Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Lucia Prieto-Torres | -- | 3999 | 2024-01-03 17:30:54 | | | |
| 2 | Jessie Wu | + 10 word(s) | 4009 | 2024-01-04 04:09:06 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Prieto-Torres, L.; Requena, L.; Rodríguez-Pinilla, S.M. Myeloproliferative Related Dermatosis with Indolent Clinical Outcomes. Encyclopedia. Available online: https://encyclopedia.pub/entry/53383 (accessed on 09 August 2026).
Prieto-Torres L, Requena L, Rodríguez-Pinilla SM. Myeloproliferative Related Dermatosis with Indolent Clinical Outcomes. Encyclopedia. Available at: https://encyclopedia.pub/entry/53383. Accessed August 09, 2026.
Prieto-Torres, Lucía, Luis Requena, Socorro Maria Rodríguez-Pinilla. "Myeloproliferative Related Dermatosis with Indolent Clinical Outcomes" Encyclopedia, https://encyclopedia.pub/entry/53383 (accessed August 09, 2026).
Prieto-Torres, L., Requena, L., & Rodríguez-Pinilla, S.M. (2024, January 03). Myeloproliferative Related Dermatosis with Indolent Clinical Outcomes. In Encyclopedia. https://encyclopedia.pub/entry/53383
Prieto-Torres, Lucía, et al. "Myeloproliferative Related Dermatosis with Indolent Clinical Outcomes." Encyclopedia. Web. 03 January, 2024.
Copy Citation
Myeloid neoplasms and acute leukemias include different entities that have been recently re-classified taking into account molecular and clinicopathological features. Two major articles were published in 2022, the ICC and the WHO classifications. The myelodysplastic syndrome/myeloproliferative neoplasm (MDS/MPN) category comprises a heterogeneous group of hybrid neoplastic myeloid diseases characterized by the co-occurrence of clinical and pathological features of both myelodysplastic and myeloproliferative neoplasms. The most frequent entity in this category is chronic myelomonocytic leukemia (CMML) which is, after acute myeloid leukemia (AML), the main myeloid disorder prone to develop cutaneous manifestations.
myelodysplastic and myeloproliferative neoplasms
CMML
skin
1. Neutrophilic Dermatoses
Neutrophilic dermatoses (ND) (Figure 1) comprise a heterogeneous group of inflammatory skin disorders that share a similar histopathology, consisting of a sterile neutrophilic infiltrate. Clinical manifestations of the ND are diverse, even with variations in the same patient. Theoretically, the histopathologic location of the neutrophilic infiltrate (epidermis, dermis, subcutis), the disease course and the clinical appearance help to make the differential diagnosis among different ND, such as Sweet syndrome (SS), pyoderma gangrenosum (PG), Behçet syndrome (BS), erythema elevatum diutinum (EED) and neutrophilic eccrine hidradenitis (NEH) [1].
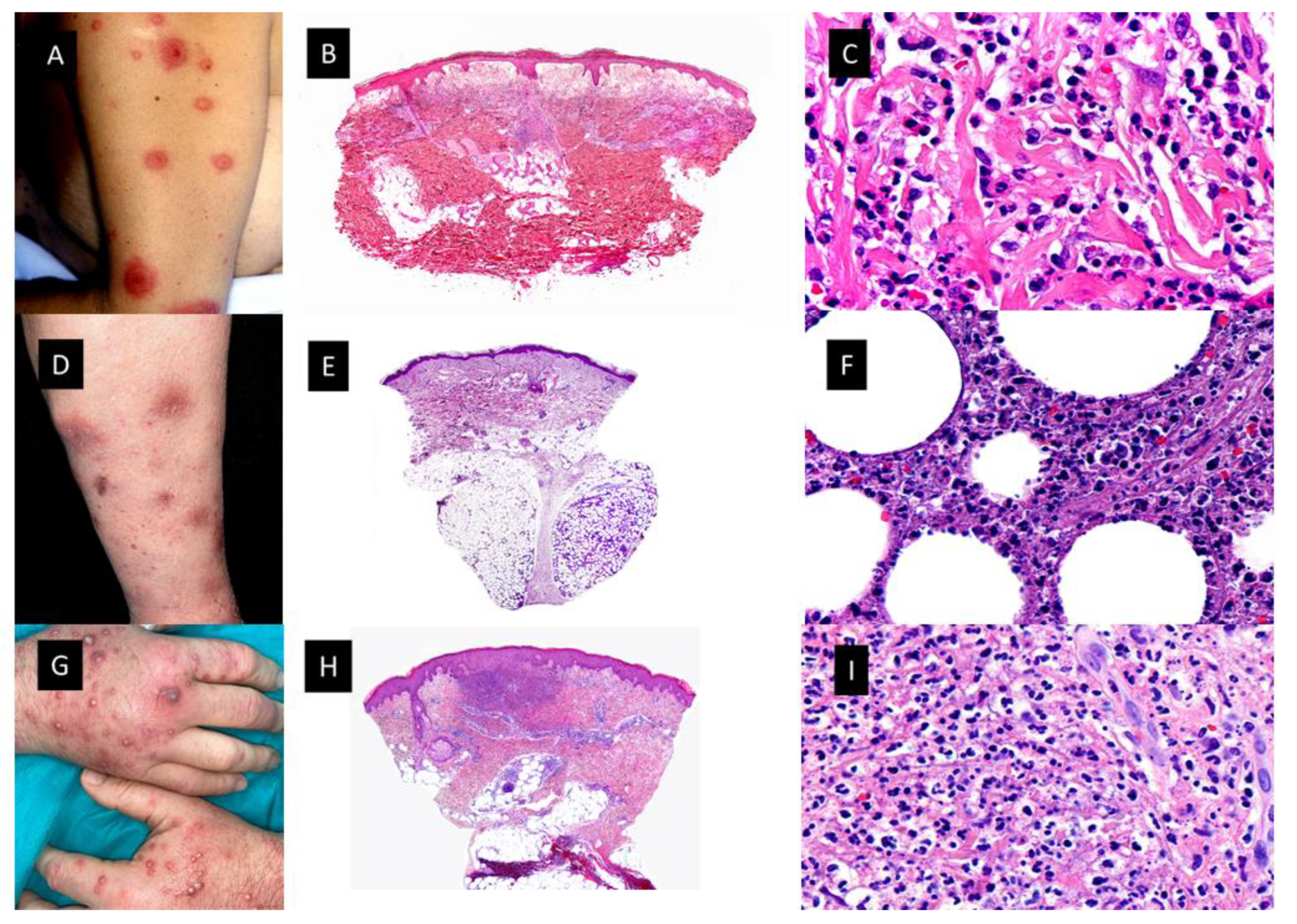
Figure 1. Neutrophilic dermatoses. (A). Classic Sweet syndrome (C-SS) presenting with edematous, erythematous and tender plaques located on the left arm. (B). At low magnification, intense edema of the papillary dermis and an underlying band-like, dense, dermal, inflammatory infiltrate mostly composed of mature neutrophils, with leukocytoclasis (HE staining 4×) (C). Detail of the neutrophils interstitially arranged in a linear way between collagen bundles (HE staining ×40) (D). Subcutaneous Sweet syndrome (S-SS) with erythematous and violaceous skin nodules located on the leg (E). Panoramic picture showing a mostly lobular subcutaneous infiltrate in the absence of vascular changes (HE staining 4×). (F). Higher magnification of the neutrophils located around fat lobules (HE staining 40×) (G). Vexas Syndrome with violaceous edematous papules and pustules with erythematous halo located on the dorsum of both hands (H). Dense dermal infiltrate located in superficial and middle dermis (HE staining 4×) (I). On higher magnification, dense infiltrate composed mainly of neutrophils (HE staining ×40).
