDF1 and fine-tunes its nuclear translocation [17]. O-GlcNAc transferase (OGT) binds to YTHDF1 and modifies Ser196/Ser197/Ser198 sites. Moreover, O-GlcNAcylation augments the cytosolic localization of YTHDF1 by strengthening its interaction with chromosomal maintenance 1 (Crm1), also recognized as exportin 1. This enhancement results in the upregulation of translation efficiency for specific downstream targets, including c-Myc, in colon cancer cells.
3. Transcriptional Activation of m6A-Related Genes
Compared with the PTMs of m6A genes, the transcriptional mechanisms that regulate the expression of these genes remain poorly understood. This suggests that the focus should shift toward the investigation of these mechanisms. In particular, gaining a deeper understanding of how the activation of specific cell signaling pathways triggers the transcription of specific m6A-related genes holds great promise for the development of novel therapeutic strategies for cancer treatment. Additionally, these mechanisms shed light on the larger picture of how the m6A methylome is shaped throughout the various stages of carcinogenesis, ranging from neoplastic transformation to drug resistance.
In gastric cancer (GC) cells, the activation of METTL3 transcription is induced by the promotion of the P300-mediated H3K27 acetylation of its promoter [18]. This, in turn, stimulates an m6A modification on hepatoma-derived growth factor (HDGF) mRNA. The m6A site on HDGF mRNA is subsequently recognized and binds to the m6A reader, insulin-like growth factor 2 mRNA-binding protein 3 (IGF2BP3), resulting in enhanced HDGF mRNA stability. Tumor angiogenesis is promoted by the secretion of HDGF, whereas the activation of GLUT4 and ENO2 expression by nuclear HDGF leads to an increase in glycolysis in GC cells. This increase in glycolysis is associated with subsequent tumor growth and liver metastasis [18].
Cigarette smoke condensation (CSC) induces the hypomethylation of the METTL3 promoter, resulting in elevated expression of METTL3 in pancreatic duct epithelial cells. Following this, the oncogenic primary microRNA-25 (miR-25) undergoes excessive maturation due to cigarette smoke condensate (CSC), facilitated by increased m6A modification mediated by nuclear factor-kappa B-associated protein (NKAP). The mature forms, miR-25 and miR-25-3p, act to suppress PH domain leucine-rich repeat protein phosphatase 2 (PHLPP2), consequently activating the oncogenic AKT-p70S6K signaling pathway and inducing malignant phenotypes in pancreatic cancer cells [19].
The master regulator of the tumorigenesis transcription factor, myelocytomatosis (MYC), binds to the promoter of the m6A reader, IGF2BP3, and activates transcription. Moreover, IGF2BP3 promotes the stability of m6A-modified KPNA2, leading to cell proliferation and metastasis in nasopharyngeal carcinoma cells [20].
These studies, although limited in number, have demonstrated that the expression of m6A-related genes may be influenced by the extracellular environment through transcriptional mechanisms. In particular, exploring how epigenetic modifications affect the expression of m6A genes and shape the epitranscriptome is an intriguing topic for further research. Such investigations would illustrate the dynamic interplay between epigenetics and the epitranscriptome in cancer cells.
4. Regulation of m6A Specificity
Despite a clear understanding of m6A methylation machinery, the mechanisms governing the specificity of m6A deposition remain elusive. m6A-IP-Seq experiments have revealed that m6A in human cells is predominantly enriched in the 3′-untreanslated (3′-UTR) region to regulate the translation efficiency and stability of mRNA [21]. In addition, m6A is highly enriched in mRNA containing long exons [22]. Furthermore, the m6A modification of specific mRNA is changeable depending on extracellular signals, nutrient availability, developmental stage, and response to chemotherapy in cancer cells [23]. The characteristic distribution pattern of m6A in the transcriptome raises questions about the mechanisms that regulate its specificity. To date, three potential mechanisms have been reported that underscore the roles of transcription factors, epigenetic modifications, and exon architecture in the pattern of m6A methylation, as summarized in Figure 2. These findings collectively indicate a close association between the transcriptional process and m6A modification. Specifically, transcription factors and co-activators have been identified as key players in recruiting the m6A methyltransferase complex to specific gene promoters, thereby promoting co-transcriptional m6A modification of the mRNA transcribed from the respective promoter. While the study of transcription factors has traditionally focused on their role in transcriptional regulation, the presented data highlight an additional role for these factors in post-transcriptional RNA processing. Consequently, their involvement in facilitating m6A modification suggests a broader impact on translation efficiency, expanding the understanding of the multifaceted functions of transcription factors beyond their well-established transcriptional regulatory roles. Moreover, epigenetic modifications, such as histone methylation, play a pivotal role as markers in recruiting the methyltransferase complex. Recent studies have highlighted the significant contribution of exon architecture in defining the distinctive pattern of m6A modification observed in mRNA. This exon-centric model precisely elucidates the reasons behind the enrichment of m6A modifications in the last exon or within long internal exons. However, the understanding of the upstream signaling pathways that influence the distribution of m6A marks across the transcriptome remains incomplete.
