Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Wang, L.; Lu, H.; Jiang, Y. Natural Polyketides. Encyclopedia. Available online: https://encyclopedia.pub/entry/53399 (accessed on 15 June 2026).
Wang L, Lu H, Jiang Y. Natural Polyketides. Encyclopedia. Available at: https://encyclopedia.pub/entry/53399. Accessed June 15, 2026.
Wang, Li, Hui Lu, Yuanying Jiang. "Natural Polyketides" Encyclopedia, https://encyclopedia.pub/entry/53399 (accessed June 15, 2026).
Wang, L., Lu, H., & Jiang, Y. (2024, January 04). Natural Polyketides. In Encyclopedia. https://encyclopedia.pub/entry/53399
Wang, Li, et al. "Natural Polyketides." Encyclopedia. Web. 04 January, 2024.
Copy Citation
Invasive fungal infections present a significant risk to human health. The current arsenal of antifungal drugs is hindered by drug resistance, limited antifungal range, inadequate safety profiles, and low oral bioavailability. Consequently, there is an urgent imperative to develop novel antifungal medications for clinical application.
polyketides
antifungal
invasive fungal infections
1. Introduction
Invasive fungal infections significantly threaten human health, resulting in approximately 1.5 million deaths annually [1][2]. The primary culprits responsible for these fatalities are Candida, Cryptococcus, and Aspergillus species [3][4]. The rise in severe underlying diseases and immunocompromised populations, such as those undergoing hematopoietic stem cell transplantation, organ transplantation, immunosuppressive therapy, acquired immune deficiency syndrome, cancer, advanced age, and preterm birth, has further exacerbated the morbidity and mortality associated with invasive fungal infections [5][6]. The current antifungal agents utilized in clinical settings are associated with drawbacks such as drug resistance, limited bioavailability, nephrotoxicity, and a restricted antifungal spectrum [7]. As a result, there is a pressing demand for developing novel antifungal agents to treat invasive fungal infections.
Among the three primary categories of antifungal medications presently accessible, amphotericin B and caspofungin are classified as polyketide compounds. Amphotericin B, a polyene macrolide polyketide, exhibits a broad spectrum of fungicidal activity against Candida, Aspergillus, and Cryptococcus species [8] and remains a preferred treatment option for severe invasive fungal infections [7]. Caspofungin, a non-ribosomal polyketide derivative, selectively targets β-1,3-glucan synthase and impedes fungal cell wall biogenesis with notable selectivity and biological safety compared to amphotericin B [9]. Amphotericin B and caspofungin, both polyketide compounds, have demonstrated clinical efficacy in treating invasive fungal infections. This suggests that developing polyketide compounds as antifungal drugs shows considerable potential.
Polyketides are synthesized through a series of Claisen decarboxylation condensation reactions, utilizing short-chain acyl starting substrates and extension units, including acetyl-CoA, propionyl-CoA, malonyl-CoA, and methylmalonyl-CoA [10]. Polyketides, categorized as secondary metabolites, demonstrate a broad spectrum of structural diversity and are generated by various organisms, including bacteria, fungi, plants, and animals. The biosynthesis of polyketides involves a sequence of condensation reactions catalyzed by three types of polyketide synthases (PKSs): type I PKSs, type II PKSs, and type III PKSs. Type I PKSs are responsible for the biosynthesis of macrolides and related polyenes [11]. Within the category of type I PKSs, there are two distinct subtypes: modular and iterative. Modular type I PKSs are composed of enzyme complexes containing multiple modules, each consisting of linear domains. Each set of domains is utilized only once during the assembly of polyketides [12]. In contrast, iterative type I PKSs possess a single reusable module, with the domains within this module being reused to catalyze multiple rounds of decarboxylation condensation reactions [13]. Type II PKSs, also called aromatic PKSs, are comprised of multiple distinct proteins that function as enzyme complexes. These complexes facilitate repeating a specific chemical reaction by using reusable domains. Typically employing malonyl-CoA as a substrate, Type II PKSs incrementally add two carbon atoms to the polyketide intermediate following each round of decarboxylation condensation reaction. Subsequently, the polyketide is transformed into an aromatic compound under ketoreductase, an aromatase, and a cyclase. The resulting preliminary aromatic polyketide is further modified by an oxygenase, a glycosyltransferase, and a methyltransferase to yield the ultimate aromatic products [14]. Type II PKSs produce aromatic polyketide compounds, including anthracyclines, anticyclones, aureolic acids, tetracyclines, anthracyclines [14], and polyenes [15]. In contrast to the other two types of PKSs, type III PKSs are comprised of a single protein that directly utilizes simple carboxylic acids as substrates, which are activated by acyl-CoA and do not require acyl carrier protein-activated acyl-CoA. Type III PKSs primarily facilitate the biosynthesis of flavonoids, stilbenes, phenylpropanoids, pyrone-type aromatic polyketides, and resorcinol-type aromatic polyketides [16][17][18].
2. Macrolide Polyketides
Macrolide polyketides are mainly synthesized by the Type I PKSs. The structural diversity of these compounds is achieved through variations in starting substrates, extension units, modules, and domains, as well as a series of post-modifications that occur after their release. Macrolide polyketides can form glycosidic bonds with one or more sugar moieties. These compounds are classified based on the number of atoms present in the macrolide ring, which includes 12-membered, 14-membered, 24-membered, 26-membered, 32-membered, 36-membered, and 38-membered variants.
Amphidinins Q (1), C (2) and E (3) (Figure 1), which are 12-membered macrolides, are derived from the endophytic dinoflagellates Amphidinium species (2012-7-4A strain) found in the marine acoel flatworm Amphiscolops species [19]. Amphidinolide Q (1) exhibits antifungal activity against C. albicans (MIC = 32 μg/mL). On the other hand, amphidinin C (2), which is the open-loop structure of amphidinolide Q (1), loses its activity against C. albicans but demonstrates antifungal activity against Aspergillus niger (MIC = 32 μg/mL). Additionally, amphidinin E (3), where the carbonyl group at the C-6 position of amphidinin C (2) is replaced by a β-hydroxyl group, enhances the antifungal activity against A. niger (MIC = 16 μg/mL) [19].
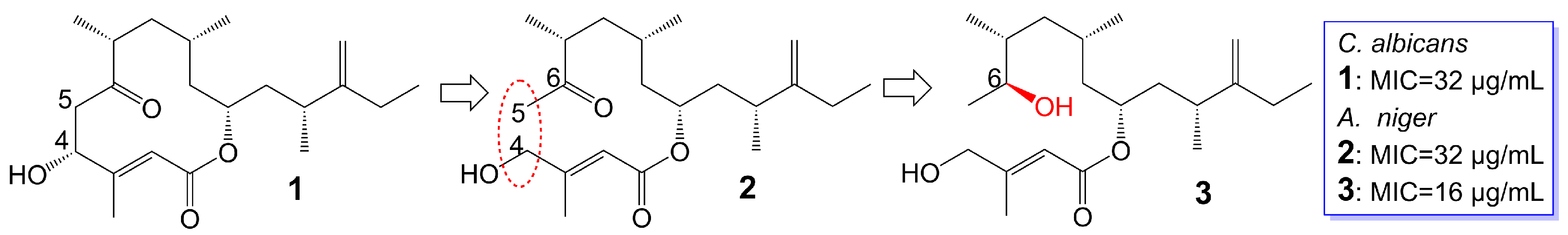
Figure 1. Chemical structures of Amphidinins Q (1), C (2), and E (3). The red dotted box marks the open-loop structure.
Rustmicin (4) (Figure 2), a 14-membered macrolide, has been obtained from the cultured broth of Micromonospora narashinoensis 980-MC [20]. This compound functions as a potent inhibitor of inositol phosphoceramide synthase, thereby impeding the transfer of inositol phosphoceramide to ceramide in synthesizing sphingolipids in fungi [21]. Rustmicin (4) exhibits remarkable antifungal activity against Cryptococcus neoformans MY2062, C. neoformans ATCC9011, C. tropicalis MY1012, and C. albicans MY1055, C. albicans ATCC90028 with MIC values of 0.0001 μg/mL, 0.063 μg/mL, 0.05 μg/mL, 6.25 μg/mL, and 4 μg/mL [22][23]. Nevertheless, the effective reduction of fungal burdens in the spleen and brain necessitates administering high doses of rustmicin (4) in C. neoformans infected mice model [21]. The reduced potency of rustmicin (4) in vivo is attributed to the acceleration of its conversion to the inactive C-2 isoform, γ-lactone, by serum, which is subsequently degraded [21]. Additionally, the antifungal activity of rustmicin (4) is diminished due to the conversion of its enol ether structure at the C-6 position to an inactive ketone structure under acidic conditions. However, substituting the methoxy group at the C-6 position with methylthio (5) (Figure 2) exhibits weaker antifungal activity against C. neoformans ATCC9011 (MIC = 0.5 μg/mL) and C. albicans ATCC90028 (MIC = 64 μg/mL) [23]. Compared to the oxygen atom, the sulfur atom exhibits lower electronegativity and cannot form hydrogen bonds. Consequently, sulfur is not an efficient hydrogen bond donor compared to oxygen. Replacing the original oxygen atom with a sulfur atom elevates the logP value and augments the lipophilicity of compound 5, consequently leading to diminished water solubility.
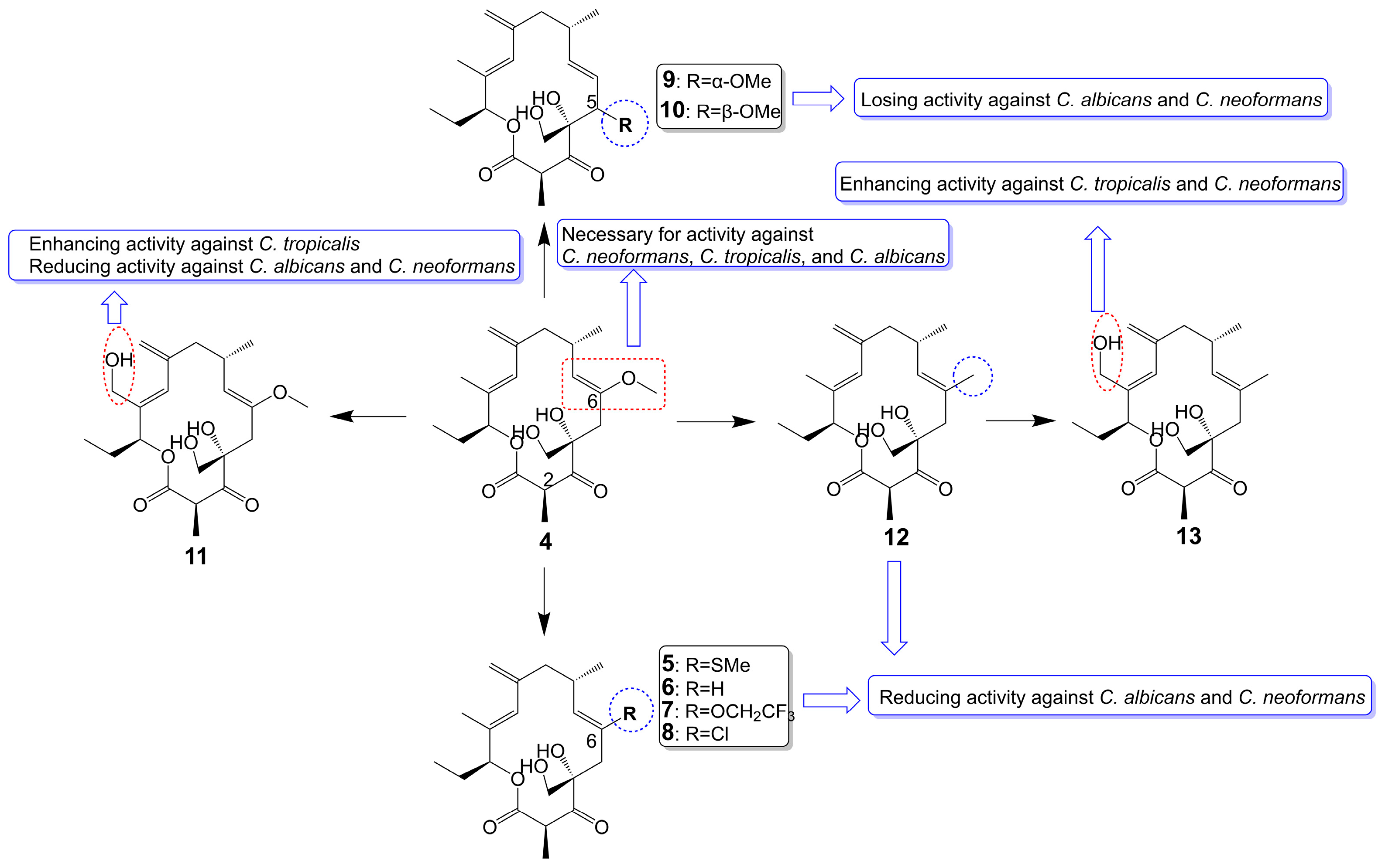
Figure 2. Chemical structures of rustmicin (4) and its analogues.
