 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Alessandro Pingitore | -- | 6832 | 2024-01-02 16:28:59 | | | |
| 2 | Jessie Wu | -195 word(s) | 6637 | 2024-01-03 02:40:43 | | | | |
| 3 | Jessie Wu | -3 word(s) | 6634 | 2024-01-03 02:44:56 | | | | |
| 4 | Jessie Wu | + 2 word(s) | 6636 | 2024-01-03 02:53:39 | | |
Video Upload Options
The thyroid–heart relationship has a long and articulated history of its own, a history that encompasses physiological and pathophysiological knowledge. Molecular biology studies, in an experimental context, have highlighted the extraordinary dialogue that exists among the two systems in the field of cardioprotection, which is an extremely important area for the treatment of cardiac diseases in both acute and chronic phases. In addition, in the last few years, several studies have been carried out on the prognostic impact of alterations in thyroid function, including subclinical ones, in heart disease, in particular in heart failure and acute myocardial infarction, with evidence of a negative prognostic impact of these and, therefore, with the suggestion to treat these alterations in order to prevent cardiac events, such as death. A comprehensive summary of the heart–thyroid relationship is provided.
1. History of the Relationship between Heart and Thyroid
The thyroid–heart relationship has a very long and articulated history, encompassing physiological and pathophysiological knowledge, especially in cases of heart disease, such as heart failure (HF), arrhythmias, and ischemic heart disease. The hypothesis of a relationship between heart and thyroid was, in fact, born well over two hundred years ago, in 1813, when Caleb Hillier Parry showed a detailed description of eight cases of thyroid gland enlargement combined with the presence of heart palpitation [1].
In 1828, the term ‘Kropfherz’ or ‘cardiac goiter’ was coined to describe the association between goiter and palpitations [2], and, in 1898, Friedrich Kraus introduced the concept of ‘toxic cardiac goiter’ [3]. Then, at the beginning of the 20th century, surgical hypothyroidism was proposed as an extracardiac approach for the treatment of ischemic heart disease [4]. This approach, however, was abandoned due to poor results.
In 1918, Symmers noted that the sympathology present in thyrotoxic heart disease was virtually identical to that of clinical cases described as ‘idiopathic dilatation and hypertrophy of the heart’ [5]. In the same year, White and Aub studied the effect of thyroid disease on the heart [6], showing hypertension, tachycardia, T-wave variations, and paroxysms of atrial (auricular) fibrillation. In 1929, Wishart reported atrial fibrillation as the most frequently encountered rhythm disturbance in thyrotoxicosis [7][8].
In 1922, Hamilton classified thyrotoxicosis of the heart into two classes: the first without cardiac damage with tachycardia as the main symptom, the second with cardiac damage and no evidence of rheumatic or other heart disease [9]. In 1924, Hamilton first described cases of adenomatous or exophthalmic goiter complicated by congestive heart failure that were reversible after thyroid removal [10]. Furthermore, Raab and Sharpey-Schafer successfully treated patients with angina pectoris and severe congestive HF using thiouracil, which reduced the heart’s sensitivity to epinephrine [11][12]. Canary et al., in 1957, instead used antiadrenergic agents in the treatment of hyperthyroidism [13]. In the 1970s, beta-adrenergic blocking drugs, represented by D,L-propranolol, were finally incorporated into treatment, improving the adrenergic signs and symptoms associated with hyperthyroidism [14]. Also in the 1970s, an interventional study called the Coronary Drug Project (CDP) demonstrated negative outcomes on the pro-arrhythmic effects of D-T4 (the inactive form of thyroxine, while L-T4 represents the active form) [15]. Indeed, the clinical outcomes of administering 6 mg/day of D-T4 were unclear (more than doubling endogenous T4 production) [16]. Only later was it discovered that the dose of D-T4 was actually contaminated with a high level of active L-T4, so the total dose administered was much higher than that expected to correct manifest hypothyroidism [17]. It is precisely for this reason, therefore, that CDP was unable to provide relevant information on TH supplementation in the treatment of coronary patients. In the early experimental studies, Tata et al. showed a TH effect on 44 protein synthesis at the transcription level [18][19], and, in 1972, it was discovered through the rat study that the basic unit of TH action is the triiodothyronine nuclear receptor complex [20][21]. In the late 1980s, it was finally demonstrated that hyperthyroidism was able to induce a 61% decrease in phospholamban mRNA level and that cardiac genes regulated by TH include sodium-potassium ATPase ion channels, voltage-gated potassium channels, sodium-calcium exchanger, and β1-adrenergic receptor genes [22][23][24].