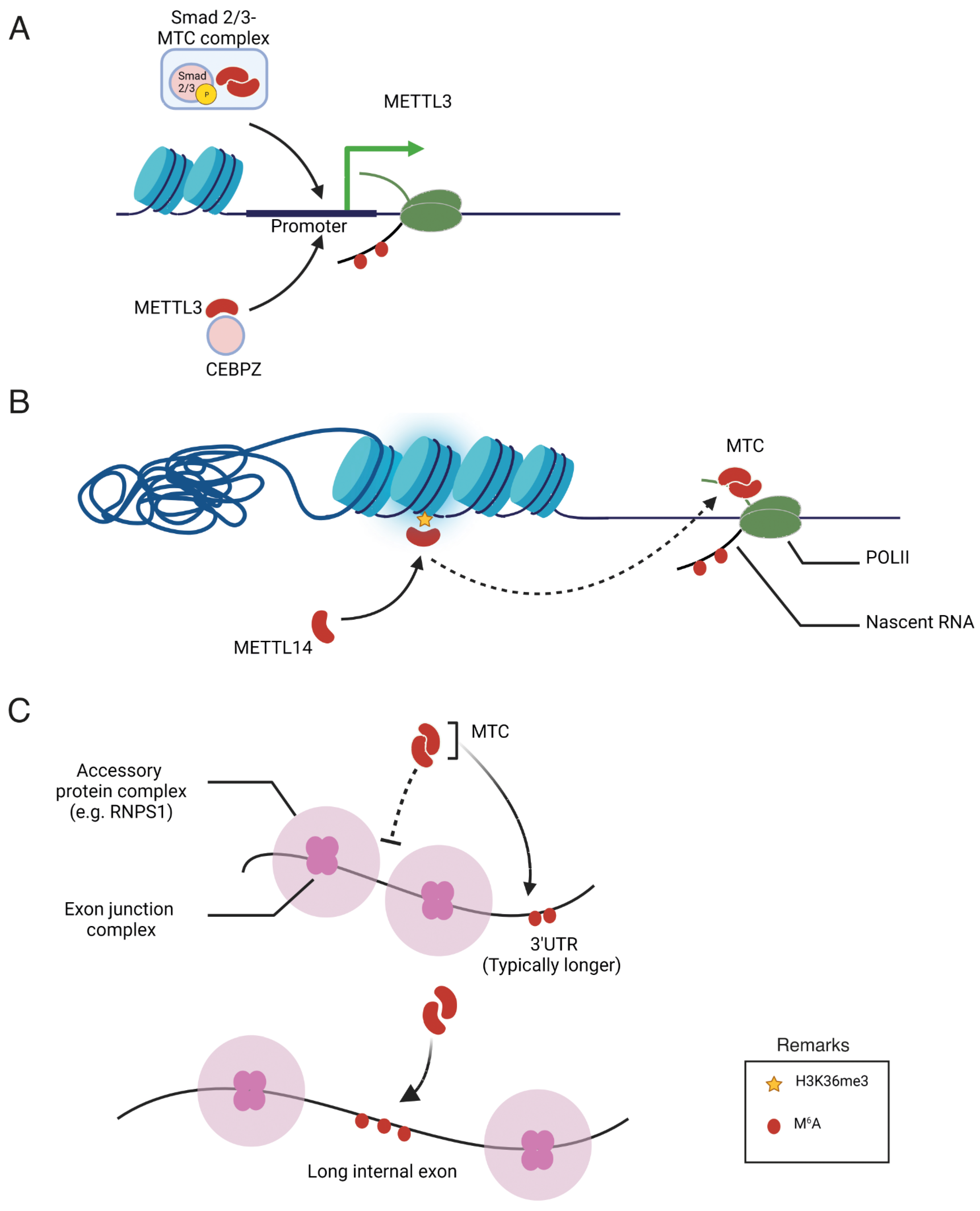
Figure 2. Illustration of proposed mechanisms regulating the specificity of m6A deposition. (A) Recruitment of the m6A methyltransferase complex (MTC) and METTL3 by Smad2/3 and CEBPZ, respectively, into the promoter of specific genes. (B) MTC recruitment to adjacent RNA polymerase II (POLII) is promoted through the interaction of METTL14 with H3K36 trimethylation (H3K36Me3). (C) Regulatory effects of exon junction complexes and accessory protein complexes on the m6A modification of specific mRNA.