In substituting the methoxy group at the C-6 position with hydrogen (6), fluoroalkoxy (7), and halogen (8) (Figure 2), the antifungal activity of the compounds is greatly reduced or even lost [23]. Compound 6 loss had a significant effect on C. neoformans ATCC9011 (MIC > 64 μg/mL) and C. albicans ATCC90028 (MIC > 64 μg/mL). Compound 7, bearing a fluoroalkoxy group, also belongs to the enol ether structure and has altered acid-base stability and lipophilicity. Although bearing a fluoroalkoxy group can enhance the stability of compound 7, it will reduce the pH value and weaken the alkalinity. Meanwhile, compared with the methyl group, the logP value of a fluoroalkoxy group increased, and its solubility in water decreased. Therefore, compound 7 exhibited attenuated antifungal activity against C. neoformans ATCC9011 (MIC = 32 μg/mL) and lost its activity against C. albicans ATCC90028 (MIC > 64 μg/mL). Compound 8 exhibited attenuated antifungal activity against C. neoformans ATCC9011 (MIC = 16 μg/mL) and lost its activity against C. albicans ATCC90028 (MIC > 64 μg/mL). Removing the methoxy group from the C-6 position to the C-5 position (9 and 10) (Figure 2), the antifungal activity of these compounds also disappeared (MIC > 64 μg/mL) [23]. Evidence supports the idea that the enol ether structure at position C-6 is essential for the antifungal activity of rustmicin. 21-Hydroxyrustmicin (11) (Figure 2) is isolated from Micromonospora sp. UV Mutant (MA 7186) and exhibits stronger antifungal activity (MIC = 0.024 μg/mL) against C. tropicalis MY1012 and weaker antifungal activity against C. albicans MY1055 (MIC = 12.5 μg/mL) and C. neoformans MY2062 (MIC = 0.1 μg/mL) [22]. Galbonolide B (12) (Figure 2) is isolated from Micromonospora species (MA 7094) and UV Mutant (MA 7186) [22]. Galbonolide B (12), substituting the methoxy group at the C-6 position with a methyl group, greatly reduces antifungal activity against C. neoformans MY2062 (MIC = 12.5 μg/mL), C. tropicalis MY1012 (MIC = 200 μg/mL), and C. albicans MY1055 (MIC > 200 μg/mL). 21-Hydroxygalbonolide B (13) (Figure 2) is isolated from Micromonospora species UV Mutant (MA 7186) greatly enhances the antifungal activity of galbonolide B (12) against C. tropicalis MY1012 and C. neoformans MY2062 with MIC values of 0.78 μg/mL and 3.1 μg/mL, respectively [22].
Preussolides A (14) and B (15) (Figure 3), 24-membered macrolides, have been extracted from the coprophilous isolates of Preussia typharum [24]. These compounds are characterized by a distinctive phosphoethanolamine substituent, with the only difference being the presence or absence of a double bond between the C-10 and C-11 positions. Preussolide B (15), with the double bond, exhibits weak antifungal activity against C. neoformans H99 (37 °C) (MIC = 32 μg/mL), C. neoformans H99 (23 °C) (MIC = 32 μg/mL), and C. albicans ATCC 10231 (MIC = 256 μg/mL). On the other hand, preussolide A (14), lacking the double bond, shows enhanced activity against C. neoformans H99 (37 °C) (MIC = 4 μg/mL), C. neoformans H99 (23 °C) (MIC = 8 μg/mL), C. albicans ATCC 10231 (MIC = 256 μg/mL) and A. fumigatus AF239 (MIC = 8 μg/mL) [24]. Preussolide A (14) exhibits stronger activity, maybe due to the C10-C11 single bond being more stable than the C10-C11 double bond. Additionally, single and double bonds can change the stereoconfiguration of compounds.
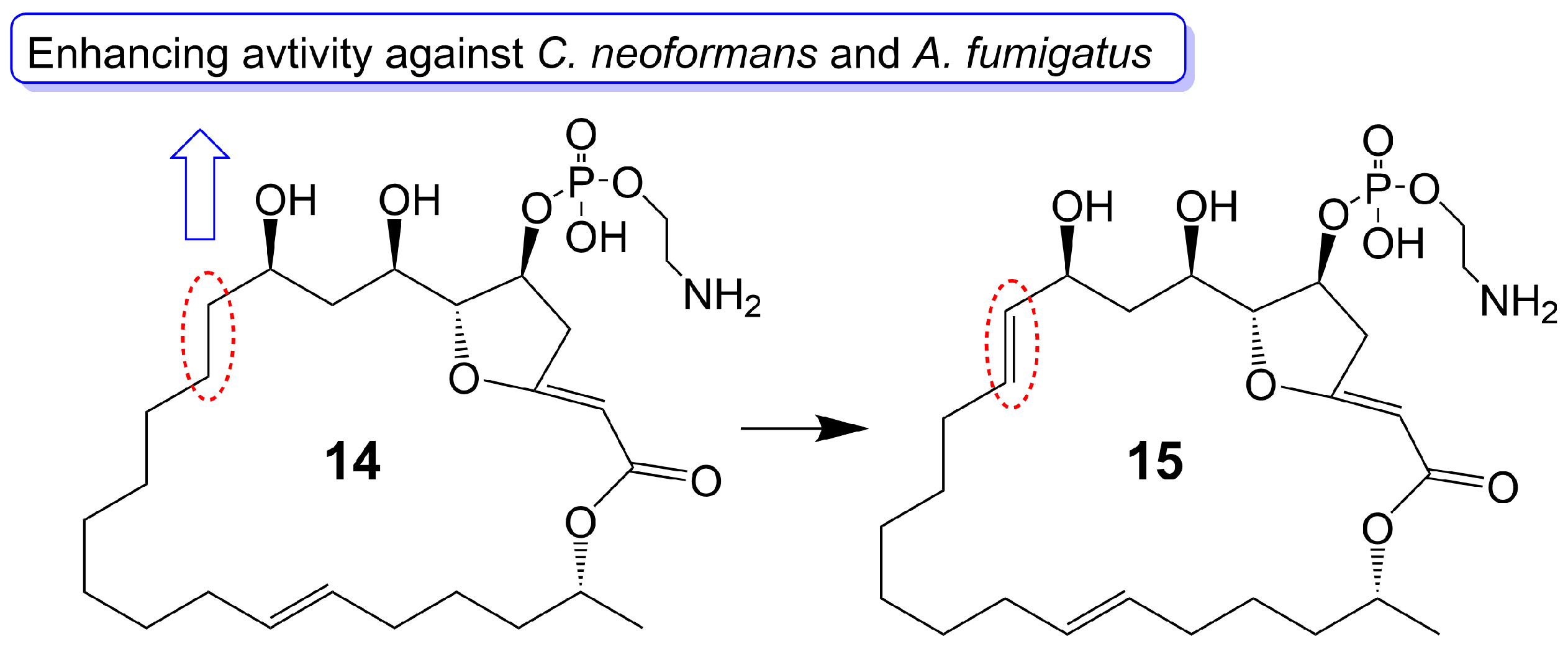
Figure 3. Chemical structures of Preussolides A (14) and B (15). The red dotted boxes mark the difference between Preussolides A (14) and B (15).
Oligomycin A, a 26-membered macrolide with a bicyclic spiroketal, is isolated from Streptomyces [25]. Oligomycin A presents potently broad spectrum antifungal activity against C. albicans ATCC 24433 (MIC = 2–4 μg/mL), Candida krusei 432M (MIC = 1–2 μg/mL), Candida parapsilosis ATCC 22019 (MIC = 2 μg/mL), Candida utilis 84 (MIC = 1 μg/mL), Candida tropicalis 3019 (MIC = 1 μg/mL), A. niger (MIC = 0.5–2 μg/mL), Cryptococcus humicolus ATCC 9949 (MIC = 2 μg/mL), and Trichophyton mentagrophytes ATCC 9533 (MIC = 10 μg/mL) [26][27][28][29][30]. The oligomycin A molecule contains a C16-C19 diene system that can adopt two conformations, namely s-Trans (16) and s-Cis (17) (Figure 4). When the s-Cis conformation of the diene system in oligomycin A is replaced with benzo-quinone (18) and N-benzyl maleimide (19 and 20) (Figure 4), these resulting cycloadducts exhibit a loss of activity against C. albicans ATCC 24433, C. parapsilosis ATCC 22019, C. krusei 432M, and A. niger 137a (MICs > 32 μg/mL). This activity loss is likely attributed to the reduced permeability of the cycloadducts across the fungal cell wall [26]. (33S)-oligomycin A (21) (Figure 4), substituted the α-hydroxy group at the C-33 position with a β-hydroxy group, exhibits comparable antifungal activity against C. parapsilosis ATCC 22019 (MIC = 1 μg/mL), C. albicans ATCC 24433 (MIC = 4 μg/mL), C. utilis 84 (MIC = 2 μg/mL), C. tropicalis 3019 (MIC = 1 μg/mL), C. krusei 432 M (MIC =4 μg/mL), and A. niger 137 a (MIC = 2 μg/mL) to oligomycin A [28]. The 8, 9 carbon bond of oligomycin A is disrupted to obtain the acyclic derivative (22) (Figure 4) of oligomycin A, and the antifungal activity of these acyclic compounds against C. albicans ATCC 14053 (MIC = 16 μg/mL) and A. niger ATCC 16404 (MIC = 4 μg/mL) is greatly reduced due to losing the inhibitory activity against F0F1 ATP synthase-containing proteoliposomes [31]. Bromo-oligomycin A (23) (Figure 4), in which the tetrahydropyrane ring contains bromine in the C-16 position, loses antifungal activity against A. niger ATCC 16404 (MIC >16 μg/mL), C. albicans ATCC 14053 (MIC > 16 μg/mL) except for C. humicolus ATCC 9949 (MIC = 2 μg/mL) may be explained that the derivative forms a webbed structure that distorts conformation in comparison with oligomycin A, and the derivative lacks the free 13-OH group that interacts with the target site [29]. Oligomycin E (24) (Figure 4), the analog of oligomycin A s-Cis (17), is extracted from Streptomyces species strain HG29 isolated from Saharan soil [32]. Oligomycin E (24), in which an α-methyl group replaces the β-methyl group at the C-4 position of oligomycin A and the bicyclic spiroketal has additional hydroxyl and carbonyl groups, exhibits comparable antifungal activity against Aspergillus carbonarius M333 (MIC = 2 μg/mL), A. westerdijkiae NRRL 3174 (MIC = 8 μg/mL), A. parasiticus CBS 100926 (MIC = 4 μg/mL), A. nidulans KE202 (MIC = 75 μg/mL), A. niger OT304 (MIC = 4 μg/mL), A. terreus CT290 (MIC = 75 μg/mL), and A. fumigatus CF140 (MIC = 100 μg/mL) in vitro [32]. Oligomycin C (25) (Figure 4), the analog of oligomycin A s-Trans (16), is extracted from Streptomyces diastaticus [30]. Oligomycin C (25), in which the β-hydrogen replaces the β-hydroxyl group at the C-12 position of oligomycin A, exhibits comparable antifungal activity against A. niger ATCC 10335 (MIC = 2 μg/mL) [30]. Oligomycin A annelates the structure of nitrone and forms a cyclic nitrone to form a new compound 26 (Figure 4), which reduces the cytotoxicity but loses the antifungal activity [33]. Compounds 27 and 28 (Figure 4), formed by oligomycin A linked to pyrazolo [1,5-a] pyridine, show reduced cytotoxicity and weaker antifungal potential against A. niger (MIC = 2 μg/mL) to oligomycin A (MIC = 0.125 μg/mL) [33]. Neomaclafungins A-I (29–37) (Figure 4), homologs of oligomycin A, have been extracted from the fermentation broth of Actinoalloteichus species NPS702 [27]. These compounds exhibit stronger antifungal activity against T. mentagrophytes ATCC 9533, with MIC values ranging from 1 to 3 μg/mL, which may be explained by the absence of the ketones in the 26-membered ring and the different substituent ation at the C-24 position [27].