2. Thyroid and Heart: A Tight Physiological Relation
THs strongly influence the growth, development, and metabolism of cells, organs, and systems. Among these, THs play a pivotal role in cardiovascular (CV) homeostasis maintenance, both directly influencing all components of the system and indirectly via regulation of the autonomous nervous system, renin–angiotensin–aldosterone system, and systemic vasculature, with involvement in cardiac contractility, electrophysiological functions, and cardiac morphology and structure [25].
These effects are mainly mediated by different signaling pathways clustered into genomic and non-genomic actions. THs influence cardiac function with genomic pathways through binding to nuclear receptors that contribute to the regulation of target gene expression. In more detail, these actions, inducing the enhancement of contractile function and diastolic relaxation, are essentially due to gene expression through specific nuclear a and bTH receptors (TRs) [26]. T3 regulates different myocyte genes related to cardiac contractile function, such as sarco-plasmic reticulum (SR)Ca2þ ATPase (SERCA2), phospholamban (PLB), and the myosin heavy chains (MHC), α and β, that encode the protein isoforms of the thick filament. In this regard, many plasma-membrane ion transporters are involved in synchronizing electrochemical functions in the myocardium, and different voltage-gated potassium channels are regulated by T3 [27]. In the cardiac myocyte and on the systemic vasculature, THs also have non-genomic effects, not involving TH response elements (TRE)-mediated transcriptional events via interactions with cytoplasmic and membrane-associated TRs. These properties are related to changes in various membrane ion channels for sodium, potassium, and calcium, both in the heart and vascular smooth muscle cells (VSM). THs exert their effects directly on myocardial structure, modifying the interstitial collagen content, promoting the development of angiogenesis, and consequently, regulate cardiac function through chronotropic, inotropic, and dromotropic effects. Also, THs, via direct action on mitochondria, influence inflammation, neuroendocrine systems, and oxidative stress pathways [28]. Usually, the non-genomic and genomic actions of T3 work in concert to regulate cardiac function and CV hemodynamics. As depicted in Figure 1, TH actions on the heart and peripheral vasculature are realized in a decreased systemic vascular resistance (SVR) and an increased heart rate at rest, left ventricular contractility, and blood volume. THs are also involved in the control of cardiac pacemaker activity influencing the action potential duration and repolarization currents in myocytes and regulating the transcription of the pacemaker-related genes [29][30]. Furthermore, although not all studies show a correlation, evidence obtained from studies on community-based populations indicate that thyroid function is involved in the control of blood pressure [31]. THs enhance basal metabolic rate in most organs and tissues with subsequent raised metabolic demands that lead to modifications in cardiac output, SVR, and blood pressure [31].