5. Dysregulation of m6A in Cancer and Chemoresistance
Because of the numerous post-translational modifications (PTMs) discussed earlier, the expression levels of m6A-related proteins and their functions are frequently disrupted in cancers. Additionally, the abnormal expression of specific m6A-related genes, such as METTL3 and ALKBH5, contributes to the development of resistance to cytotoxic chemotherapy, targeted drugs, and immunotherapy. Moreover, METTL3, an m6A methyltransferase, plays a positive role in double-strand break repair, further promoting resistance to radiotherapy. Target mRNAs subject to m6A methylation in cancer appear to be critical for cancer cell proliferation and survival, encompassing genes related to autophagy, the cell cycle, and proliferation, among others. Despite the abundance of information on m6A-modified mRNA, the mechanisms underlying the specificity of m6A modifications remain unclear. While the exon architecture and epigenetic mechanisms discussed earlier explain the patterns of m6A methylation across the transcriptome, they do not address how cell signaling pathways regulate the m6A modification of specific mRNAs. Recruitment of m6A methyltransferase subunits such as METTL3 by specific transcription factors may be a possible mechanism governing the specific and dynamic m6A modification of mRNA. However, the underlying mechanisms remain poorly understood in cancer. Additionally, a comprehensive understanding of the interactome of m6A-releated proteins may reveal novel mechanisms regulating the functions of m6A-related genes. A recent study on the protein interactome of m6A methyltransferases and demethylases revealed numerous novel binding partners for these proteins; however, the functional characterization of these interactions remains to be studied [24].
References
- Kumar, S.; Mohapatra, T. Deciphering Epitranscriptome: Modification of mRNA Bases Provides a New Perspective for Post-transcriptional Regulation of Gene Expression. Front. Cell Dev. Biol. 2021, 9, 628415.
- Liu, J.; Yue, Y.; Han, D.; Wang, X.; Fu, Y.; Zhang, L.; Jia, G.; Yu, M.; Lu, Z.; Deng, X.; et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014, 10, 93–95.
- Zaccara, S.; Ries, R.J.; Jaffrey, S.R. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 608–624.
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Lo Muzio, L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int. J. Mol. Med. 2017, 40, 271–280.
- Sun, H.L.; Zhu, A.C.; Gao, Y.; Terajima, H.; Fei, Q.; Liu, S.; Zhang, L.; Zhang, Z.; Harada, B.T.; He, Y.Y.; et al. Stabilization of ERK-Phosphorylated METTL3 by USP5 Increases m6A Methylation. Mol. Cell 2020, 80, 633–647.e7.
- Perez-Pepe, M.; Desotell, A.W.; Li, H.; Li, W.; Han, B.; Lin, Q.; Klein, D.E.; Liu, Y.; Goodarzi, H.; Alarcón, C.R. 7SK methylation by METTL3 promotes transcriptional activity. Sci. Adv. 2023, 9, eade7500.
- Yu, F.; Wei, J.; Cui, X.; Yu, C.; Ni, W.; Bungert, J.; Wu, L.; He, C.; Qian, Z. Post-translational modification of RNA m6A demethylase ALKBH5 regulates ROS-induced DNA damage response. Nucleic Acids Res. 2021, 49, 5779–5797.
- Biggar, K.K.; Li, S.S.C. Non-histone protein methylation as a regulator of cellular signalling and function. Nat. Rev. Mol. Cell Biol. 2015, 16, 5–17.
- Liu, X.; Wang, H.; Zhao, X.; Luo, Q.; Wang, Q.; Tan, K.; Wang, Z.; Jiang, J.; Cui, J.; Du, E.; et al. Arginine methylation of METTL14 promotes RNA N6-methyladenosine modification and endoderm differentiation of mouse embryonic stem cells. Nat. Commun. 2021, 12, 3780.