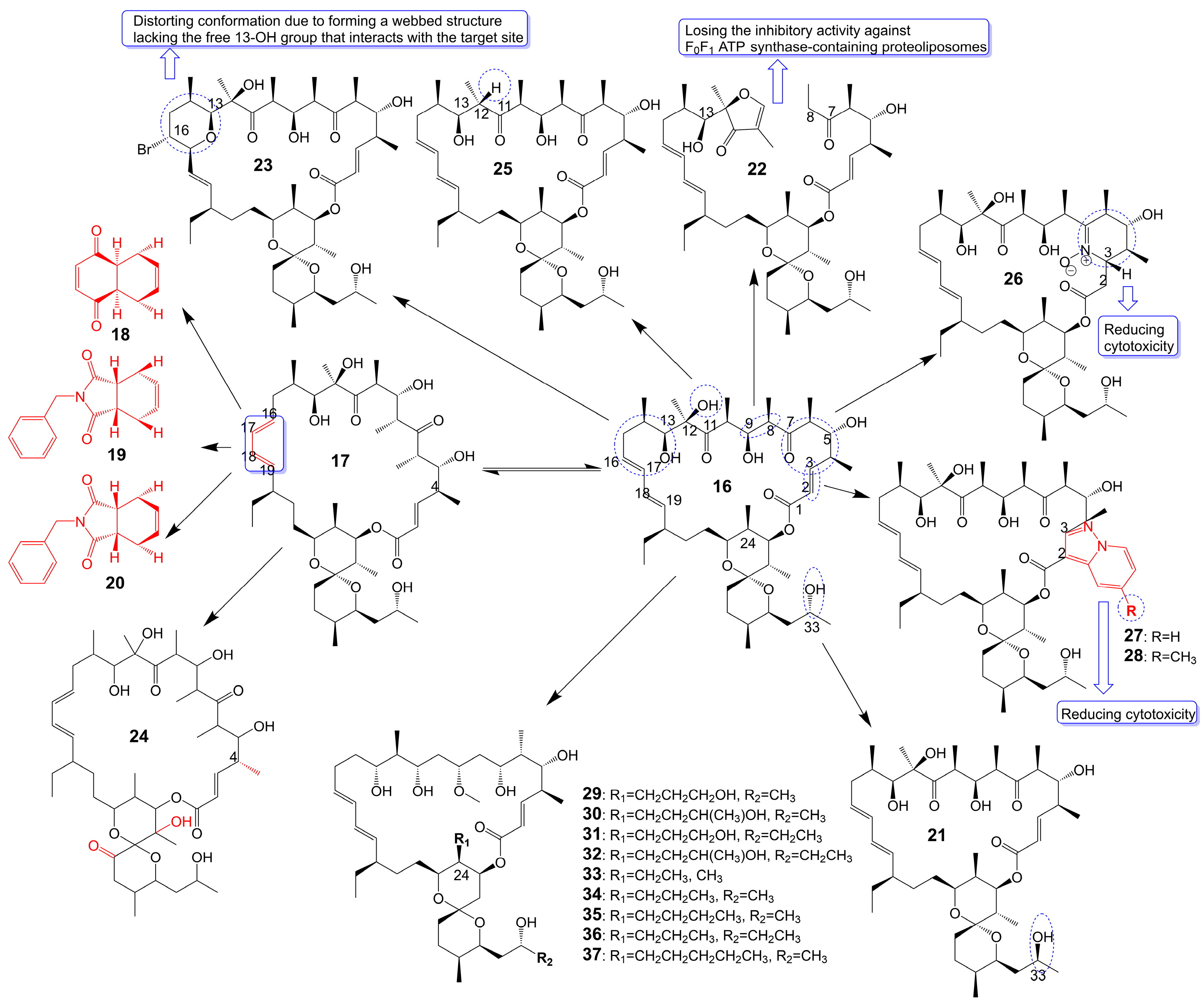
Figure 4. Chemical structures of analogues of Oligomycin A.
Brasilinolides A (38) and B (39) (Figure 5), 32-membered macrolides with a tetrahydropyran ring and a 2-deoxyfucopyranose, are obtained through the fermentation of Nocardia brasiliensis IFM0406 [34][35]. Brasilinolide A (38) exhibited selective inhibition of A. niger IFM 40406 (MIC = 3.13 μg/mL) [34]. The malonyl side chain of brasilinolide A (38) is not essential for its antifungal activity [36]. Brasilinolide B (39), changing the malonyl side chain and the sugar moiety of brasilinolide A (38), has broad-spectrum antifungal activity against A. niger (MIC = 12.5 μg/mL), A. fumigatus IFM 41219 (MIC = 12.5 μg/mL), C. albicans ATCC 90028 (MIC = 25 μg/mL), C. albicans IFM 40007 (MIC = 12.5 μg/mL), C. albicans 94–2530 (MIC = 25 μg/mL), C. krusei M 1005 (MIC = 25 μg/mL), C. parapsilosis ATCC 90018 (MIC = 12.5 μg/mL), C. glabrata ATCC 90030 (MIC = 25 μg/mL), C. neoformans ATCC 90112 (MIC = 12.5 μg/mL), and C. neoformans 145 A (MIC = 25 μg/mL) [35]. Compared with brassinolide A (38), copiamycin (40) (Figure 5) replaces the malonyl side chain from the C-23 position to the C-21 position without the sugar moiety, and copiamycin shows antifungal activity against C. albicans Yu 1200 (MIC = 25 μg/mL) [36]. Methylcopiamycin (41) (Figure 5), the 15-OH methylation product of copiamycin, has the same antifungal activity against C. albicans Yu 1200 (MIC = 25 μg/mL) [36]. Demalonylmethylcopiamycin (42) (Figure 5), removing the malonyl side chain connected to C-21 of methylcopiamycin, shows stronger antifungal activity against C. albicans Yu 1200 (MIC = 6.25 μg/mL) compared to copiamycin and methylcopiamycin [36]. Langkolide (43) (Figure 5), a compound obtained from the mycelium of Streptomyces species Acta 3062, replacing the malonyl side chain connected to C-23 of brassinolide A (38) with acetyl side chain connected to C-23 position and having disaccharide moiety with 1,4-naphthoquinone connected to C-37 of the aglycone moiety, has been found to exhibit inhibitory effects on the growth of Candida glabrata and C. albicans, with half maximal inhibitory concentration (IC50) values of 1.00 ± 0.02 and 1.23 ± 0.10 μM, respectively [37].
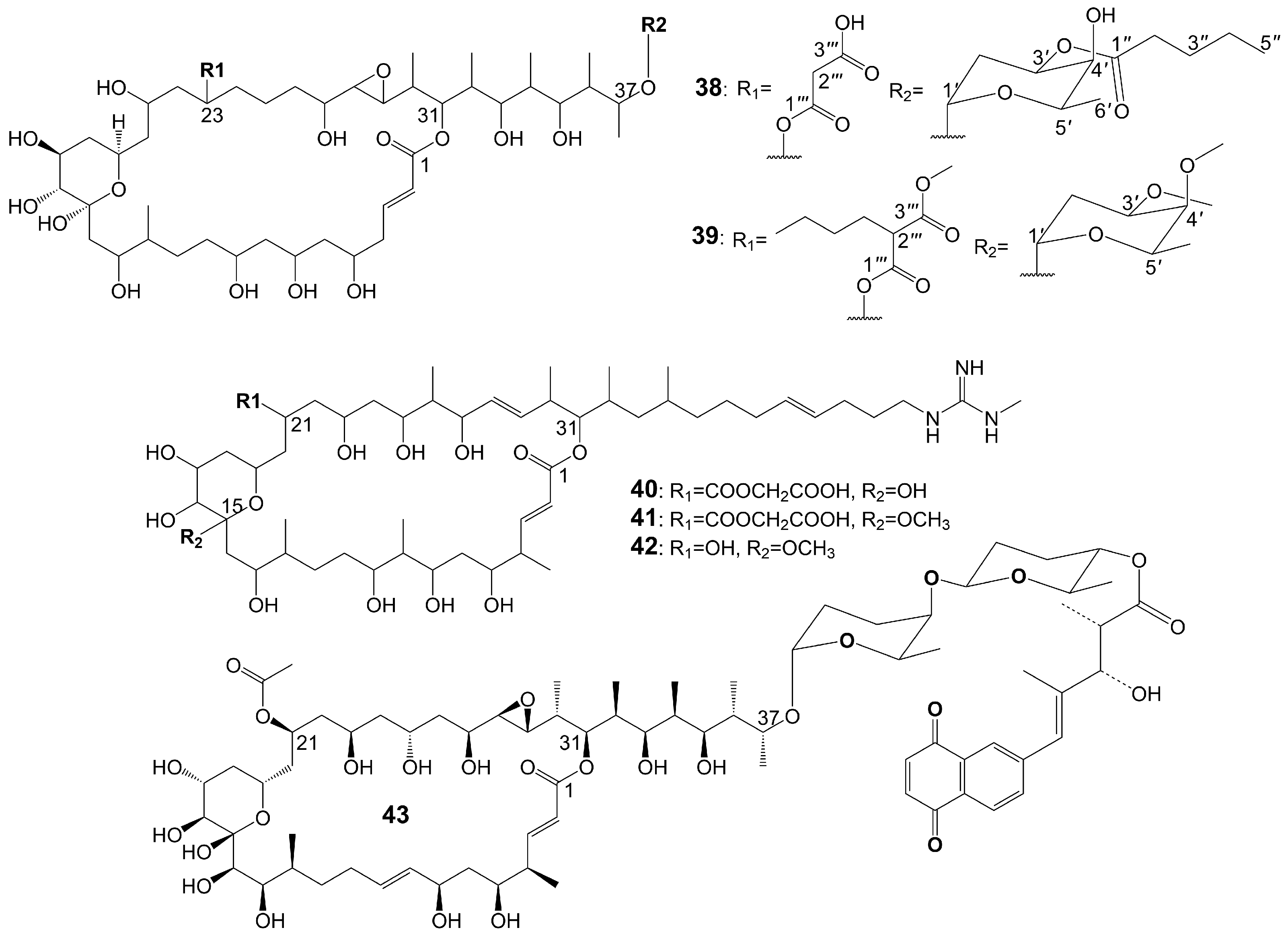
Figure 5. Chemical structures of Brasilinolides A (38) and B (39) and their analogues.