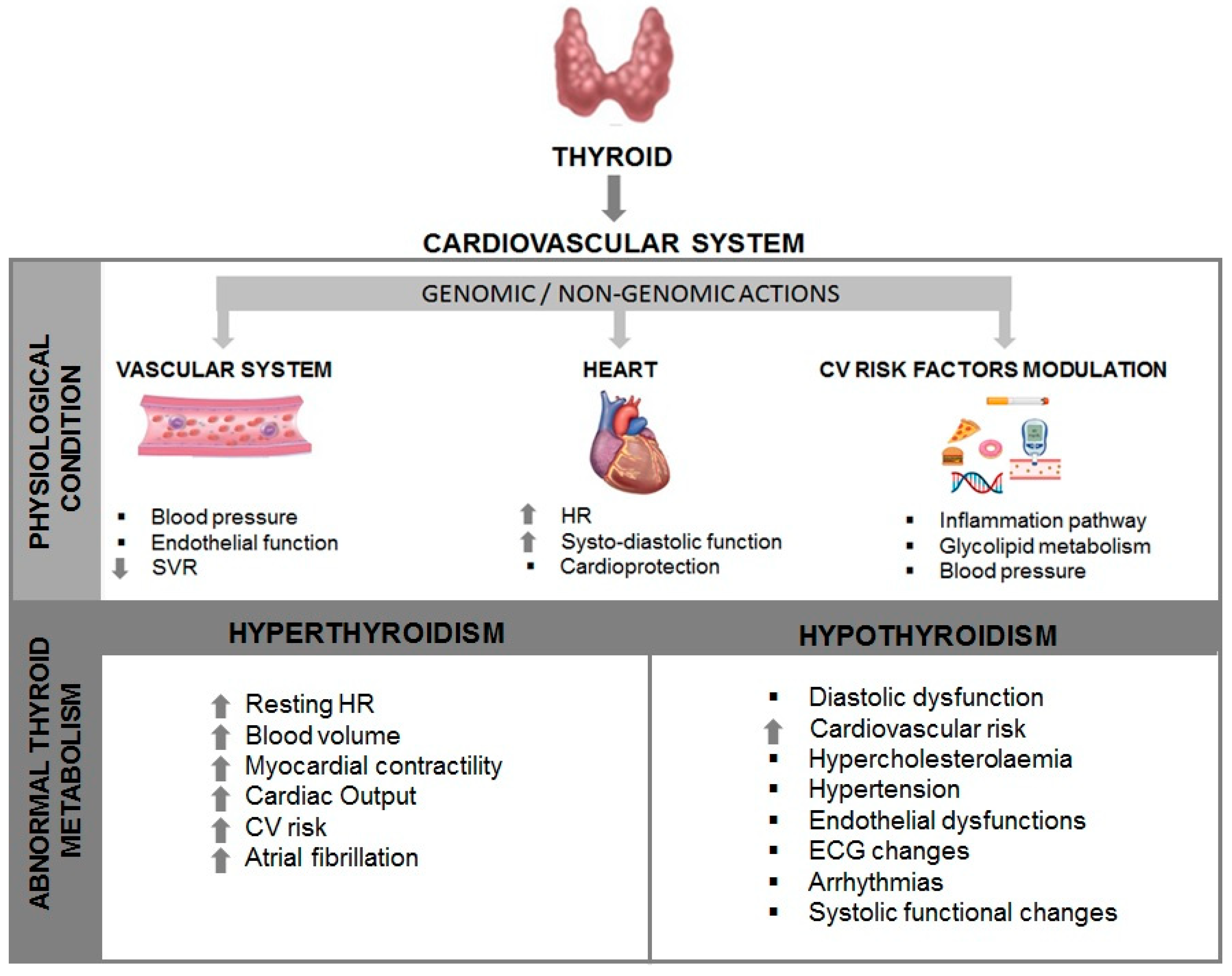
Figure 1. Interaction of the Thyroid and the Heart. SVR: systemic vascular resistance; HR: heart rate; CV: cardiovascular; ECG: electrocardiogram.
3. Hyperthyroidism, Hypothyroidism, and Subclinical Conditions
It is now recognized that clinical characterization of abnormal thyroid metabolism is linked to the TH effects on the heart and CV system [32] (see Figure 1). Both pathological conditions, hyperthyroidism and hypothyroidism, cause alterations in cardiac patterns, such as contractility, myocardial oxygen consumption, cardiac output, blood pressure, and SVR [33].
Thyroid diseases are quite common, with a prevalence in the female population between 9% and 15%, while in the male population, the incidence is lower, probably due to autoimmune mechanisms for the most widespread forms, including both Graves’ and Hashimoto’s disease [34].
Hyperthyroidism is a condition affected by stimulation of the TSH receptors by autoantibodies (Graves’ disease) or as a consequence of the autonomous production of THs by thyroid nodules [35]. In patients with hyperthyroidism, many of the clinical consequences are related to the heart and CV system, including palpitations, exercise intolerance, dyspnea on exertion, widened pulse pressure, and occasionally atrial fibrillation (see Figure 1). In addition, peripheral vascular resistance is reduced, while cardiac contractility and heart rate at rest are improved, resulting in an increase of 50% to 300% of the normal cardiac output [33]. The reduction of SVR determines low renal perfusion, which, in turn, sets the renin–angiotensin–aldosterone system (RAAS) into action, leading to a growth of total blood volume. Cardiac arrhythmias or electrocardiogram alterations, sinus tachycardia, atrial fibrillation, and reduced PR and QT intervals are documented in patients with hyperthyroidism and, in rare instances, also atrio-ventricular blockage [36][37]. Hyperthyroidism has been shown to result in hemodynamic changes with a lowered ejection fraction (EF) and cardiac output due to a decrease in myocardial contractile reserve [38]. Diametrically opposed CV signs and symptoms, such as bradycardia, diastolic hypertension, cold intolerance, and fatigue, are typical for hypothyroidism. Hypothyroidism occurs in 3% of the adult female population and is linked to an increase in SVR, a reduction in cardiac contractility and cardiac output, and an enhanced atherosclerosis [39]. In contrast to hyperthyroidism, in which atrial arrhythmias are well documented, hypothyroidism is characterized by a prolonged QT interval that, in turn, affects ventricular irritability [40].