1.1. Sweet Syndrome
In 1964, Robert Douglas Sweet reported eight patients with an “acute febrile neutrophilic dermatosis” that afterwards was renamed SS [2]. Clinically, patients presented with fever and edematous, erythematous tender cutaneous plaques located on any area of the skin surface, but mainly involving the upper trunk. The usual histopathological findings consisted of intense edema of the papillary dermis and an underlying band-like, dense, dermal, inflammatory infiltrate mostly composed of mature neutrophils, with leukocytoclasis but without features of vasculitis. (Figure 1B,C) In rare instances, however, typical lesions of SS may show histopathological features of leukocytoclastic vasculitis, which seems to be a secondary epiphenomenon. Approximately 21% of patients with SS have an associated malignancy [3]. Malignancy-associated SS is more commonly reported with hematological malignancies and myelodysplastic syndrome/myeloproliferative neoplasm (MDS/MPN) compared to solid-organ malignancies. The most common associations are acute myeloid leukemia (AML) followed by MDS. SS may precede or follow a diagnosis of malignancy, and it has even been described as a signal of cancer recurrence [4]. There are several clinical and histopathological variants of SS, including localized SS, histiocytoid SS (H-SS), subcutaneous SS, acute necrotizing SS and xanthomatized neutrophilic dermatosis. One clinicopathological variant that deserves special attention in patients with myeloid neoplasms is H-SS.
Histiocytoid SS (H-SS)
This histopathologic variant of SS, first described by Requena et al. in 2005 [5], is characterized by a dermal infiltrate of mononuclear cells of histiocytic appearance, but strongly positive for myelocytic and promyelocytic markers, as demonstrated by the double immunostain for myeloperoxidase (MPO) and myeloid cell nuclear differentiation antigen (MNDA) [6]. As in classic SS, patients with H-SS present with tender erythematous plaques and nodules on the extremities and trunk accompanied by systemic symptoms such as fever and arthralgia [5][6]. (Figure 2A–D) Association with malignancy ranges from 30% to 61% and the most frequent associated neoplasia is MDS.
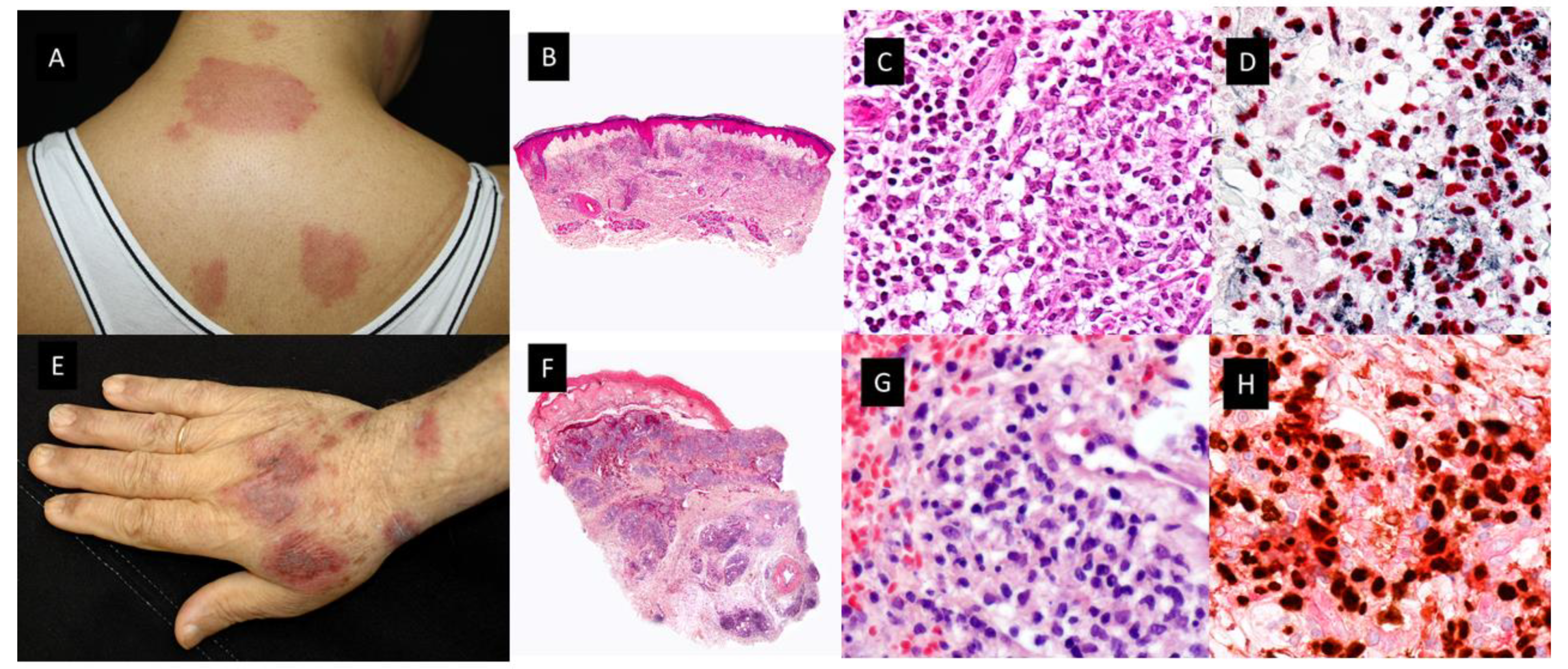
Figure 2. Clinicopathological features of Histiocytoid Sweet syndrome(H-SS) and Myelodysplasia cutis (MDS-cutis). (A) H-SS clinical picture showing erythematous and edematous plaques on the back and posterior region of the neck (B) Scanning power shows edema and nodular infiltrates in the superficial dermis. (HE staining 4×). (C) The dermal nodules are mostly composed of mononuclear cells with twisted vesicular nuclei and scant eosinophilic cytoplasm. (D) Double immunostained specimen with nuclear myeloid nuclear differentiation antigen [MNDA, black] and cytoplasmic myeloperoxidase [MPO, red]) (40×). (E) MDS-cutis showing edematous plaques on the dorsum of the hand and arm, with raised erythematous border and a slightly depressed violaceous center. (F) Histopathologic features consist of edema in the papillary dermis and multiple nodular infiltrates in the superficial and deep dermis; there are also perivascular infiltrates around deep dermal vascular plexus (HE 4×). (G) On higher magnification, atypical, dermal hematolymphoid infiltrate and some red cells, the atypical cells demonstrate reniform nuclei and eosinophilic cytoplasm and include some pseudo-Pelger–Huet anomaly (HE 40×). (H) Double immunostained specimen with myeloid nuclear differentiation antigen [MNDA, black] and cytoplasmic CD123 [CD123, red].