The structures of 1,4-naphthoquinone, glycosylated moiety, and large polyol macrolide aglycones are important in cyphomycin and its derivatives, which are 36-membered macrolides [38]. Cyphomycin (44) (Figure 6), isolated from the microbiome of the fungus-growing ant Cyphomyrmex species, a Brazilian Streptomyces ISID311, exhibits potent antifungal activity against resistant triazole A. fumigatus 11628 (MIC = 0.5 μg/mL), resistant echinocandin C. glabrata 4720 (MIC = 0.5 μg/mL), and resistant triazole, echinocandin, and amphotericin B C. auris B11211 (MIC = 4 μg/mL) [38]. Furthermore, cyphomycin has demonstrated in vivo antifungal efficacy [38]. The neutropenic mouse model of candidiasis is subjected to treatment with cyphomycin, resulting in attenuation of the renal fungal burden in mice compared to the zero-hour control [38]. Caniferolides (45), B (46), C (47), and D (48) (Figure 6) are isolated from the marine Streptomyces caniferus CA-271066 [39]. Caniferolide C (47) shows antifungal activity against A. fumigatus ATCC46645 (MIC = 4–8 μg/mL) and C. albicans MY1005 (MIC = 0.5–1 μg/mL). Caniferolide C (47) is a compound that converts the C32-C33 double bond of cyphomycin to an epoxy group. Caniferolide B (46) shows antifungal activity against A. fumigatus ATCC46645 (MIC = 2–4 μg/mL) and C. albicans MY1005 (MIC = 1–2 μg/mL). Caniferolide B (46) is a compound that adds a hydroxyl group to C-18 of caniferolide C. Caniferolide A (45) shows antifungal activity against A. fumigatus ATCC46645 (MIC = 2–4 μg/mL) and C. albicans MY1005 (MIC = 0.5–1 μg/mL). The hydroxyl group linked to C-19 of caniferolide B is replaced by a sulfate ester group to give the compound caniferolide A. Caniferolide D shows antifungal activity against A. fumigatus ATCC46645 (MIC = 4–8 μg/mL) and C. albicans MY1005 (MIC = 0.5–1 μg/mL). Unlike caniferolide A, Caniferolide D has no hydroxyl group at the C-15 position. Iseolides A (49), B (50), and C (51) (Figure 6) identified from the culture extract of Streptomyces species DC4-5, isolated from a stony coral Dendrophyllia, exhibits potent antifungal activity against C. albicans NBRC0197 with the MIC values of 0.39 μg/mL, 6.25 μg/mL, and 3.16 μg/mL [40]. Iseolide B and cyphomycin differ in that a deoxysugar is attached at C-52 of the sugar moiety, and iseolide B shows antifungal activity against C. albicans with MIC value of 6.25 μg/mL [40]. Iseolide A (49), which links an α-hydroxyl group at the C-18 position of the aglycone of Iseolide B (50), shows 16 times higher antifungal activity (MIC = 0.39 μg/mL) than iseolide B [40]. Iseolide C (51) does not attach to anything but contains this methyl group, which has greatly reduced antifungal activity (MIC = 3.16 μg/mL) compared to iseolide A (49) [40]. Astolides A (52) and B (53) (Figure 6) are isolated from Streptomyces hygroscopicus and are collected from alkaline soil in the Saratov region of Russia [41]. Astolide A (52), removing the α-methyl group at position C-4 of the aglycone moiety of iseolide A (49), is found to be effective for C. albicans ATCC 14053 (MIC = 2.5 μg/mL), A. niger ATCC 16404 (MIC = 1.25 μg/mL), C. albicans 1582 (MIC = 2.53 μg/mL), C. tropicales 1402 (MIC = 5.06 μg/mL), and A. niger 219 (MIC = 2.53 μg/mL) [41]. Astolide B (53), adding a hydroxyl group at the C-3” position of the deoxysugar of astolide A (52), shows enhanced antifungal activity against C. albicans ATCC 14053 (MIC = 1.25 μg/mL), A. niger ATCC 16404 (MIC = 0.6 μg/mL), C. albicans 1582 (MIC = 2.51 μg/mL), C. tropicales 1402 (MIC = 5.01 μg/mL), and A. niger 219 (MIC = 2.51 μg/mL) compared with astolide A (52) [41].
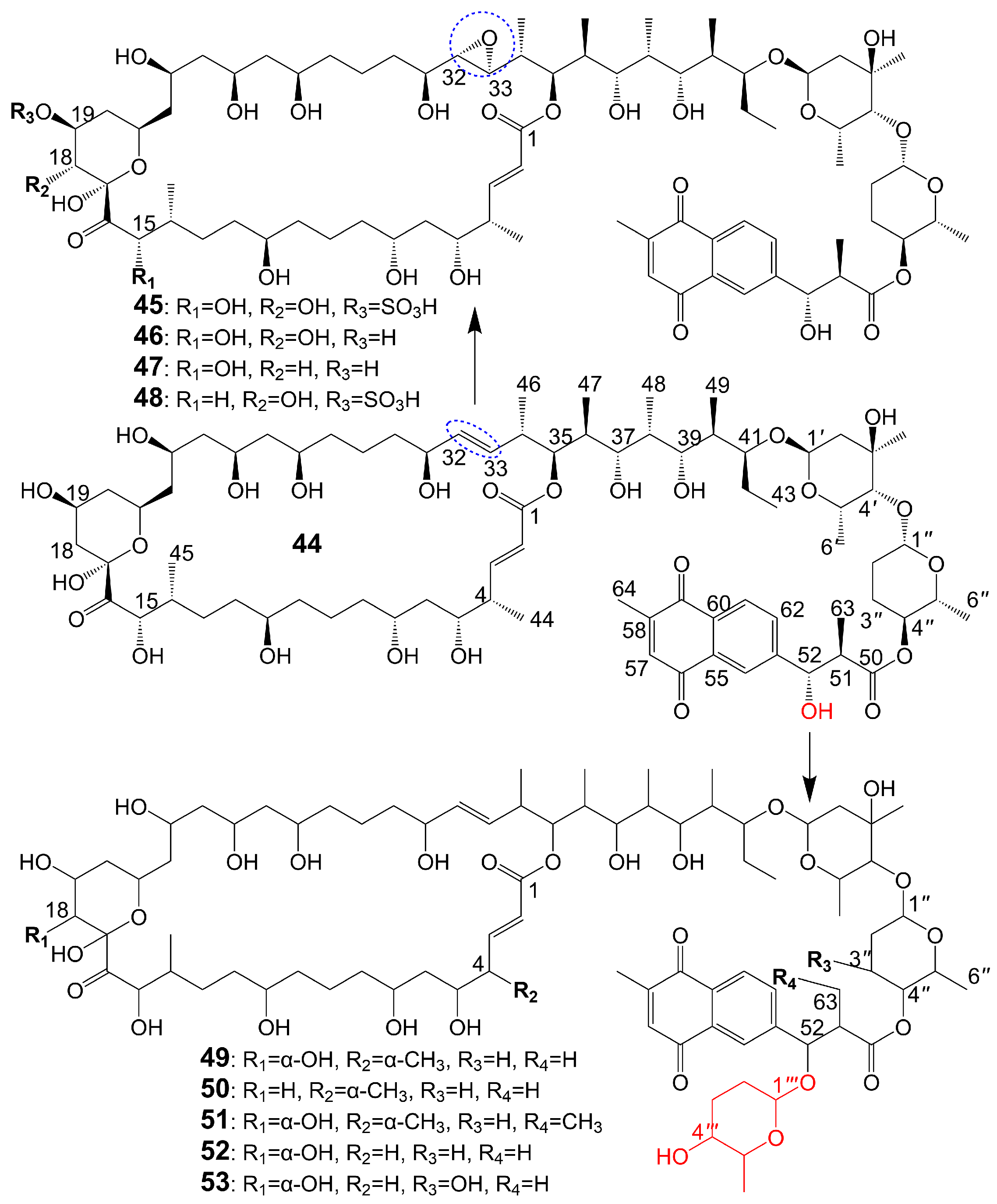
Figure 6. Chemical structures of cyphomycin (44) and its analogues. The blue dotted boxs mark the difference between cyphomycin (44) and caniferolide C (47).
Guanidylfungin A (54) (Figure 7), a 36-membered macrolide, is isolated from the mycelia of Streptomyces hygroscopicus No. 662 and exhibits antifungal activity against C. albicans IAM 4888 (MIC = 12.5 μg/mL), C. albicans Yu 1200 (MIC = 50 μg/mL), and A. fumigatus IAM 2153 (MIC = 25 μg/mL) [36][42]. Structural modification of guanidylfungin A gives alkyl products, including methylguanidylfungin A (55), ethylguanidylfungin A (56), butylguanidylfungin A (57), and allylguanidylfungin A (58) (Figure 7), the activity of these compounds is comparable to or slightly lower than that of guanidylfungin A (54) [36]. Methylguanidylfungin A (55) shows antifungal activity against C. albicans IAM 4888 (MIC = 25 μg/mL), C. albicans Yu 1200 (MIC = 50 μg/mL), and A. fumigatus IAM 2153 (MIC = 12.5 μg/mL). Ethylguanidylfungin A (56) shows antifungal activity against C. albicans IAM 4888 (MIC = 25 μg/mL) and A. fumigatus IAM 2153 (MIC = 25 μg/mL). Butylguanidylfungin A (57) shows antifungal activity against C. albicans IAM 4888 (MIC = 25 μg/mL) and A. fumigatus IAM 2153 (MIC = 50 μg/mL). Allylguanidylfungin A (58) shows antifungal activity against C. albicans IAM 4888 (MIC = 25 μg/mL) and A. fumigatus IAM 2153 (MIC = 25 μg/mL). Compound 59 (Figure 7), removing the malonyl side chain at the C-23 position of methylguanidylfungin A (55), increases antifungal activity against C. albicans IAM 4888 (MIC = 3.12 μg/mL), C. albicans Yu 1200 (MIC = 6.25 μg/mL), and A. fumigatus IAM 2153 (MIC = 3.12 μg/mL) due to increased solubility in water [36]. Compound 60 (Figure 7), the ring-opening structure of the tetrahydropyran ring of guanidylfungin A (54), loses antifungal activity against C. albicans IAM 4888 (MIC > 100 μg/mL), C. albicans Yu 1200 (MIC > 100 μg/mL), and A. fumigatus IAM 2153 (MIC = 50 μg/mL). Compound 61 (Figure 7), removing the malonyl side chain at the C-23 position of compound 60, cannot restore antifungal activity against C. albicans IAM 4888 (MIC = 100 μg/mL), C. albicans Yu 1200 (MIC = 100 μg/mL), and A. fumigatus IAM 2153 (MIC = 12.5 μg/mL) despite the increased water solubility [36]. These lines of evidence suggest that the tetrahydropyran ring is necessary for guanidylfungin A (54) activity, but the malonyl group is not.