Subclinical hyperthyroidism is a condition characterized by low serum TSH levels and normal levels of serum T3 and T4. Usually, its prevalence increases in advancing age with concomitant risk for CV mortality and atrial fibrillation [41][42]. Clinical evidence has documented a relationship between subclinical hyperthyroidism and incident CV death, HF, and atrial fibrillation [43][44][45]. Subclinical hypothyroidism is instead the condition associated with serum TSH levels higher than normal range but normal TH levels. Although subclinical disease is frequently “asymptomatic,” cardiac characterization of patients with subclinical hypothyroidism displays alterations, such as impaired ventricular filling, systolic functional changes, and an increased C-Reactive protein, as risk factors [46][47][48]. Atherosclerosis, coronary heart disease, and myocardial infarction risk in the female population (aged 60 and older) were also documented [49].
4. Thyroid and Cardioprotection
Cardioprotection has been defined as including any intervention (and as such, both physiological adaptive and compensatory mechanisms as well as therapeutic strategies) that contribute to heart preservation by reducing or even preventing myocardial damage [50]. TH exerts recognized cardioprotective effects through modulation of different key cellular pathways, including preservation of mitochondrial activity and morphology, antifibrotic and proangiogenic actions, and promotion of cell regeneration and growth after the postischemic injury [51]. Specifically, the large amount of available experimental data suggest multiple TH effects such as regulation of prosurvival pathways (e.g., activation of PI3K)/Akt and PKC, and inhibition of p38MAPK signaling), remodeling of the myocardial interstitium (e.g., TGF-β1 signaling), proangiogenic effects on coronary circulation (e.g., mediated by integrin αVβ3 and HIF-1a expression), anti-inflammatory and anti-oxidative properties (e.g., cytokine reduction, direct/indirect antioxidant action as iodide, can act scavenger of free radicals, or enhancement or inhibition of the activity of antioxidant enzymes and free radical scavengers), effects on the neuroendocrine system (e.g., deactivation of the neuroendocrine system, characterized by a reduction in plasma levels of noradrenaline, NTproBNP, and aldosterone), epigenetic effects and chromatin changes (e.g., regulation of α and β-MHC genes), post-transcriptional regulation of miRNA (e.g., miR-214, miR-208) [28].
In particular, 3,5,3′-triiodothyronine (T3) and its precursor thyroxine (T4), are considered key regulators of mitochondrial function [52]. Accordingly, reduced levels of these hormones induce imbalance of cardiac mitochondrial activity and subsequently, lead to adverse consequences at the cardiac muscle level within the myocardium [53]. Thus, the low T3 syndrome (LT3S), a status characterized by decreased total serum T3 and free T3 (fT3) with normal levels of thyroxine (T4) and thyrotropin (TSH), observed in acute post-ischemic and chronic heart pathologies and generally considered an adaptive response, can actually retain negative effects, favoring the progressive worsening of cardiac function and myocardial remodeling and representing a strong predictor of mortality and adverse CV events in these patients [54].
In view of the beneficial effects of TH on CV function, several attempts have been made utilizing TH replacement therapy in short-term and long-term administration, T3 or T4 at different doses and modalities of administration [54]. However, there are great differences in the population cohorts in which these treatments are given, including HF patients with a normal or abnormal TH profile, stable or unstable HF, or acute myocardial infarction (AMI) patients [54]. Moreover, attention must be paid to avoid important adverse effects due to possible pharmacological-induced hyperthyroidism. Consequently, a shared consensus on the use of TH replacement therapy has not been achieved yet by the scientific community. Nonetheless, the American Association of Clinical Endocrinologists, in conjunction with the American Thyroid Association, guidelines propose TH treatment in patients with HF and TSH levels exceeding the high normality reference limit up to 10 μIU/mL for restoration and maintenance of a normal TH status, monitoring status in order to prevent over-treatment [55].
Recently, a number of additive underlying mechanisms related to the cardioprotective effects of TH have been further emerging, detailing more and more the possible key cellular pathways involved.
One recent advancement concerns the opening of the ATP-sensitive mitochondrial potassium channel (mitoK-ATP), with an important antioxidant and protective role against post-ischemia/reperfusion mitochondrial dysfunction and cell loss [56]. In particular, data obtained in experimental (rats) and in vitro (rat cardiomyocytes) models suggested that T3 administration (given to restore the physiological concentration) was able to modulate the expression level of both the channel subunits (mitoK and mitoSur), downregulated under the stress conditions [57]. These data identify the post-ischemic LT3S as a permissive condition for the inhibition of mitoK and mitoSur. Translated to a clinical setting, it may suggest that restoring T3 plasma and myocardial levels to euthyroidism could represent a beneficial tool to limit the evolution of post-ischemic left ventricular dysfunction, because it is carried out in a window where the activation of dangerous mitochondrial regulatory events can be still prevented or mitigated, at least in part, by derepression of the mitoK-ATP channel.