- Wang, J.; Wang, Z.; Inuzuka, H.; Wei, W.; Liu, J. PRMT1 methylates METTL14 to modulate its oncogenic function. Neoplasia 2023, 42, 100912.
- Jia, Y.; Yu, X.; Liu, R.; Shi, L.; Jin, H.; Yang, D.; Zhang, X.; Shen, Y.; Feng, Y.; Zhang, P.; et al. PRMT1 methylation of WTAP promotes multiple myeloma tumorigenesis by activating oxidative phosphorylation via m6A modification of NDUFS6. Cell Death Dis. 2023, 14, 512.
- Shang, S.; Liu, J.; Hua, F. Protein acylation: Mechanisms, biological functions and therapeutic targets. Signal Transduct. Target. Ther. 2022, 7, 396.
- Li, Y.; He, X.; Lu, X.; Gong, Z.; Li, Q.; Zhang, L.; Yang, R.; Wu, C.; Huang, J.; Ding, J.; et al. METTL3 acetylation impedes cancer metastasis via fine-tuning its nuclear and cytosolic functions. Nat. Commun. 2022, 13, 6350.
- Zhang, G.; Huang, R.; Zhao, H.; Xia, Y.; Huang, H.; Qian, M.; Fu, Y.; Cui, Y. ACAT1-mediated METTL3 acetylation inhibits cell migration and invasion in triple negative breast cancer. Genes Immun. 2023, 24, 99–107.
- Zhang, X.-L.; Chen, X.-H.; Xu, B.; Chen, M.; Zhu, S.; Meng, N.; Wang, J.-Z.; Zhu, H.; Chen, D.; Liu, J.-B.; et al. K235 acetylation couples with PSPC1 to regulate the m6A demethylation activity of ALKBH5 and tumorigenesis. Nat. Commun. 2023, 14, 3815.
- Du, Y.; Hou, G.; Zhang, H.; Dou, J.; He, J.; Guo, Y.; Li, L.; Chen, R.; Wang, Y.; Deng, R.; et al. SUMOylation of the m6A-RNA methyltransferase METTL3 modulates its function. Nucleic Acids Res. 2018, 46, 5195–5208.
- Li, J.; Ahmad, M.; Sang, L.; Zhan, Y.; Wang, Y.; Yan, Y.; Liu, Y.; Mi, W.; Lu, M.; Dai, Y.; et al. O-GlcNAcylation promotes the cytosolic localization of the m6A reader YTHDF1 and colorectal cancer tumorigenesis. J. Biol. Chem. 2023, 299, 104738.
- Wang, Q.; Chen, C.; Ding, Q.; Zhao, Y.; Wang, Z.; Chen, J.; Jiang, Z.; Zhang, Y.; Xu, G.; Zhang, J.; et al. METTL3-mediated m6A modification of HDGF mRNA promotes gastric cancer progression and has prognostic significance. Gut 2020, 69, 1193–1205.
- Zhang, J.; Bai, R.; Li, M.; Ye, H.; Wu, C.; Wang, C.; Li, S.; Tan, L.; Mai, D.; Li, G.; et al. Excessive miR-25-3p maturation via N6-methyladenosine stimulated by cigarette smoke promotes pancreatic cancer progression. Nat. Commun. 2019, 10, 1858.
- Du, M.; Peng, Y.; Li, Y.; Sun, W.; Zhu, H.; Wu, J.; Zong, D.; Wu, L.; He, X. MYC-activated RNA N6-methyladenosine reader IGF2BP3 promotes cell proliferation and metastasis in nasopharyngeal carcinoma. Cell Death Discov. 2022, 8, 53.
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive Analysis of mRNA Methylation Reveals Enrichment in 3′ UTRs and near Stop Codons. Cell 2012, 149, 1635–1646.
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206.
- Jang, K.H.; Heras, C.R.; Lee, G. m6A in the Signal Transduction Network. Mol. Cells 2022, 45, 435–443.
- Covelo-Molares, H.; Obrdlik, A.; Poštulková, I.; Dohnálková, M.; Gregorová, P.; Ganji, R.; Potěšil, D.; Gawriyski, L.; Varjosalo, M.; Vaňáčová, Š. The comprehensive interactomes of human adenosine RNA methyltransferases and demethylases reveal distinct functional and regulatory features. Nucleic Acids Res. 2021, 49, 10895–10910.
More
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
707
Revisions:
2 times
(View History)
Update Date:
08 Jan 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No