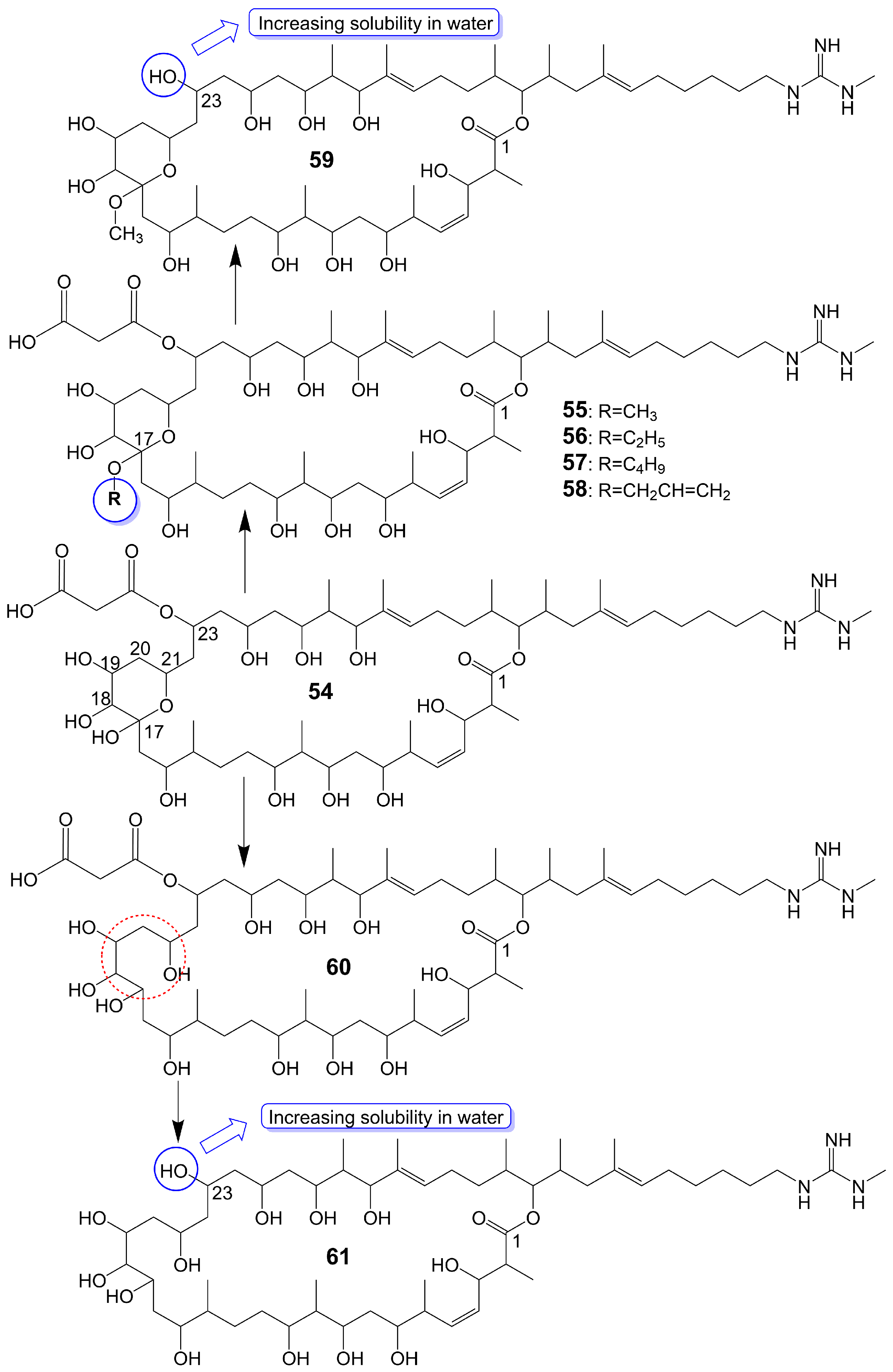
Figure 7. Chemical structures of Guanidylfungin A (54) and its analogues. The red dotted box marks the ring-opening structure of compound 60, which differs from the tetrahydropyran ring guanidylfungin A (54).
In the 1950s, amphotericin B (62) (Figure 8), a 38-membered macrolide isolated from Streptomyces nodosus, was introduced to the clinic as a natural antifungal agent, demonstrating broad-spectrum antifungal activity against various invasive fungi [7][8]. Its mechanism of action involves acting as a “sterol sponge”, forming aggregates outside the cell membrane to extract ergosterol from the bilayer and kill yeasts [43]. However, amphotericin B (62) exhibits dose-dependent renal and hematopoietic toxicity by targeting and extracting cholesterol from host cell membranes, damaging host cells [44]. The introduction of lipid-based formulations of amphotericin B (62) during the mid-1990s significantly reduced its nephrotoxicity [44]. However, the clinical application of amphotericin B (62) is restricted due to its requirement for intravenous administration. A new nanoparticle crystal encapsulated formulation of amphotericin B (62) called cochleated amphotericin B (CAmB) is a novel oral formulation of amphotericin B [45]. CAmB demonstrates in vitro activity against C. neoformans, Candida species, and A. fumigatus [46]. Intraperitoneal injection of CAmB significantly increases the survival rate of mice infected with C. albicans [47]. Using a systemic aspergillosis model, survival is 70% after 14 days at oral doses of 20 mg/kg and 40 mg/kg of CAmB and the fungal burden of lung, liver and kidney is reduced by more than 100 times [48]. Using a 3-day delayed model of murine cryptococcal meningoencephalitis and a large inoculum of a highly virulent strain of serotype A C. neoformans, CAmB, in combination with flucytosine, is found to have efficacy equivalent to parental amphotericin B deoxycholate with flucytosine and superior to oral fluconazole without untoward toxicity [49]. In a Phase I trial, the safety and tolerability of CAmB treatment for cryptococcal meningitis in HIV-infected patients were assessed, revealing that oral CAmB was well tolerated and not nephrotoxic when compared to intravenous CAmB (NCT04031833) [46]. Furthermore, a Phase II trial examined the efficacy of CAmB in patients with azole-resistant chronic mucocutaneous candidiasis, and subsequent clinical trials demonstrated favorable tolerance and safety outcomes (NCT02629419) [50].
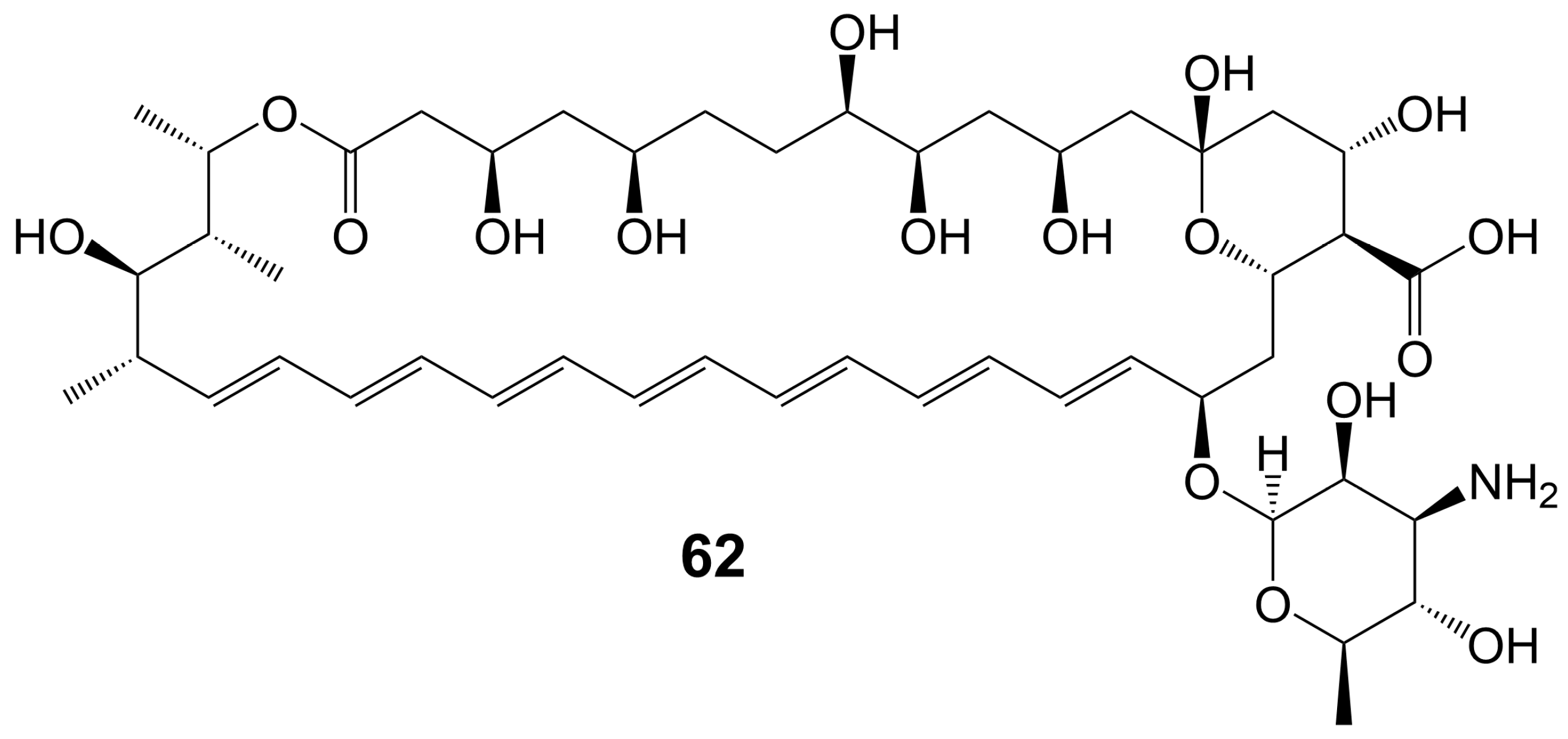
Figure 8. Chemical structures of amphotericin B (62).
3. Polyether Polyketides
Type I PKSs catalyze the decarboxylation reaction of the substrate to generate a polyketide skeleton. This skeleton then is undergone a series of post-modifications, including epoxidation, epoxide ring opening cascade to ether, hydroxylation, methylation, and glycosylation, ultimately forming polyether polyketides. Polyether polyketides are natural polyketide products with multiple asymmetric centers and two or more tetrahydrofuran and tetrahydropyran rings. Polyether polyketides can be categorized into three groups based on their distinct chemical structures: polyethers with long-chain and multi-hydroxyl groups, macrolide polyethers, and ladder-like polyethers.
3.1. Polyethers with Long-Chain and Multi-Hydroxyl Groups
Polyethers with long-chain and multi-hydroxyl groups are water-soluble polyethers whose linear polyketide skeletons only partially form ether rings, and the ether rings and polyketide skeletons are highly hydroxylated.