Another additive key underlying mechanism involves the nuclear factor erythroid 2-related factor (Nrf2) pathway, one of the main endogenous antioxidant responses, effector of the cardioprotective-related TH effects [58]. Nrf2 is a crucial factor in TH-related cardioprotection, which affects different pathways related to oxidative stress, inflammation, cell growth, and energy supply [58]. Interestingly, different natural antioxidants (e.g., vitamin E, curcumin, and quercetin) may modulate Nrf2 signaling, improving altered TH level conditions [58]. This fact opens the possibility of targeting the Nrf2 pathway for thyroid disease prevention or improvement. However, there are difficulties related to the involvement of this factor in a number of diseases and the possibility of eliciting unwanted collateral adverse effects, equally harmful to other pathophysiological conditions.
An exciting development in this field is instead the issue regarding the role of vitamin D and the possibility to use 25-Hydroxyvitamin D (25(OH)D) administration to modulate cardioprotection, also through effects linked to TH modulation. Vitamin D has a number of beneficial effects on cardiometabolic disease (e.g., through modulation of endothelial and smooth muscle cell activity, renin–angiotensin–aldosterone system, nitric oxide, oxidative stress, and inflammatory response) [59][60]. Moreover, the administration of vitamin D3 in diabetic rats increases deiodinase (DIO) 2 expression and, consequently, leads to an increase in fT3 and a decrease in fT4 levels [61]. In vitro studies evidenced that 1,25(OH)2 administration may increase D2 activity, suggesting a cross-talk between TH and vitamin D [62][63]. Other in vitro data suggested a role of vitamin D through central and peripheral activities in the modulation of TSH and THs, as 1,25-(OH)2D3 inhibits TSH-stimulated adenylyl cyclase activity and iodide uptake in rat thyroid cells, whereas it increases TRH-induced TSH release in the normal pituitary cells [64][65]. Moreover, vitamin D administration may increase the serum levels of fT3 and fT4 in offspring of female rats administered 100 times the normal dose of iodide (100 HI; 750 μg/d) during pregnancy and lactation, as well as the protein expression levels of TRα1 and TRβ1 in their thyroid cells. In addition, 1,25(OH)2D3 supplementation reverses the imbalance in proinflammatory and anti-inflammatory cytokines (IL-17A, IFN-γ, and IL-10) [66]. At clinical levels, there is evidence of a relationship between vitamin D [25(OH)D] deficiency and autoimmune thyroid diseases or cancer, as well as between 25(OH)D levels and titers of antibodies and thyroid autoimmunity replacement, although some studies reported a lack of association [67][68]. Many factors may contribute to this high variability between the available results, including the use of different methods to assess circulating 25(OH)D levels, cut-off used to define normality, and the confounding effects of sex, age, obesity, and seasonal blood withdrawal. Researchers previously observed a relationship between LT3S and reduced 25(OH)D levels in AMI [69]. These findings suggest that 25(OH)D might serve as an additional tool to counteract LT3S in acute settings, and, as the association was more evident in those with severe 25(OH)D hypovitaminosis, this particular subgroup of patients may benefit more from vitamin D administration [64].
In the majority of studies evaluating the relationship between vitamin D supplementation and anti-thyroid antibodies (generally in patients with autoimmune thyroid diseases), a decrease of TPOAb and TgAb levels has been observed. Available findings about TSH and TH were less clear, also reporting null association [68]. Nonetheless, one study enrolled a general population of more than 11.000 subjects receiving vitamin D supplementation (in order to achieve blood 25(OH)D levels over 100 nmol/L at 12-month follow-up) in a health and wellness program; hypothyroidism was found in 2% (23% including subclinical hypothyroidism) of subjects at baseline decreasing to 0.4% (or 6% with subclinical) at follow-up. Moreover, 25(OH)D levels ≥125 nmol/L were associated with a 30% reduced risk of hypothyroidism and a 32% reduced risk of elevated anti-thyroid antibodies [70].
Actually, the strategy of T3 replacement, conducted in order to replace fT3 blood levels without eliciting major systemic adverse effects, represents the best tool to be utilized in clinical practice. New insights may identify additional determinants to better understand key mechanisms related to the replacement of T3 (e.g., the mitoK-ATP channel). At the same time, other possibilities, such as those emerging in the oxidative stress field (e.g., modulation of the Nrf2 pathway), may represent alternative/additive therapeutic targets, although this knowledge is still in its infancy. Instead, for what concerns vitamin D, if it is true that clinical data are controversial and far from definitive, it would be wrong to consider irrelevant a hypothesis highly plausible in view of many experimental findings supporting a relationship between vitamin D and TH. Ultimately, vitamin D supplementation is safe as very rarely toxic, even at high doses, unlike thyroid hormones, which can cause hyperthyroidism with all the consequences that may arise from this condition; thus its use alone, but especially in combination with TH treatment in the CV clinical settings, may be of interest and merits further investigation, representing an interesting issue for future research (Figure 2).