In the first 41-patient-series published by Requena et al., there were 7 patients with associated malignancies, including chronic myelomonocytic leukemia (CMML) (case 3) “lymphoma” (no more specific diagnosis could be obtained) (case 21), monoclonal gammopathy of undetermined significance (case 24), renal carcinoma (case 29), breast carcinoma (case 28), chronic B lymphocytic leukemia (case 38), and multiple myeloma (case 39) [5]. In a series of nine patients, Vignon-Pennamen et al. suggested for the first time that this histopathological variant could be more frequently associated with AML and MDS than classic SS [7]. Later, in 2015, Ghoufi et al. published a larger series, which included 62 patients with a histopathologic diagnosis of SS, 22 cases with H-SS and 40 cases with the classical neutrophilic variant (N-SS) [8]. They found that MDS was diagnosed in 7 of 22 patients with H-SS and only in 1 of 40 patients with N-SS (p < 0.001). In three H-SS patients with MDS, the cutaneous lesions preceded the hematological diagnosis by more than 6 months. Therefore, they proposed that a complete hematological assessment and close follow-up should be mandatory in patients with H-SS [8]. In contrast, Alegría et al. found that H-SS was not more frequently related to hematologic malignancies than N-SS [6]. In their series of 33 patients with H-SS, 8 suffered from hematological malignancies, including 3 with MDS and 2 with CMML. In these patients, a CD163+/MPO+ population was absent in the infiltrate, whereas double immunostain with MNDA/MPO was intensely positive in most cells of the infiltrate, whereas a minor proportion of cells expressed the double immunostain for MNDA/CD163. The skin lesions appeared 5 and 2 years before the diagnosis of the hematological condition [6].
The initial presumption of the reactive nature of SS, including variants like H-SS, was supported by the acute onset of the skin lesions and the complete response to steroid therapy. However, the improvement in molecular techniques has demonstrated clonally related cells in both the skin and bone marrow of hematological patients with N-SS and H-SS [9]. In 2020, a molecular study including 10 patients with SS and AML, myelodysplastic syndrome, myeloproliferative neoplasm or MDS/MPN was published. Eight patients had N-SS and 2 H-SS. They performed Next Generation Sequencing (NGS) analysis of paired skin-lesion biopsies and detected 35 out of the 37 mutations found in hematopoietic samples, without additional mutations. Seven patients exhibited identical mutational profiles in both tissues. In all mutated cases (n 9), mutations of the major clone of the myeloid neoplasm were also found in the cells of cutaneous infiltrates. CD34 and myeloperoxidase immunohistochemical staining of skin biopsies revealed very few immature myeloid cells in only 2 out of 10 patients, ruling out leukemia cutis. Most mutations detected in skin-lesion samples were at relatively high allelic burden (median 20%, at least one mutation with variant allele frequencies of 10% for each patient) indicating that they did not relate to rare infiltrating blast cells or blood contamination of skin biopsies. The authors assumed that the process of migration and infiltration of the skin in SS may be related to the tumoral phenotype of myeloid cells, rather than only triggered by cell extrinsic factors [9].
These NGS studies in the skin of patients with MDS and ND, especially H-SS, also support the concept of MDS-cutis [10].
Other Morphological Variants of the Sweet Syndrome
There have been other clinicopathogical variants reported in the literature in the setting of MDS/MPN mentioned above [11]. Among all of these variants, it is worth highlighting the subcutaneous Sweet syndrome (S-SS) or neutrophilic lobular panniculitis (Figure 1D–F) [12][13]. Other names employed in the literature include “Sweet’s panniculitis”, “Sweet´s-like neutrophilic panniculitis” and “neutrophilic panniculitis associated with myeloid disorders”. Primary neutrophilic panniculitis, defined as neutrophilic infiltrates confined to the fat lobules of the subcutis as opposed to extension of a dermal neutrophilic infiltrate into the subcutis (secondary neutrophilic panniculitis) has been reported in the literature as a histological pattern seen in a heterogeneous group of disorders including infectious panniculitis, “reactive panniculitis reaction”, which is associated with some bacterial antigens and usually has vascular changes, and leukemia cutis [14]. Some of these diseases may be readily diagnosed, but others are less distinct clinically and histopathologically. For example, it may be difficult to distinguish between S-SS and infectious panniculitis [14].
S-SS in the setting of myeloid disorders has been described in scattered case reports and small series of cases as erythematous skin nodule(s) generally located in the extremities with the trunk and the face affected less frequently. Cutaneous lesions are usually accompanied by fever and they resolve within days or a few weeks of oral steroid treatment like classic SS (C-SS). Histopathologically, these nodules are characterized by a dense neutrophilic infiltrate confined to the subcutis in the absence of vascular changes. Unlike C-SS, papillary dermal edema is minimal. Both septal and lobular panniculitic patterns have been reported, the lobular one being the commonest form. Interestingly, occasional large epithelioid cells consistent with reactive stromal fibroblasts and abnormal nuclear segmentation (hyposegmentation or hypersegmentation) of neutrophils have been described in these myeloid-related cases [14]. AML and MDS are the myeloid neoplasms more frequently related to S-SS in the reported cases [14]. A variant of subcutaneous H-SS has also been reported heralding transformation of MDS into AML [15] in a relapsed myeloblastic leukemia [16] and in MDS-refractory anemia [17].