Amphidinols possess a bis-tetrahydropyran parent structure linked by a C6 alkyl chain with a hydrophilic polyhydroxyl chain and a hydrophobic polyene tail. The hairpin-shaped structure of amphidinols is believed to be crucial for their antifungal activity [51][52]. Amphidinols exhibit potent antifungal activity by interacting with the membrane integration protein glycophorin A, thereby increasing cell membrane permeability [53]. Amphidinol 3 (63) (Figure 9) is isolated from a marine dinoflagellate, Amphidinium klebsii [53]. Amphidinol 3 (63) has antifungal activity against A. niger by disk diffusion method with a minimum effective concentration (MEC) of 8 μg/disk [52]. The presence of the C1 to C20 polyol moiety in amphidinol 3 (63) does not contribute to its antifungal activity, while the C21 to C30 moiety is crucial for its antifungal activity [52]. When the C21-C67 section (64) (Figure 9) of amphidinol 3 (63) is retained, it exhibits a MEC of 20 μg/disk against A. niger. However, when only the C31-C67 moiety (65) (Figure 9) of amphidinol 3 (63) is retained, the compound loses its activity against A. niger [52]. The hydrophobic polyene tail of amphidinol 3 (63) inserts into the lipid bilayer membrane and the hairclip amphidinol 3 (63) results in pore formation that increases the permeability of the cell membrane in a sterol-dependent manner [52]. Amphidinol 3 (63) is believed to exhibit two distinct mechanisms of action: the barrel stave model and the toroidal model. According to the barrel stave model, amphidinol 3 (63) permeates the lipid bilayer directly, forming pores. Conversely, the toroidal model involves the insertion of the hydrophobic polyene tail of amphidinol 3 (63) into the lipid bilayer, while the hydrophilic polyol portion interacts with the cell membrane surface, ultimately leading to pore formation [52]. Amphidinol 18 (66) (Figure 9) is isolated from a marine dinoflagellate, Amphidinium carterae [54]. Amphidinol 18 (66), which has a carbonyl group added to the polyol structure compared to amphidinol 3 (63), shows antifungal activity against C. albicans with MIC values of 9 μg/mL [54]. Amphidinol A (67) (Figure 9) is isolated from a marine dinoflagellate, Amphidinium carterae [51]. Amphidinol A (67), replacing the C54-C63 polyene chain of amphidinol 18 (66) with alkyl chains, exhibits lower antifungal activity against C. albicans, with MIC values of 19 μg/mL [51]. Karatungiol A (68) (Figure 9) is isolated from marine dinoflagellates [55]. Karatungiol A (68), replaced with a longer polyol moiety than amphidinol A (67), exhibits potent antifungal activity against A. niger [55]. Amphidinol 6 (69) (Figure 9) is isolated from a marine dinoflagellate, Amphidinium klebsii [53]. Amphidinol 6 (69), which has a longer polyol moiety and a shorter polyene tail than amphidinol 3 (63), exhibits similar antifungal activity against A. niger with a MEC value of 6 μg/disk [53]. Amphidinols 2 (70) and 7 (71) (Figure 9) are isolated from a marine dinoflagellate, Amphidinium klebsii [53]. Amphidinols 2 (70) and 7 (71), having shorter polyol moieties than amphidinol 6 (69), show similar antifungal activity against A. niger with MEC values of 6 and 10 μg/disk [53]. Desulfurization amphidinol 7 (72) (Figure 9), where the 2-hydroxyl group replaces the sodium sulfonate of amphidinol 7, exhibits stronger antifungal activity against A. niger with a MEC value of 8 μg/disk [53]. Amphidinols 20 (73) and 21 (74) (Figure 9) are isolated from a marine dinoflagellate, Amphidinium carterae [56]. Amphidinols 20 (73) and 21 (74), having longer polyol moieties than amphidinol 6 (69), exhibit negligible antifungal activity against A. niger [56]. This could be attributed to the elongated polyol chains, which hinder their integration into the lipid bilayer, disrupting the barrel stave model and diminishing their membrane destruction activity [56]. Another possible explanation for the diminished bioactivity of amphidinols 20 (73) and 21 (74) is their increased solubility, which hinders the polyol from attaching to the membrane surface [56]. Carteraol E (75) (Figure 9) is isolated from marine dinoflagellates [57]. Carteraol E (75), having a different polyol moiety compared to amphidinol 6 (69), exhibits lower antifungal activity against A. niger with an MEC value of 15 μg/disk [57].
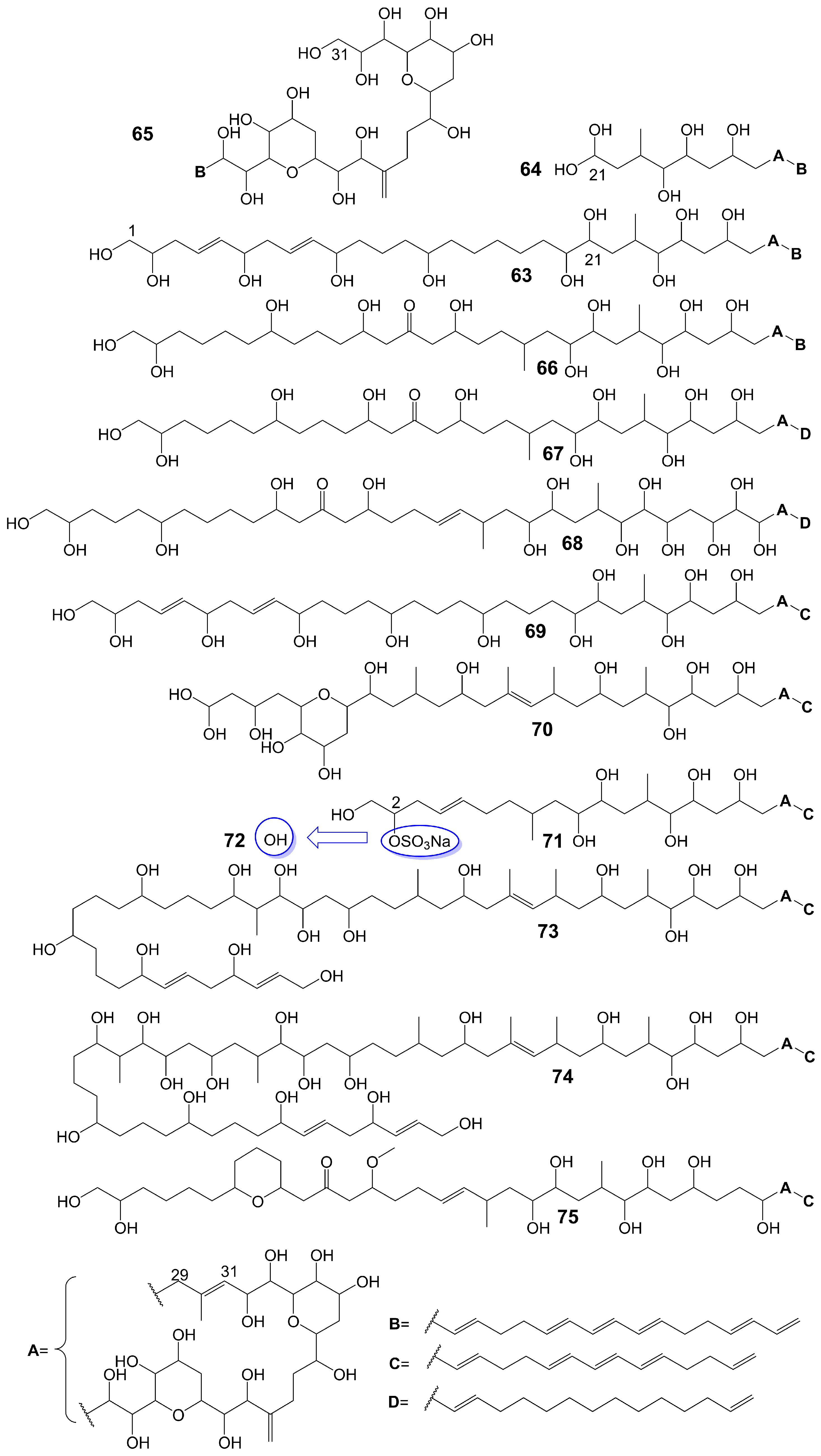
Figure 9. Chemical structures of amphidinols.
3.2. Ladder-like Polyethers
Ladder-like polyethers are composed of ether rings, which are composed mainly of six-membered rings. The ether rings are arranged into ladder-like structures by trans configuration. The oxygen atoms of the adjacent ether rings are alternately located at the upper and lower ends of the ring. Ladder-like polyethers have low polarity and are lipid-soluble compounds.
Yessotoxin (76) (Figure 10), a compound derived from the dinoflagellate Protoceratium reticulatum found in Mutsu Bay, Japan, has been investigated regarding its structure–activity relationship (SAR) [58]. Desulfated yessotoxin (77) and hydrogen-desulfated yessotoxin (78) (Figure 10), two derivatives of yessotoxin, have been synthesized for this purpose [58]. Desulfated yessotoxin (77) has been found to exhibit reduced hydrophilicity and increased antifungal activity against A. niger [58]. Hydrogen-desulfated yessotoxin (78) is a product of the hydrogenation of the polyene side chain of desulfated yessotoxin (77), and its antifungal activity is comparable to that of desulfated yessotoxin (77), indicating that the ladder-shaped polyether structure, as opposed to the polyene side chain, is critical for the antifungal activity of yessotoxin. Desulfated yessotoxin (77) has been found to bind to the transmembrane α-helix motif of the membrane integral protein glycophorin A, thereby inducing the dissociation of glycophorin A oligomers into dimers and monomers [58]. Despite its antifungal activity, yessotoxin (76) has been observed to induce subacute cardiotoxicity [59]. In vitro studies have shown that human ether-a-go-go related gene (hERG) Chinese hamster ovary cells treated with 100 nM yessotoxin for 12 or 24 h exhibit increased hERG potassium channels on the cell surface [59]. In vivo experimentation involves the intraperitoneal injection of rats with either 50 μg/kg or 70 μg/kg yessotoxin (76) every 4 days, resulting in significant physiological changes such as bradycardia, hypotension, cardiac structural alterations, and elevated levels of plasma tissue metalloproteinase-1 inhibitor after 15 days [59]. Additional studies on its structure–activity relationship are warranted to improve the antifungal efficacy of yessotoxin (76) while mitigating its cardiotoxicity.
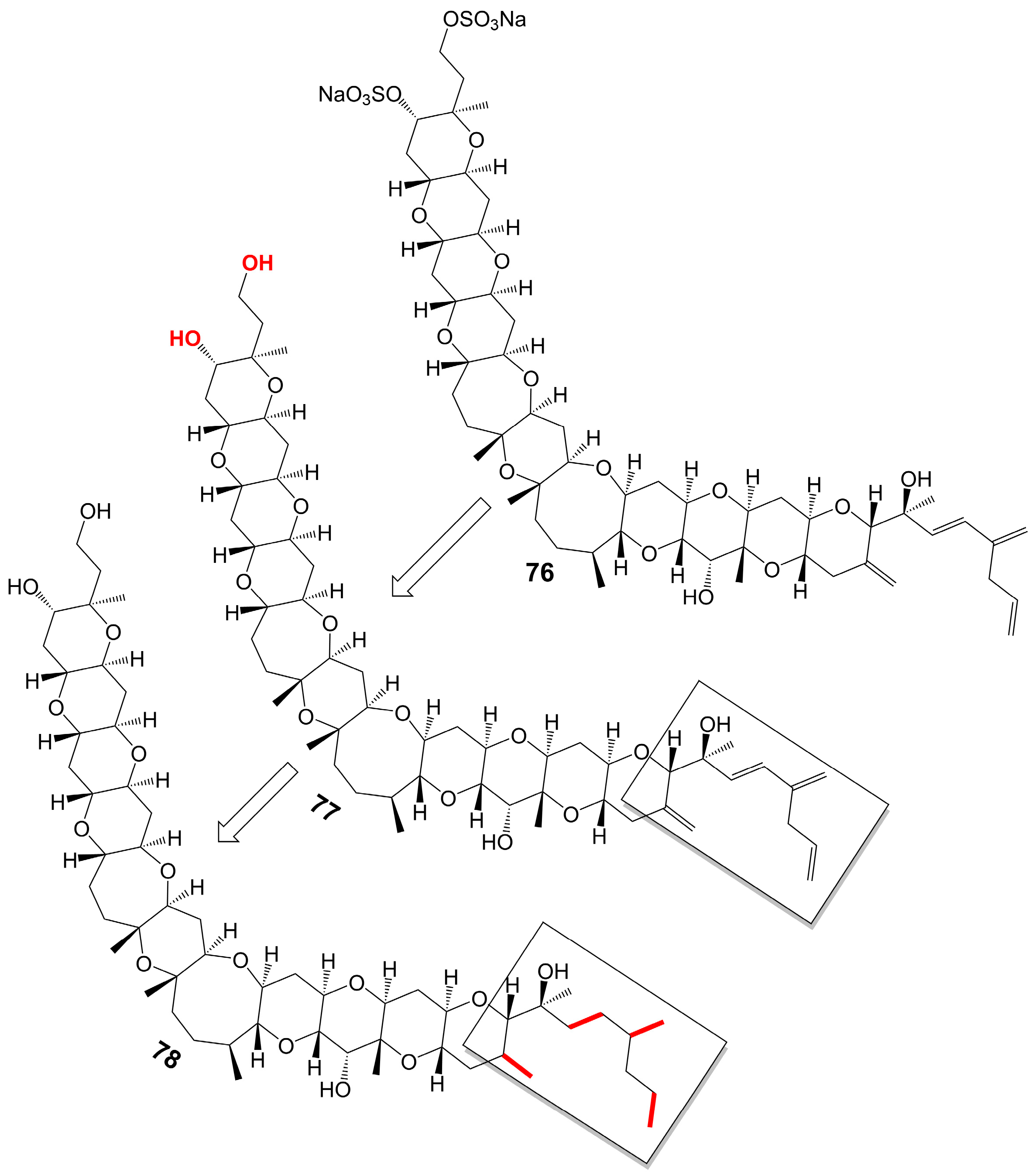
Figure 10. Chemical structures of yessotoxin (76), desulfated yessotoxin (77), and hydrogen-desulfated yessotoxin (78).