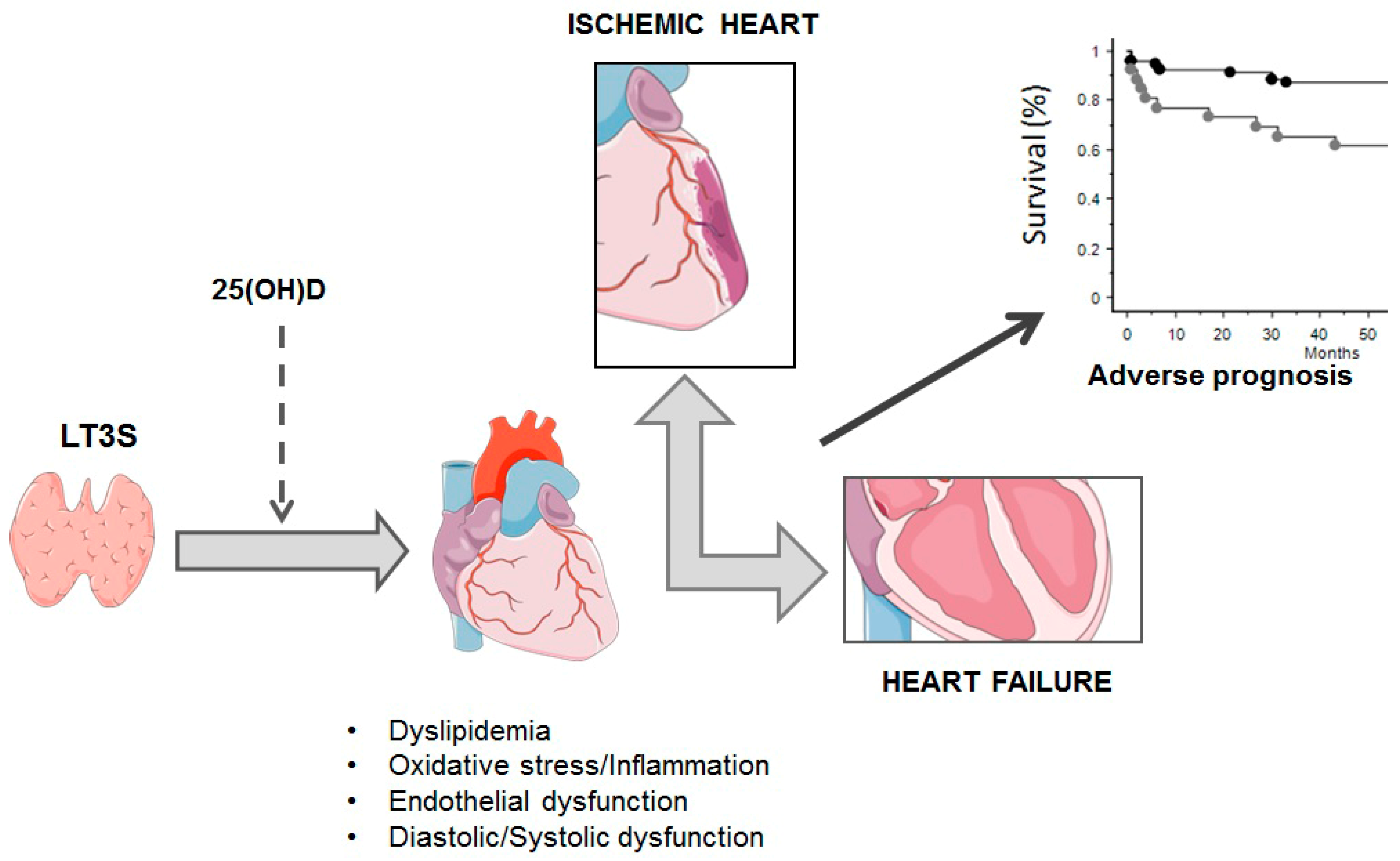
Figure 2. Main possible vitamin D-related mechanisms in thyroid functioning. LT3S: low T3 syndrome; 25(OH)D: 25-HydroxyvitaminD.