1.2. Vexas Syndrome
In 2020, a new adult-onset autoinflammatory syndrome with overlapping features with MDS or multiple myeloma (MM) and inflammatory diseases such as relapsing polychondritis, SS, polyarteritis nodosa or giant-cell arteritis was described in 25 men with somatic mutations in p.Met41 of UBA1, the major E1 enzyme that initiates ubiquitylation [18]. This enzyme is encoded by the gene UBA1 located on the X chromosome. Its estimated prevalence is about 1 in every 13591 adults [19].
The acronym, VEXAS, stands for vacuoles, E1 enzyme, X-linked, autoinflammatory and somatic syndrome. Somatic mutations, first acquired and then clonally selected, have been implicated as previously referred to in neoplastic hematological diseases [20]. With VEXAS syndrome, the authors of the original description used a genotype-driven approach to identify a genetic cause of an autoinflammatory disease. All patients with UBA1 mosaic mutations had predominantly wild-type lymphocytes (T and B cells) and mutant myeloid cells (neutrophils and monocytes) in peripheral blood, although a patient with vacuolated lymphoid precursors was also reported [21]. Hematopoietic stem cells and multipotent progenitors isolated from BM had abundant mutant cells. The patients began to develop the inflammatory syndromes in the fifth to seventh decade of life. Patients had progressive hematological abnormalities, including macrocytic anemia, thrombocytopenia and myeloid dyspoiesis. Co-occurrence of VEXAS with hematologic neoplasms has been described with MDS and plasma cell dyscrasia, and recently a male patient with chronic myeloid leukemia (CML) and VEXAS [22] has also been reported. The dermatological manifestations include neutrophilic dermatoses similar to SS with a poor response to conventional therapies and frequent relapses, similar to the previously described MDS-cutis patients [23].
1.3. Myelodysplasia Cutis
Osio et al. identified in a series of 150 patients with MDS and cutaneous lesions 24 patients with non-blastic tumor cells in the dermal infiltrate, defined as medium-sized immature myeloid cells with abundant eosinophilic cytoplasm and twisted nuclei or the pseudo-Pelger–Huet anomaly (see Figure 2E–H) [24]. They found that these cells had a combined myeloid and monocytic immunophenotype, with the expression of both MPO and CD163 or CD68 antigens and negativity for CD34, CD56 or CD117. The proliferative index was low in 56% of cases (<10% of positive Mib-1 cells) or intermediate in 44% of cases (10 to 66% positive Mib-1 cells). In addition, the cutaneous infiltrate was rich in mature neutrophils and normal CD3+ T-lymphocytes. The presence of edema in the superficial dermis was a frequent finding (67% of samples) [24]. Fluorescent in situ hybridization (FISH) analyses showed common cytogenetic abnormalities in the skin and the BM of 4/6 patients. Previously, only Sujobert et al. had demonstrated a clonal relationship of this kind in neutrophilic dermatoses associated with AML [25]. These authors postulated that some cases previously reported as “H-SS” in the course of MDS, especially those with a poor response to treatments such as hydroxychloroquine, dapsone, colchicine or thalidomide or patients with steroid dependence or in need of high dose oral prednisone could have MDS-cutis. Furthermore, they compared the survival rate and other clinicopathologic characteristics of patients with MDS-cutis and leukemia cutis, and they found significant differences between the two groups [24]. Regarding overall survival (OS) after the skin diagnosis, it was significantly longer in MDS-cutis patients than in patients who suffered from leukemia cutis (62 vs. 5 months, (p < 0.001)). In addition, there were other discriminant histopathological features related to survival influence, such as positivity for CD34, CD56 or CD117 for shorter survival in patients with leukemia cutis (p < 0.059), and the presence of CD3+ T lymphocytes (p < 0.001), edema (p < 0.01) and a lower Mib-1 proliferative index (p < 0.05) for longer survival in patients with MDS-cutis. Clinically, leukemia cutis patients usually have persistent skin nodules, whereas in MDS-cutis patients there are flares and relapses of cutaneous lesions. These lesions were nodules in leukemia cutis (p < 0.01) and erythematous plaques (p < 0.001), with a frequent annular pattern (p < 0.05) accompanied by fever, or arthralgia (p < 0.01) in MDS-cutis patients [24].
Finally, there has been a recent report of seven patients with MDS-cutis, one of them with UBA1 mutation (VEXAS syndrome), in which NGS analysis found one to five mutated genes both in the BM (with a median VAF of 30%) and in the cutaneous lesions of the same patients (with a median VAF of 11%) [10]. In addition, there were some mutations detected in one tissue but not in the other, supporting the possibility of clonal evolution in one of the tissues [10]. They suggested that MDS-cutis should be considered as differential diagnosis of leukemia cutis, classical SS and MDS-associated vasculitis. MDS-cutis may potentially be underdiagnosed if a molecular investigation is not performed, especially in patients with H-SS. Unlike what happens in patients with N-SS, MDS-cutis patients seem to be more resistant to steroid therapy, and they may respond to hypomethylating agents, like Azacytidine, although this sensitivity still must be confirmed in larger prospective series [10].
2. Granulomatous Dermatoses
Interstitial granulomatous dermatitis (IGD) was first described by Gottlieb et al. in 1995 as a granulomatous dermatitis with interstitially disposed dermal infiltrates composed mainly of histiocytes in a palisaded arrangement around the foci of degenerated collagen bundles, with scattered neutrophilis and eosinophilis [26]. Shortly after, Chu et al. proposed the term “palisaded neutrophilic and granulomatous dermatitis (PNGD)” for the papular eruption on the extensor surface of extremities described in the context of collagen vascular diseases [27]. Both terms, IGD and PNGD, as well as the overlapping between IGD and granuloma annulare, are now included by some authors in the spectrum of the so-called reactive granulomatous dermatitis [28].
These cutaneous lesions have been mainly related to rheumatologic diseases and drug reactions. However, the association of PNGD or IGD with hematological malignancies, including CMML and MDS, has been reported firstly as a reactive dermatitis in the setting of the hematological condition and lately as a possible skin manifestation of the disease [29][30].