3.3. Macrolide Polyethers
Macrolide polyethers are end-to-end polyether products in the form of ester bonds. Forazoline A (79) (Figure 11) is obtained from Actinomadura species strain WMMB-499 isolated from the ascidian Ecteinascidia turbinate [60]. Forazoline A (79) exhibits favorable water solubility, with a concentration of approximately 5 mg/mL [60]. The chemogenomic approach suggests that forazoline A (79) may interfere with the integrity of the cell membrane by disrupting phospholipid homeostasis [60]. Forazoline A (79) exhibits growth inhibition of C. albicans K1 with a MIC value of 16 μg/mL [60]. In a mouse model of C. albicans infection, forazoline A (79) demonstrates comparable in vivo efficacy to amphotericin B (62) without toxicity [60]. The administration of forazoline A (79) at a dose of 0.125 mg/kg reduced the colony-forming unit more than 10 times in the fungal burden of mice kidneys after 8 h, compared to the control group [60].
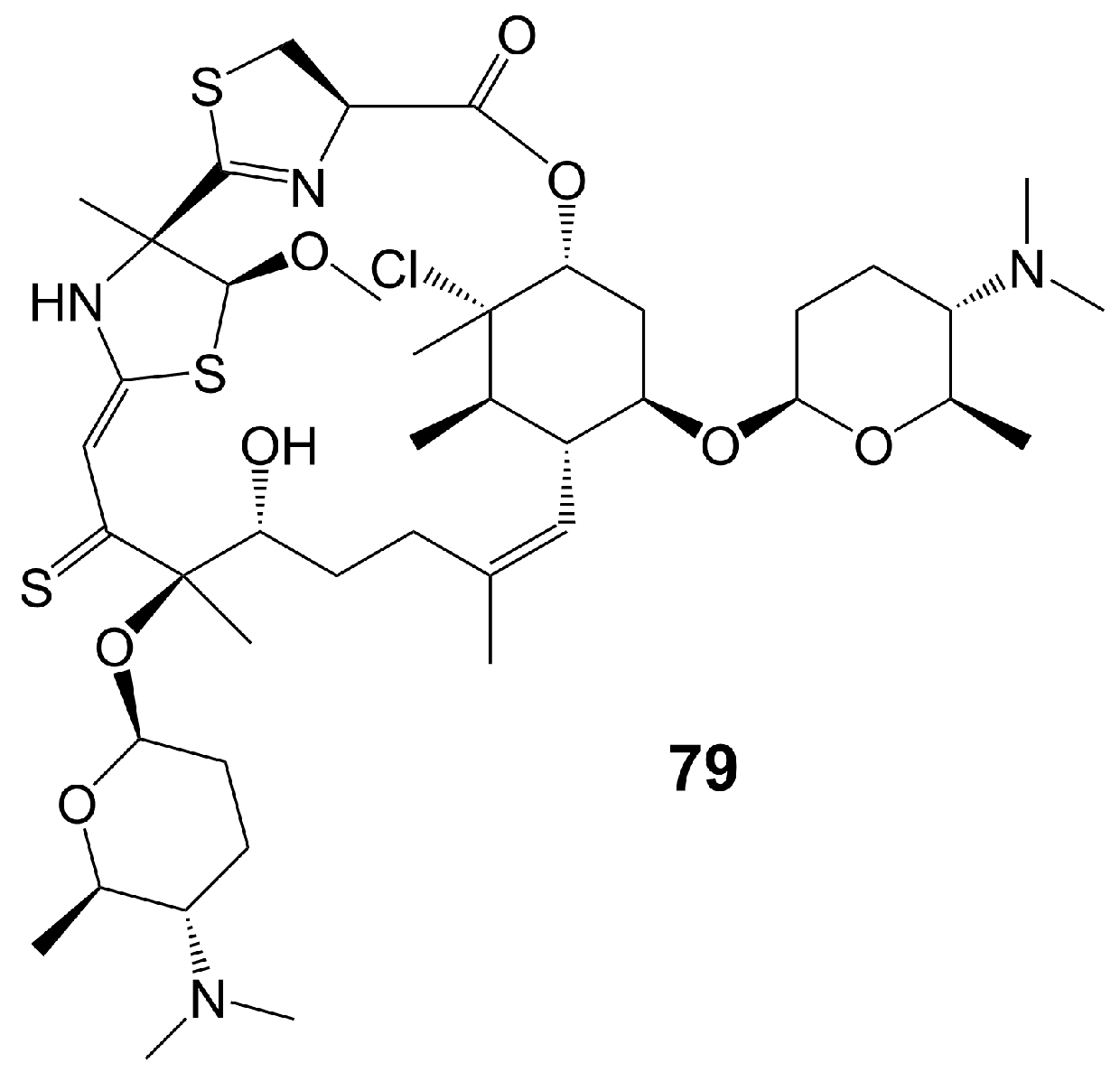
Figure 11. Chemical structures of forazoline A (79).
References
- Lee, Y.; Puumala, E.; Robbins, N.; Cowen, L.E. Antifungal Drug Resistance: Molecular Mechanisms in Candida albicans and Beyond. Chem. Rev. 2021, 121, 3390–3411.
- Zhao, Y.; Ye, L.; Zhao, F.; Zhang, L.; Lu, Z.; Chu, T.; Wang, S.; Liu, Z.; Sun, Y.; Chen, M.; et al. Cryptococcus neoformans, a global threat to human health. Infect. Dis. Poverty 2023, 12, 20.
- Iyer, K.R.; Revie, N.M.; Fu, C.; Robbins, N.; Cowen, L.E. Treatment strategies for cryptococcal infection: Challenges, advances and future outlook. Nat. Rev. Microbiol. 2021, 19, 454–466.
- Lu, H.; Hong, T.; Jiang, Y.; Whiteway, M.; Zhang, S. Candidiasis: From cutaneous to systemic, new perspectives of potential targets and therapeutic strategies. Adv Drug Deliv Rev 2023, 199, 114960.
- Pfaller, M.A.; Diekema, D.J. Epidemiology of invasive mycoses in North America. Crit. Rev. Microbiol. 2010, 36, 1–53.
- Enoch, D.A.; Yang, H.; Aliyu, S.H.; Micallef, C. The Changing Epidemiology of Invasive Fungal Infections. Methods Mol. Biol. 2017, 1508, 17–65.
- Robbins, N.; Wright, G.D.; Cowen, L.E. Antifungal Drugs: The Current Armamentarium and Development of New Agents. Microbiol. Spectr. 2016, 4, 4–5.
- Ostrosky-Zeichner, L.; Casadevall, A.; Galgiani, J.N.; Odds, F.C.; Rex, J.H. An insight into the antifungal pipeline: Selected new molecules and beyond. Nat. Rev. Drug Discov. 2010, 9, 719–727.
- Denning, D.W. Echinocandin antifungal drugs. Lancet 2003, 362, 1142–1151.
- Shimizu, Y.; Ogata, H.; Goto, S. Type III Polyketide Synthases: Functional Classification and Phylogenomics. Chembiochem A Eur. J. Chem. Biol. 2017, 18, 50–65.
- Walsh, C.T. Polyketide and nonribosomal peptide antibiotics: Modularity and versatility. Science 2004, 303, 1805–1810.
- Khosla, C.; Tang, Y.; Chen, A.Y.; Schnarr, N.A.; Cane, D.E. Structure and mechanism of the 6-deoxyerythronolide B synthase. Annu. Rev. Biochem. 2007, 76, 195–221.
- Wang, B.; Guo, F.; Huang, C.; Zhao, H. Unraveling the iterative type I polyketide synthases hidden in Streptomyces. Proc. Natl. Acad. Sci. USA 2020, 117, 8449–8454.
- Hertweck, C.; Luzhetskyy, A.; Rebets, Y.; Bechthold, A. Type II polyketide synthases: Gaining a deeper insight into enzymatic teamwork. Nat. Prod. Rep. 2007, 24, 162–190.
- Xie, S.; Zhang, L. Type II Polyketide Synthases: A Bioinformatics-Driven Approach. Chembiochem A Eur. J. Chem. Biol. 2023, 24, e202200775.
- Austin, M.B.; Noel, J.P. The chalcone synthase superfamily of type III polyketide synthases. Nat. Prod. Rep. 2003, 20, 79–110.
- Hashimoto, M.; Nonaka, T.; Fujii, I. Fungal type III polyketide synthases. Nat. Prod. Rep. 2014, 31, 1306–1317.
- Kirimura, K.; Watanabe, S.; Kobayashi, K. Heterologous gene expression and functional analysis of a type III polyketide synthase from Aspergillus niger NRRL 328. Biochem. Biophys. Res. Commun. 2016, 473, 1106–1110.
- Kubota, T.; Iwai, T.; Sakai, K.; Gonoi, T.; Kobayashi, J. Amphidinins C-F, Amphidinolide Q analogues from marine dinoflagellate Amphidinium sp. Org. Lett. 2014, 16, 5624–5627.
- Takatsu, T.; Nakayama, H.; Shimazu, A.; Furihata, K.; Ikeda, K.; Furihata, K.; Seto, H.; Otake, N. Rustmicin, a new macrolide antibiotic active against wheat stem rust fungus. J. Antibiot. 1985, 38, 1806–1809.
- Mandala, S.M.; Thornton, R.A.; Milligan, J.; Rosenbach, M.; Garcia-Calvo, M.; Bull, H.G.; Harris, G.; Abruzzo, G.K.; Flattery, A.M.; Gill, C.J.; et al. Rustmicin, a potent antifungal agent, inhibits sphingolipid synthesis at inositol phosphoceramide synthase. J. Biol. Chem. 1998, 273, 14942–14949.
- Harris, G.H.; Shafiee, A.; Cabello, M.A.; Curotto, J.E.; Genilloud, O.; Göklen, K.E.; Kurtz, M.B.; Rosenbach, M.; Salmon, P.M.; Thornton, R.A.; et al. Inhibition of fungal sphingolipid biosynthesis by rustmicin, galbonolide B and their new 21-hydroxy analogs. J. Antibiot. 1998, 51, 837–844.
- Sakoh, H.; Sugimoto, Y.; Imamura, H.; Sakuraba, S.; Jona, H.; Bamba-Nagano, R.; Yamada, K.; Hashizume, T.; Morishima, H. Novel galbonolide derivatives as IPC synthase inhibitors: Design, synthesis and in vitro antifungal activities. Bioorganic Med. Chem. Lett. 2004, 14, 143–145.
- Perlatti, B.; Lan, N.; Xiang, M.; Earp, C.E.; Spraker, J.E.; Harvey, C.J.B.; Nichols, C.B.; Alspaugh, J.A.; Gloer, J.B.; Bills, G.F. Anti-cryptococcal activity of preussolides A and B, phosphoethanolamine-substituted 24-membered macrolides, and leptosin C from coprophilous isolates of Preussia typharum. J. Ind. Microbiol. Biotechnol. 2021, 48, kuab022.
- Xiao, L.; Niu, H.J.; Qu, T.L.; Zhang, X.F.; Du, F.Y. Streptomyces sp. FX13 inhibits fungicide-resistant Botrytis cinerea in vitro and in vivo by producing oligomycin A. Pestic. Biochem. Physiol. 2021, 175, 104834.