5. The Prognostic Impact of Thyroid and Heart Abnormalities in Heart Failure and Acute Myocardial Infarction
There is a large amount of data showing that subclinical TH abnormalities have a negative prognostic impact in patients with heart disease. In this context, HF and AMI were the main cardiac diseases in which the prognostic impact of TH mild abnormalities was assessed. Iervasi et al. showed that LT3S is a strong predictor of cardiac death [71]. In this research, fT3 was the main independent predictor of overall death. Interestingly, fT3 increased prognostic prediction of cumulative death after considering all of the other conventional variables (age/sex, cardiac risk factors, historical and clinical data) [71]. LT3S is counted among the subclinical TH disorders, although it is not the expression of a primary thyroid disorder but rather the result of an acute or chronic disease. Instead, it is considered an adaptive TH metabolic phenomenon to minimize energy expenditure. However, experimental data, as shown above, and clinical data questioned this theory because of the negative structural, histological, cellular, and functional effects experimentally documented in line with prognostic data. Therefore, the new hypothesis is that LT3S is initially adaptive but, if persistent, can shift into a maladaptive mechanism, favoring HF progression. This hypothesis comes up from the pathophysiological evidence that the continuous activation of other systems, such as the inflammatory and the sympathetic ones, causes the shift from adaptive to maladaptive and, thus, toxic effects in chronic diseases, such as HF. In addition, this hypothesis finds its reason in the pathophysiology of allostatic overload determined by the continuous activation of the stress-dedicated systems to physiological response, whose main efficiency is the up and down activation. In this context, a reduction in peripheral production of biologically active T3, probably linked to increased proinflammatory markers (interleukin 6), has been documented in asymptomatic or mildly symptomatic patients with non-ischemic dilated cardiomyopathy [72].
Iervasi et al. also showed that the primary subclinical TH abnormalities have negative prognostic implications in cardiac patients. Specifically, after adjustment for several risk factors, hazard ratios for cardiac death were significantly higher in SCH, (HR, 2.40), SCT (HR, 2.32), and LT3 (1.63) in comparison to euthyroidism [43]. The same impact has also been shown in a setting of patients with acute cardiac disease, including mainly patients with acute HF and acute coronary syndrome [73][74]. In particular, overt hypothyroidism and low fT3 levels were independent in-hospital predictors of all-cause mortality. Actually, a major part of the studies on the effects of TH abnormalities in patients with cardiac diseases are focused on HF and AMI. The studies on TH prognostic impact in HF patients are reassumed in Table 1 [75][76][77][78][79][80][81][82][83][84][85][86][87][88][89][90][91][92][93][94][95][96][97][98]. In general, in HF patients, all three types of subclinical TH abnormalities are predictors of cardiac events, including both hard and soft ones. When researchers clustered patients according to left ventricular ejection fraction (LVEF), low T3 circulating levels identified patients with a higher probability of overall death in the presence of an LVEF value lower than 20%. In fact, the survival of patients with an LVEF ≤ 20% and LT3S (total T3 level ≤ 1.2 pmol/L) was significantly lower than that in patients with similar LVEF and without LT3S (61 vs. 83%). Similarly, the probability of survival of patients with an LVEF >20% and without LT3s was significantly greater than that in patients with the same LVEF value and with LT3S (90 vs. 73%) [97] Also, LT3S showed additive prognostic stratification power when associated with BNP value for overall and cardiac death: Patients with LT3S and high BNP (>cut-off value 165 ng/L for cardiac death) had a survival probability of 46% in comparison to those normal T3 values and lower BNP (90%) [94]. A relationship between TH and inflammatory markers has also been documented in HF patients, with IL-6, TNFα, and CRP, correlating inversely with fT3 [99]. Interestingly, Shen et al. showed that FT3 and inflammatory patterns (neutrophil-to-lymphocyte ratio) were independently associated with all-cause mortality or HF rehospitalization [100]. Cittadini et al. showed that the occurrence of multiple hormonal and metabolic deficiency syndrome, encompassing several anabolic systems, the somatotropic axis (growth hormone and its tissue effector insulin-like growth factor-1), anabolic steroids (testosterone and DHEA-S), and THs, was associated with increased overall mortality and CV hospitalization in HF [101]. More recently, the prognostic impact of altered thyroid metabolism has also been documented in patients with HF and preserved LVEF (HFpEF). In particular, a low FT3/FT4 ratio was associated with clinical and cardiac instrumental changes and predicted a higher risk of diuretic intensification, urgent HF visits, HF hospitalization, or cardiac death in HFpEF.