Federmann et al. published in 2017 the findings regarding three patients with widespread papular eruptions resistant to conventional skin-directed therapies, which histopathologically showed interstitial granulomas composed of epithelioid histiocytes in the reticular dermis arranged around the foci of degenerated collagen and variable neutrophilic inflammation consistent with PNGD (see Figure 3A–F) [31]. The granulomas showed a similar immunohistochemical profile, with positivity for CD14, CD68 (PG-M1) and CD123, whereas only very few cells were positive for TCL1 and CD303. CD56, S100 protein and CD1a were negative. The most interesting contribution of this research was the finding of SRSF2 P95 hotspot mutations, found in 40–50% of patients suffering from CMML, which were retrospectively detected in both the skin and BM biopsies of the three patients. In addition, in one of them, these mutations were already found 5 years before the diagnosis of the hematologic neoplasia [31]. Since then, only a few more cases have been reported [30][32][33]. All but one were patients with CMML, in all cases where molecular techniques were performed, SRSF2 mutations were found in the BM and in the cutaneous infiltrates of the patients. For that reason, Enescu et al. proposed that this kind of dermatitis is specific for CMML patients with this molecular alteration [33]. The molecular technique used for the study of mutations in the BM and skin of these patients was variable, including NGS, pyrosequencing or RFLP analysis by BsaJI digestion and/or sequencing of SRSF2 polymerase chain reaction products containing the hotspot region in codon 95 (2 p.Pro95His;1 p.Pro95Leu) [30][31][33].
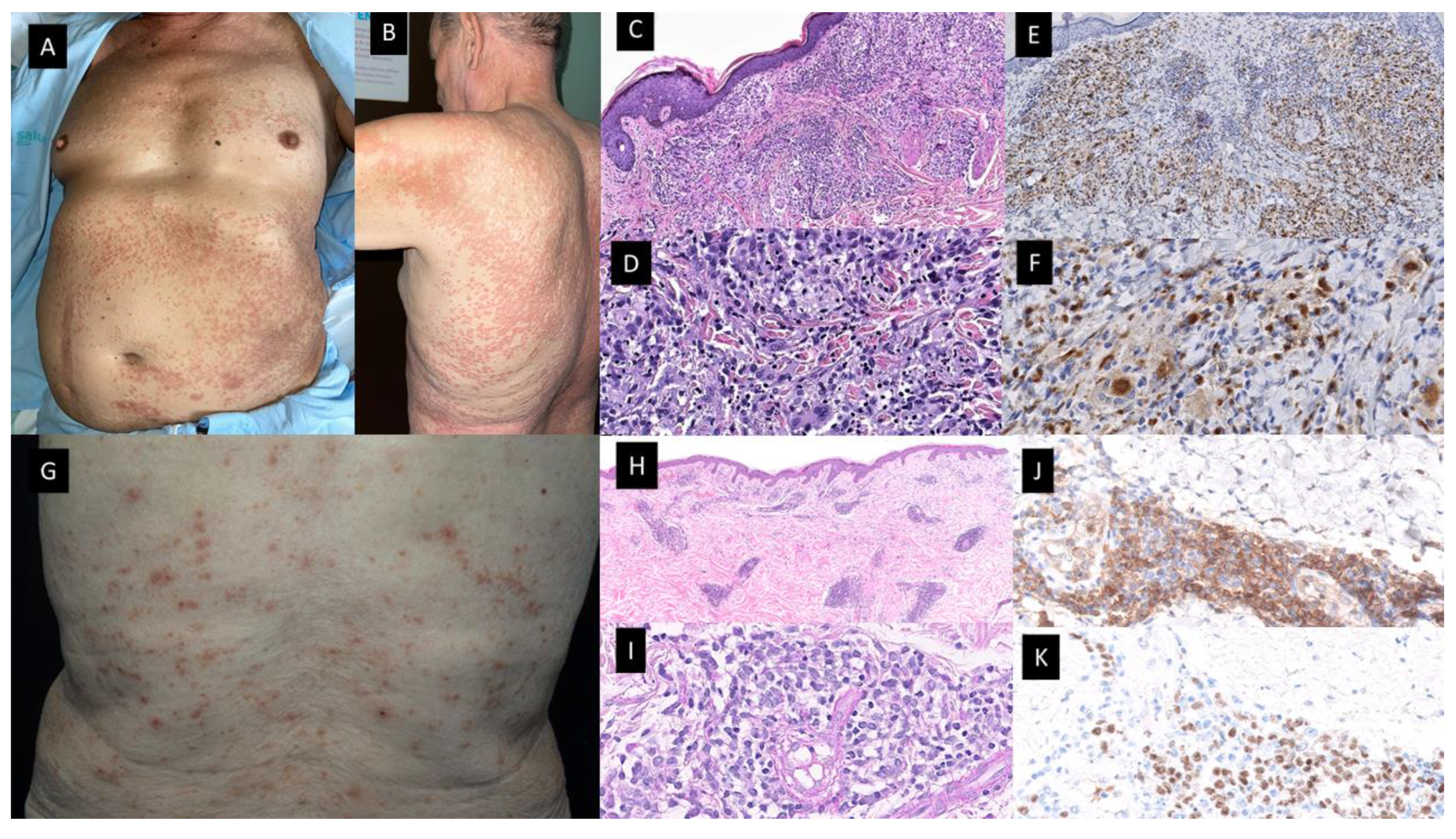
Figure 3. Granulomatous dermatitis (A–F) and mature plasmacytoid dendritic cell dermatosis (G–K). Patient with a diagnosis of CMML presented with multiple 1–2 mm erythematous-brownish papules that coalesce on (A). His anterior trunk and abdomen as well as on (B) Back and upper arm and extremities (not shown). (C,D) Hematoxylin-eosin-stained specimen. (C) Histopathological features consist of a dermal inflammatory infiltrate composed of epithelioid histiocytes, giant cells and lymphocytes, with rare neutrophils, configuring ill-defined granulomas (20×) (D) On higher magnification, the detail of the granulomas with multinucleated giant cells (40×). (E,F) On immunohistochemical study, CD68 stained the histiocytes [(E) (20×), (F) (40×)]. (G) Multiple erythematous and purpuric papules on the back. (H,I) Hematoxylin-eosin-stained specimen. (H) Dermal nodular infiltrate with perivascular and periadnexal arrangement (20×) (I). Detail of lymphocytic infiltration as well as interspersed larger, paler cells corresponding to mature PDC (40×). (J) Cytoplasmic CD123 immunostaining highlighting clusters of plasmacytoid dendritic cells (PDC) (40×) (K) Nuclear SPIB stain in PDC (40×).
Before the description of the Federmann cases in 2017, this type of granulomatous reaction had been published in association with MDS, and the authors argued that it could be a manifestation of the disease, because the cutaneous lesions did not respond to conventional treatments for reactive dermatoses, but they improved with the specific chemotherapy treatment for the myeloid neoplasia [29].