- Omelchuk, O.A.; Malyshev, V.I.; Medvedev, M.G.; Lysenkova, L.N.; Belov, N.M.; Dezhenkova, L.G.; Grammatikova, N.E.; Scherbakov, A.M.; Shchekotikhin, A.E. Stereochemistries and Biological Properties of Oligomycin A Diels-Alder Adducts. J. Org. Chem. 2021, 86, 7975–7986.
- Sato, S.; Iwata, F.; Yamada, S.; Katayama, M. Neomaclafungins A-I: Oligomycin-class macrolides from a marine-derived actinomycete. J. Nat. Prod. 2012, 75, 1974–1982.
- Lysenkova, L.N.; Saveljev, O.Y.; Omelchuk, O.A.; Zatonsky, G.V.; Korolev, A.M.; Grammatikova, N.E.; Bekker, O.B.; Danilenko, V.N.; Dezhenkova, L.G.; Mavletova, D.A.; et al. Synthesis, antimicrobial and antiproliferative properties of epi-oligomycin A, the (33S)-diastereomer of oligomycin A. Nat. Prod. Res. 2020, 34, 3073–3081.
- Lysenkova, L.N.; Turchin, K.F.; Korolev, A.M.; Danilenko, V.N.; Bekker, O.B.; Trenin, A.S.; Shtil, A.A.; Preobrazhenskaya, M.N. Synthesis and properties of a novel brominated oligomycin A derivative. J. Antibiot. 2012, 65, 223–225.
- Yang, P.W.; Li, M.G.; Zhao, J.Y.; Zhu, M.Z.; Shang, H.; Li, J.R.; Cui, X.L.; Huang, R.; Wen, M.L. Oligomycins A and C, major secondary metabolites isolated from the newly isolated strain Streptomyces diastaticus. Folia Microbiol. 2010, 55, 10–16.
- Lysenkova, L.N.; Turchin, K.F.; Korolev, A.M.; Bykov, E.E.; Danilenko, V.N.; Bekker, O.B.; Trenin, A.S.; Elizarov, S.M.; Dezhenkova, L.G.; Shtil, A.A.; et al. A novel acyclic oligomycin A derivative formed via retro-aldol rearrangement of oligomycin A. J. Antibiot. 2012, 65, 405–411.
- Khebizi, N.; Boudjella, H.; Bijani, C.; Bouras, N.; Klenk, H.P.; Pont, F.; Mathieu, F.; Sabaou, N. Oligomycins A and E, major bioactive secondary metabolites produced by Streptomyces sp. strain HG29 isolated from a Saharan soil. J. De Mycol. Medicale 2018, 28, 150–160.
- Lysenkova, L.N.; Turchin, K.F.; Danilenko, V.N.; Korolev, A.M.; Preobrazhenskaya, M.N. The first examples of chemical modification of oligomycin A. J. Antibiot. 2010, 63, 17–22.
- Tanaka, Y.; Komaki, H.; Yazawa, K.; Mikami, Y.; Nemoto, A.; Tojyo, T.; Kadowaki, K.; Shigemori, H.; Kobayashi, J. Brasilinolide A, a new macrolide antibiotic produced by Nocardia brasiliensis: Producing strain, isolation and biological activity. J. Antibiot. 1997, 50, 1036–1041.
- Mikami, Y.; Komaki, H.; Imai, T.; Yazawa, K.; Nemoto, A.; Tanaka, Y.; Gräefe, U. A new antifungal macrolide component, brasilinolide B, produced by Nocardia brasiliensis. J. Antibiot. 2000, 53, 70–74.
- Takesako, K.; Beppu, T.; Nakamura, T.; Obayashi, A. Demalonyl derivatives of guanidylfungin A and copiamycin: Their synthesis and antifungal activity. J. Antibiot. 1985, 38, 1363–1370.
- Helaly, S.E.; Kulik, A.; Zinecker, H.; Ramachandaran, K.; Tan, G.Y.; Imhoff, J.F.; Süssmuth, R.D.; Fiedler, H.P.; Sabaratnam, V. Langkolide, a 32-membered macrolactone antibiotic produced by Streptomyces sp. Acta 3062. J. Nat. Prod. 2012, 75, 1018–1024.
- Chevrette, M.G.; Carlson, C.M.; Ortega, H.E.; Thomas, C.; Ananiev, G.E.; Barns, K.J.; Book, A.J.; Cagnazzo, J.; Carlos, C.; Flanigan, W.; et al. The antimicrobial potential of Streptomyces from insect microbiomes. Nat. Commun. 2019, 10, 516.
- Pérez-Victoria, I.; Oves-Costales, D.; Lacret, R.; Martín, J.; Sánchez-Hidalgo, M.; Díaz, C.; Cautain, B.; Vicente, F.; Genilloud, O.; Reyes, F. Structure elucidation and biosynthetic gene cluster analysis of caniferolides A-D, new bioactive 36-membered macrolides from the marine-derived Streptomyces caniferus CA-271066. Org. Biomol. Chem. 2019, 17, 2954–2971.
- Zhang, Z.; Zhou, T.; Harunari, E.; Oku, N.; Igarashi, Y. Iseolides A-C, antifungal macrolides from a coral-derived actinomycete of the genus Streptomyces. J. Antibiot. 2020, 73, 534–541.
- Alferova, V.A.; Novikov, R.A.; Bychkova, O.P.; Rogozhin, E.A.; Shuvalov, M.V.; Prokhorenko, I.A.; Sadykova, V.S.; Kulko, A.B.; Dezhenkova, L.G.; Stepashkina, E.A.; et al. Astolides A and B, antifungal and cytotoxic naphthoquinone-derived polyol macrolactones from Streptomyces hygroscopicus. Tetrahedron 2018, 74, 7442–7449.
- Takesako, K.; Beppu, T. Studies on new antifungal antibiotics, guanidylfungins A and B.I. Taxonomy, fermentation, isolation and characterization. J. Antibiot. 1984, 37, 1161–1169.
- Anderson, T.M.; Clay, M.C.; Cioffi, A.G.; Diaz, K.A.; Hisao, G.S.; Tuttle, M.D.; Nieuwkoop, A.J.; Comellas, G.; Maryum, N.; Wang, S.; et al. Amphotericin forms an extramembranous and fungicidal sterol sponge. Nat. Chem. Biol. 2014, 10, 400–406.
- Perfect, J.R. The antifungal pipeline: A reality check. Nat. Rev. Drug Discov. 2017, 16, 603–616.
- Aigner, M.; Lass-Flörl, C. Encochleated Amphotericin B: Is the Oral Availability of Amphotericin B Finally Reached? J. Fungi 2020, 6, 66.
- Skipper, C.P.; Atukunda, M.; Stadelman, A.; Engen, N.W.; Bangdiwala, A.S.; Hullsiek, K.H.; Abassi, M.; Rhein, J.; Nicol, M.R.; Laker, E.; et al. Phase I EnACT Trial of the Safety and Tolerability of a Novel Oral Formulation of Amphotericin B. Antimicrob. Agents Chemother. 2020, 64, 10–1128.
- Zarif, L.; Graybill, J.R.; Perlin, D.; Najvar, L.; Bocanegra, R.; Mannino, R.J. Antifungal activity of amphotericin B cochleates against Candida albicans infection in a mouse model. Antimicrob. Agents Chemother. 2000, 44, 1463–1469.
- Delmas, G.; Park, S.; Chen, Z.W.; Tan, F.; Kashiwazaki, R.; Zarif, L.; Perlin, D.S. Efficacy of orally delivered cochleates containing amphotericin B in a murine model of aspergillosis. Antimicrob. Agents Chemother. 2002, 46, 2704–2707.
- Lu, R.; Hollingsworth, C.; Qiu, J.; Wang, A.; Hughes, E.; Xin, X.; Konrath, K.M.; Elsegeiny, W.; Park, Y.D.; Atakulu, L.; et al. Efficacy of Oral Encochleated Amphotericin B in a Mouse Model of Cryptococcal Meningoencephalitis. mBio 2019, 10, 10–1128.
- Desai, J.V.; Urban, A.; Swaim, D.Z.; Colton, B.; Kibathi, L.W.; Ferrè, E.M.N.; Stratton, P.; Merideth, M.A.; Hunsberger, S.; Matkovits, T.; et al. Efficacy of Cochleated Amphotericin B in Mouse and Human Mucocutaneous Candidiasis. Antimicrob. Agents Chemother. 2022, 66, e0030822.
- Cutignano, A.; Nuzzo, G.; Sardo, A.; Fontana, A. The Missing Piece in Biosynthesis of Amphidinols: First Evidence of Glycolate as a Starter Unit in New Polyketides from Amphidinium carterae. Mar. Drugs 2017, 15, 157.
- Wakamiya, Y.; Ebine, M.; Matsumori, N.; Oishi, T. Total Synthesis of Amphidinol 3: A General Strategy for Synthesizing Amphidinol Analogues and Structure-Activity Relationship Study. J. Am. Chem. Soc. 2020, 142, 3472–3478.
- Morsy, N.; Konoki, K.; Houdai, T.; Matsumori, N.; Oishi, T.; Murata, M.; Aimoto, S. Roles of integral protein in membrane permeabilization by amphidinols. Biochim. Biophys. Acta 2008, 1778, 1453–1459.
- Nuzzo, G.; Cutignano, A.; Sardo, A.; Fontana, A. Antifungal amphidinol 18 and its 7-sulfate derivative from the marine dinoflagellate Amphidinium carterae. J. Nat. Prod. 2014, 77, 1524–1527.
- Washida, K.; Koyama, T.; Yamada, K.; Kita, M.; Uemura, D. Karatungiols A and B, two novel antimicrobial polyol compounds, from the symbiotic marine dinoflagellate Amphidinium sp. Tetrahedron Lett. 2006, 47, 2521–2525.
- Satake, M.; Cornelio, K.; Hanashima, S.; Malabed, R.; Murata, M.; Matsumori, N.; Zhang, H.; Hayashi, F.; Mori, S.; Kim, J.S.; et al. Structures of the Largest Amphidinol Homologues from the Dinoflagellate Amphidinium carterae and Structure-Activity Relationships. J. Nat. Prod. 2017, 80, 2883–2888.
- Huang, S.J.; Kuo, C.M.; Lin, Y.C.; Chen, Y.M.; Lu, C.K. Carteraol E, a potent polyhydroxyl ichthyotoxin from the dinoflagellate Amphidinium carterae. Tetrahedron Lett. 2009, 50, 2512–2515.
- Mori, M.; Oishi, T.; Matsuoka, S.; Ujihara, S.; Matsumori, N.; Murata, M.; Satake, M.; Oshima, Y.; Matsushita, N.; Aimoto, S. Ladder-shaped polyether compound, desulfated yessotoxin, interacts with membrane-integral alpha-helix peptides. Bioorganic Med. Chem. 2005, 13, 5099–5103.
- Ferreiro, S.F.; Vilariño, N.; Carrera, C.; Louzao, M.C.; Cantalapiedra, A.G.; Santamarina, G.; Cifuentes, J.M.; Vieira, A.C.; Botana, L.M. Subacute Cardiotoxicity of Yessotoxin: In Vitro and in Vivo Studies. Chem. Res. Toxicol. 2016, 29, 981–990.
- Wyche, T.P.; Piotrowski, J.S.; Hou, Y.; Braun, D.; Deshpande, R.; McIlwain, S.; Ong, I.M.; Myers, C.L.; Guzei, I.A.; Westler, W.M.; et al. Forazoline A: Marine-derived polyketide with antifungal in vivo efficacy. Angew. Chem. 2014, 53, 11583–11586.
More
Information
Subjects:
Infectious Diseases
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
621
Revisions:
2 times
(View History)
Update Date:
04 Jan 2024
Table of Contents




Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No