3. Mature Plasmacytoid Dendritic Cell Dermatoses
Plasmacytoid dendritic cells (PDC) represent a subset of cells within the immune system with distinctive morphology, immunophenotype and which develop specific functions [34]. Within the bone marrow, the total representation of mature plasmacytoid dendritic cells is less than 1% of the total nucleated cells. There are some inflammatory diseases in which an increased number of PDC has been described. They include Castleman disease, lupus erythematosus and Kikuchi-Fujimoto lymphadenopathy [35]. In addition, it is already known that the number of PDC increases in both CMML and MDS BM biopsies [34][36][37]. The presence of CD123-positive monocyte nodules had been reported only in CMML, whereas they were not in BM samples from CML and atypical CML. Besides, their amount has been related to prognosis in previous publications [38]. To date, two extremely rare PDC neoplasms have been diagnosed in patients with other hematological diseases: on the one hand a very aggressive one, blastic plasmacytoid dendritic cells neoplasms (BPDCN) and on the other hand a more indolent one consisting of mature PDC proliferation.
In 2012, Vitte et al. published an article describing a series of 42 patients with CMML and skin infiltrates [39]. They classified them in four different groups depending on the nature of the skin cells: (1) skin tumors composed of myelomonocytic cells; (2) skin tumors of mature PDC; (3) cutaneous blastic PDC tumors (BPDCT); and finally (4) skin tumors formed by blastic indeterminate dendritic cells. Their study included 16 patients with tumors of mature PDC. These patients were 15 males and only 1 female, whose median age at presentation was 76,35 years. Clinical features consisted of erythematous and itching maculo-papules. Generally, cutaneous lesions appeared concomitantly with the diagnosis of CMML or after a period of time. There are some other myeloid neoplasms that have been related to both mature PDC proliferations and BPDCN in previous literature which are MDS and acute leukemia with monocytic differentiation [33][35][36][37].
In 2022, Machan et al. published the findings regarding six patients with pruritic papular eruptions histopathologically characterized by dermal infiltrates of mature T-lymphocytes with large clusters of CD123+ and SPIB+ cells and with a lack of myeloid cell nuclear differentiation antigen (MNDA). (See Figure 3G–K) These cells were also negative for CD4, myeloperoxidase, TDT, CD56, BCL2, and cytotoxic markers. Neither CD117 nor CD34 positive cells were present on the infiltrates. It should be noted that immunostaining with S-100 protein and CD1a highlighted an increased number of S-100 protein and CD1a positive cells, which were mainly localized in the epidermis and superficial dermis. However, they found only scattered double positive cells for both S-100 CD1a markers. Four patients have been diagnosed with CMML and two had MDS (AREB-I and MDS with 5q deletion). The skin lesions developed in all cases coincidentally with either progression or full-establishment of their hematological disease [40]. Clinically, the skin lesions were more heterogeneous than in the case of the granulomatous dermatoses previously described, and unlike the previous ones, most cutaneous lesions disappeared spontaneously or after oral or topical corticosteroid treatment. One patient presented with multiple erythematous macules and papules in the same stage of development, which were spread on the trunk and extremities with sporadic paler halos, and a clinical diagnosis of drug eruption versus vasculitis was established. Another patient presented with isolated erythematous nodules located on both the left flank and breast, which were clinically interpreted as suggestive of primary cutaneous lymphoma. Finally, small papules on the extremities mimicking insect bites were also described. Biopsy samples in these patients showed focal epidermotropism in three patients, with vacuolar degeneration of the dermo–epidermal interface and folliculotropism of the infiltrate, but without mucin deposition. If researchers compare histopathologically the Vitte et al. reported cases with the cases of Machan et al., clusters of mature PDC, lymphocytes, plasma cells and indeterminate dendritic cells were present in the dermis in a similar proportion in both series [38][39].
This histopathology with features mimicking lupus erythematosus was previously reported in a patient with CMML type 2 diagnosed simultaneously with the appearance of the cutaneous lesions [41]. In this research, PDC aggregates were positive for CD4, CD68, granzyme B, CD123, BDCA-2/CD303, TCL1 and the IFN-α inducible protein, myxovirus A (MxA). In addition, PDC were aberrantly positive for CD5 and CD7, two antigens usually not found on normal PDC. CD56 staining was negative. They did not find an abnormal number of Langerhans and dermal dendritic cells expressing CD1a, S100 protein or langerin/CD207. Besides, the patient shared the same chromosomal abnormality (chromosome 13 trisomy) detected by Fluorescence in situ hybridization (FISH) analysis in his cutaneous lesions and BM. The cutaneous lesions resolved with chemotherapy for the hematological neoplasm and the patient died one year later after progression to AML without the recurrence of the cutaneous rash [40].
From a molecular point of view, in the Machan et al. series, patient number 2 had mutations in TET2, SRSF2 and ASXL1 genes in both BM and skin. They performed NGS techniques and found an allele frequency of TET2 gene mutations of 90.3% and 15.4% in BM and skin, respectively; 45.8% and 7.3% for SRSF2 gene in BM and skin, respectively; and 39.9% and 5.9% of the ASXL1 gene in either BM or skin. The combination of TET2, SRSF2 and ASXL1 gene mutations is highly specific (98%) for the diagnosis of CMML and has proven to have prognostic implications [42]. Furthermore, the BM of their number 3 patient, who had a diagnosis of AREB-I, showed mutations of genes DNMT3A and IDH1 with an allele frequency of 38.5% and 32.9%, respectively, via NGS. They also performed pyrosequencing of the IDH1 gene on paraffin-embedded tissue from cutaneous lesions of this patient and discovered the same mutation [40]. It is worth mentioning that both TET2 and DNMT3A gene mutations have been related to clonal hematopoiesis of indeterminate potential (CHIP) in myeloid neoplasms [42], and also that IDH1 mutations have been related to a poor outcome in MDS [43].
References
- Weiss, E.H.; Ko, C.J.; Leung, T.H.; Micheletti, R.G.; Mostaghimi, A.; Ramachandran, S.M.; Rosenbach, M.; Nelson, C.A. Neutrophilic Dermatoses: A Clinical Update. Curr. Dermatol. Rep. 2022, 11, 89–102.
- Sweet, R.D. An Acute Febrile Neutrophilic Dermatosis. Br. J. Dermatol. 1964, 76, 349–356.
- Ferea, C.R.; Mihai, S.N.; Balan, G.; Badescu, M.C.; Tutunaru, D.; Tatu, A.L. Sweet Syndrome Associated with Myelodysplastic Syndrome-A Review of a Multidisciplinary Approach. Life 2023, 13, 809.
- Cohen, P.R. Sweet’s syndrome—A comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J. Rare Dis. 2007, 2, 34.
- Requena, L.; Kutzner, H.; Palmedo, G.; Pascual, M.; Fernández-Herrera, J.; Fraga, J.; García-Díez, A.; Yus, E.S. Histiocytoid Sweet syndrome: A dermal infiltration of immature neutrophilic granulocytes. Arch. Dermatol. 2005, 141, 834–842.
- Alegría-Landa, V.; Rodríguez-Pinilla, S.M.; Santos-Briz, A.; Rodríguez-Peralto, J.L.; Alegre, V.; Cerroni, L.; Kutzner, H.; Requena, L. Clinicopathologic, Immunohistochemical, and Molecular Features of Histiocytoid Sweet Syndrome. JAMA Dermatol. 2017, 153, 651–659.
- Vignon-Pennamen, M.D.; Juillard, C.; Rybojad, M.; Wallach, D.; Daniel, M.T.; Morel, P.; Verola, O.; Janin, A. Chronic recurrent lymphocytic Sweet syndrome as a predictive marker of myelodysplasia: A report of 9 cases. Arch. Dermatol. 2006, 142, 1170–1176.
- Ghoufi, L.; Ortonne, N.; Ingen-Housz-Oro, S.; Barhoumi, W.; Begon, E.; Haioun, C.; Pautas, C.; Beckerich, F.; Robin, C.; Wolkenstein, P.; et al. Histiocytoid Sweet Syndrome Is More Frequently Associated with Myelodysplastic Syndromes Than the Classical Neutrophilic Variant: A Comparative Series of 62 Patients. Medicine 2016, 95, e3033.
- Passet, M.; Lepelletier, C.; Vignon-Pennamen, M.D.; Chasset, F.; Hirsch, P.; Battistella, M.; Duriez, P.; Sicre de Fontbrune, F.; Boissel, N.; Legrand, O.; et al. Next-Generation Sequencing in Myeloid Neoplasm-Associated Sweet’s Syndrome Demonstrates Clonal Relation between Malignant Cells and Skin-Infiltrating Neutrophils. J. Invest. Dermatol. 2020, 140, 1873–1876.e1875.
- Delaleu, J.; Kim, R.; Zhao, L.P.; de Masson, A.; Vignon-Pennamen, M.D.; Cassius, C.; Ram-Wolff, C.; Bagot, M.; Clappier, E.; Adès, L.; et al. Clinical, pathological, and molecular features of myelodysplasia cutis. Blood 2022, 139, 1251–1253.
- Kamimura, A.; Yanagisawa, H.; Tsunemi, Y.; Kusano, T.; Arai, E.; Tsuchida, T.; Nakamura, K. Normolipemic xanthomatized Sweet’s syndrome: A variant of Sweet’s syndrome with myelodysplastic syndrome. J. Dermatol. 2021, 48, 695–698.
- Sutra-Loubet, C.; Carlotti, A.; Guillemette, J.; Wallach, D. Neutrophilic panniculitis. J. Am. Acad. Dermatol. 2004, 50, 280–285.
- Jagdeo, J.; Campbell, R.; Long, T.; Muglia, J.; Telang, G.; Robinson-Bostom, L. Sweet’s syndrome—Like neutrophilic lobular panniculitis associated with all-trans-retinoic acid chemotherapy in a patient with acute promyelocytic leukemia. J. Am. Acad. Dermatol. 2007, 56, 690–693.
- Chan, M.P.; Duncan, L.M.; Nazarian, R.M. Subcutaneous Sweet syndrome in the setting of myeloid disorders: A case series and review of the literature. J. Am. Acad. Dermatol. 2013, 68, 1006–1015.
- Srisuttiyakorn, C.; Reeve, J.; Reddy, S.; Imaeda, S.; Lazova, R. Subcutaneous histiocytoid Sweet’s syndrome in a patient with myelodysplastic syndrome and acute myeloblastic leukemia. J. Cutan. Pathol. 2014, 41, 475–479.
- Lee, J.; Cornejo, K.M.; Rork, J.; Rothman, K.; Deng, A. Subcutaneous Histiocytoid Sweet Syndrome in a Patient With Relapsed Acute Myeloblastic Leukemia. Am. J. Dermatopathol. 2018, 40, 459–462.
- Lin, J.; Zhang, Q.; Chen, M. Subcutaneous histiocytoid Sweet’s syndrome in a patient associated with myelodysplastic syndrome-refractory anemia. J. Dermatol. 2012, 39, 99–101.
- Beck, D.B.; Ferrada, M.A.; Sikora, K.A.; Ombrello, A.K.; Collins, J.C.; Pei, W.; Balanda, N.; Ross, D.L.; Ospina Cardona, D.; Wu, Z.; et al. Somatic Mutations in UBA1 and Severe Adult-Onset Autoinflammatory Disease. N. Engl. J. Med. 2020, 383, 2628–2638.
- Beck, D.B.; Bodian, D.L.; Shah, V.; Mirshahi, U.L.; Kim, J.; Ding, Y.; Magaziner, S.J.; Strande, N.T.; Cantor, A.; Haley, J.S.; et al. Estimated Prevalence and Clinical Manifestations of UBA1 Variants Associated with VEXAS Syndrome in a Clinical Population. JAMA 2023, 329, 318–324.
- Cooper, J.N.; Young, N.S. Clonality in context: Hematopoietic clones in their marrow environment. Blood 2017, 130, 2363–2372.
- Haines, P.; Pullarkat, S.; Said, J. VEXAS syndrome: Vacuoles in myeloid, erythroid, and lymphoid lineages. Int. J. Lab. Hematol. 2023.
- Djerbi, N.; Zimmermann, K.; Balabanov, S.; Manz, M.G.; Becker, M.O.; Roncador, M. Intra-patient competition of VEXAS syndrome and CML clones. Blood Adv. 2023, 7, 6815–6818.
- Nguyen, J.K.; Routledge, D.; van Der Weyden, C.; Blombery, P.; Angel, C.M.; Johnson, D.; Goh, M.S.; Lee, A. VEXAS syndrome: A dermatological perspective. Australas. J. Dermatol. 2022, 63, 488–492.
- Osio, A.; Battistella, M.; Feugeas, J.P.; Cuccuini, W.; Noguera, M.E.; Petrella, T.; Raffoux, E.; Janin, A.; Pennamen, V. Myelodysplasia Cutis Versus Leukemia Cutis. J. Invest. Dermatol. 2015, 135, 2321–2324.
- Sujobert, P.; Cuccuini, W.; Vignon-Pennamen, D.; Martin-Garcia, N.; Albertini, A.F.; Uzunov, M.; Redjoul, R.; Dombret, H.; Raffoux, E. Evidence of differentiation in myeloid malignancies associated neutrophilic dermatosis: A fluorescent in situ hybridization study of 14 patients. J. Invest. Dermatol. 2013, 133, 1111–1114.
- Gottlieb, G.J.; Duve, R.S.; Ackerman, A.B. Interstitial granulomatous dermatitis with cutaneous cords and arthritis: Linear subcutaneous bands in rheumatoid arthritis revisited. Dermatopathol. Pract. Concept. 1995, 1, 3–6.
- Chu, P.; Connolly, M.K.; LeBoit, P.E. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch. Dermatol. 1994, 130, 1278–1283.
- Rosenbach, M.; English, J.C., 3rd. Reactive Granulomatous Dermatitis: A Review of Palisaded Neutrophilic and Granulomatous Dermatitis, Interstitial Granulomatous Dermatitis, Interstitial Granulomatous Drug Reaction, and a Proposed Reclassification. Dermatol. Clin. 2015, 33, 373–387.
- Yoneta, K.; Fujimoto, N.; Teramura, K.; Takayama, S.; Tanaka, T. Disseminated granulomatous skin lesions associated with myelodysplastic syndrome treated successfully with tranilast: A case report and review of the literature. Eur. J. Dermatol. 2016, 26, 398–400.
- Prieto-Torres, L.; Santonja, C.; Chamizo, C.; García-Gil, M.F.; Lezcano, V.; Arranz-Sánchez, D.M.; Rodríguez-Pinilla, S.M.; Azaceta, G.; García-García, M. Granulomatous Dermatitis Heralding Myelodisplastic/Myeloproliferative Neoplasms. Neoplastic or Reactive Cells? A Study of 2 Cases. Am. J. Dermatopathol. 2022, 44, 456–460.
- Federmann, B.; Bonzheim, I.; Yazdi, A.S.; Schmidt, J.; Fend, F.; Metzler, G. Generalized palisaded neutrophilic and granulomatous dermatitis-a cutaneous manifestation of chronic myelomonocytic leukemia? A clinical, histopathological, and molecular study of 3 cases. Hum. Pathol. 2017, 64, 198–206.
- Kyriakou, A.; Patsatsi, A.; Papadopoulos, V.; Kioumi, A.; Efstratiou, I.; Lazaridou, E. A case of palisaded neutrophilic granulomatous dermatitis with subsequent development of chronic myelomonocytic leukemia. Clin. Case Rep. 2019, 7, 695–698.
- Enescu, C.D.; Patel, A.; Friedman, B.J. Unique Recognizable Histopathologic Variant of Palisaded Neutrophilic and Granulomatous Dermatitis that Is Associated With SRSF2-Mutated Chronic Myelomonocytic Leukemia: Case Report and Review of the Literature. Am. J. Dermatopathol. 2022, 44, e33–e36.
- Tzankov, A.; Hebeda, K.; Kremer, M.; Leguit, R.; Orazi, A.; van der Walt, J.; Gianelli, U. Plasmacytoid dendritic cell proliferations and neoplasms involving the bone marrow: Summary of the workshop cases submitted to the 18th Meeting of the European Association for Haematopathology (EAHP) organized by the European Bone Marrow Working Group, Basel 2016. Ann. Hematol. 2017, 96, 765–777.
- Wang, P.; Feng, Y.; Deng, X.; Liu, S.; Qiang, X.; Gou, Y.; Li, J.; Yang, W.; Peng, X.; Zhang, X. Tumor-forming plasmacytoid dendritic cells in acute myelocytic leukemia: A report of three cases and literature review. Int. J. Clin. Exp. Pathol. 2017, 10, 7285–7291.
- Facchetti, F.; Cigognetti, M.; Fisogni, S.; Rossi, G.; Lonardi, S.; Vermi, W. Neoplasms derived from plasmacytoid dendritic cells. Mod. Pathol. 2016, 29, 98–111.
- Orazi, A.; Chiu, R.; O’Malley, D.P.; Czader, M.; Allen, S.L.; An, C.; Vance, G.H. Chronic myelomonocytic leukemia: The role of bone marrow biopsy immunohistology. Mod. Pathol. 2006, 19, 1536–1545.
- Lucas, N.; Duchmann, M.; Rameau, P.; Noël, F.; Michea, P.; Saada, V.; Kosmider, O.; Pierron, G.; Fernandez-Zapico, M.E.; Howard, M.T.; et al. Biology and prognostic impact of clonal plasmacytoid dendritic cells in chronic myelomonocytic leukemia. Leukemia 2019, 33, 2466–2480.
- Vitte, F.; Fabiani, B.; Bénet, C.; Dalac, S.; Balme, B.; Delattre, C.; Vergier, B.; Beylot-Barry, M.; Vignon-Pennamen, D.; Ortonne, N.; et al. Specific skin lesions in chronic myelomonocytic leukemia: A spectrum of myelomonocytic and dendritic cell proliferations: A study of 42 cases. Am. J. Surg. Pathol. 2012, 36, 1302–1316.
- Machan, S.; Alonso-Dominguez, J.M.; Sánchez García, F.J.; Nieves Salgado, R.; Soto, C.; Castro, Y.; Pajares, R.; Manso, R.; Santonja, C.; Serrano Del Castillo, C.; et al. Plasmacytoid Dendritic Cell Dermatosis Associated to Myeloproliferative/Myelodysplastic Neoplasms. Am. J. Surg. Pathol. 2022, 46, 1623–1632.
- Dargent, J.L.; Henne, S.; Pranger, D.; Balzarini, P.; Sartenaer, D.; Bulliard, G.; Rack, K.; Facchetti, F. Tumor-forming plasmacytoid dendritic cells associated with myeloid neoplasms. Report of a peculiar case with histopathologic features masquerading as lupus erythematosus. J. Cutan. Pathol. 2016, 43, 280–286.
- Jian, J.; Qiao, Y.; Li, Y.; Guo, Y.; Ma, H.; Liu, B. Mutations in chronic myelomonocytic leukemia and their prognostic relevance. Clin. Transl. Oncol. 2021, 23, 1731–1742.
- Wang, N.; Wang, F.; Shan, N.; Sui, X.; Xu, H. IDH1 Mutation Is an Independent Inferior Prognostic Indicator for Patients with Myelodysplastic Syndromes. Acta Haematol. 2017, 138, 143–151.
More
Information
Subjects:
Dermatology; Hematology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
707
Revisions:
2 times
(View History)
Update Date:
04 Jan 2024
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No