Table 1. Prognostic impact of altered thyroid metabolism in HF patients.
|
TH Dysfunction |
Events (n) |
N° PTS (W,%) |
Age (yy) |
LVEF (%) |
NYHA Class III-IV |
Prognostic Weight |
Ref. |
|
fT3/fT4 ratio < 2.15 |
Cardiac death, Transplantation, LV device implantation |
3257 (18) |
57 |
ND |
ND |
HR values of FT3/FT4 ratio predicting the risk of composite endpoint in pts with LVEF <40%, 40–49%, and ≥50% were 0.91, 0.83, and 0.65, respectively |
[75] |
|
fT3/fT4 cutoff 0.233 |
CV death (29%), Overall death (25%) |
8887 (46) |
69 |
50 |
85% |
HR of all-cause mortality and CV death for pts with a high FT3/FT4 ratio was 0.841 and 0.844 times less than that in pts with a low FT3/FT4 ratio |
[76] |
|
TSH > 4.70 mIU/l, TSH < 0.35 mIU/l. |
Overall death |
4992 (45) |
74 |
ND |
34% |
Hypothyroidism (HR 1.259) and hyperthyroidism (HR 1.21) had a greater risk of death compared to euthyroidism. |
[77] |
|
LT3, Hypothyroidism |
Overall death |
762 |
ND |
ND |
ND |
Independent association with death significant in pts with TSH >10 mIU/L. LT3 was independently associated with HF hospitalization and death |
[78] |
|
SCH: (TSH 4-10 microUI/mL), HYPO: (TSH > 10 microUI/mL), LT3: (fT3 < 1.8 pg/mL) |
In-hospital death |
1018 (55) |
81 |
ND |
80% |
Mortality rate was 27% among HYPO pts, 17% in SCH pts, and 11% among euthyroid pts. HYPO (HR 2.1) and fT3 levels (HR 3.4) were associated with an increased likelihood of in-hospital death. |
[74] |
|
TSH quartiles (≤1.3; 1.4-2.2; 2.3-3.5; ≥3.6 mlU/L) |
Cardiac death (28), Non-Cardiac death (30) HF impairment (40), Cardiac transplantation (11), Ventricular arrhythmias (24) |
180 (21) |
37 |
28 |
35% |
Serum TSH levels (>2.67 nIU/L) may provide help for the stratification of the risk of ventricular arrhythmias |
[79] |
|
LT3:fT3 ≤ 2.03 pg/ml |
CV death (88), non-CV death (105) |
911 (41) |
68 |
60 |
3% |
LT3 at discharge is associated with higher cardiac and all cause-mortality, accompanied by high central venous pressure, lower nutritional status, and impaired exercise capacity |
[80] |
|
SCH:TSH > 4.51 mlU/L ; LT3: total T3 < 80 ng/dl |
Cardiac transplantation (104), VAD replacement (31), Overall death (327) |
1,365 (35) |
57 |
34 |
No data |
SCH (HR 1.82) and LT3 (HR 2.12) were associated with increased risk of composite endpoint |
[81] |
|
SCH:TSH > 4 µlU/mL |
Worsening HF (232), CV death (108), non-CV death (128) |
1043 (41) |
67 |
42 |
3% |
SCH is an independent predictor of cardiac event (HR 1.42) and all-cause mortality (1.421) after adjustment with other confounders |
[82] |
|
fT3 < 3.00 pmol/L |
CV death (30), non-CV death (6), Hospitalization (45) |
113 (3.5) |
61 |
31 |
64% |
Patients with fT3 < 3.00 pmol/L had higher overall mortality and HF hospitalization |
[83] |
|
SCH: TSH of 4.5 to 19.9mIU/L; SHY: TSH < 0.45 mIU/L |
CV death (27), Rehospitalization (80) |
274 (70) |
70 |
39 |
100% |
Higher TSH is independently associated with composite CV events. SCH is an independent predictor (HR: 2.31) of composite CV events |
[84] |
|
fT3 < 2.77 pg/mL |
CV death (19), non-CV death (4) |
71 (34) |
54 |
26 |
No data |
FT3 < 2.77 pg/mL was identified as predictor of events (HR: 8.623) |
[85] |
|
LT3:fT3 ≤ 2.05 pg/ml |
CV death (16), non-CV death (10) |
270 (31) |
68 |
67 |
100% |
LT3 on admission is associated with higher in-hospital all-cause, cardiac, and non-cardiac death rates and with increased 1-year death |
[86] |
|
LT3:fT3 < 1.79 pg/mL; SCH:TSH > 4.78 mlU/L normal fT3 and or fT4; SHY: TSH < 0.55 mlU/L and normal fT3 and or fT4; HYPO: TSH > 4.78 mlU/L and < fT3 and or Ft4 |
non-CV death (ND) |
458 (29) |
51 |
32 |
ND |
HYPO was the strongest predictor of death (HR 4.189), followed by LT3 (HR 3.147) and SHYPO (HR 2.869) |
[87] |
|
TSH quartiles (≤1.3; 1.4-2.2; 2.3-3.5; ≥3.6 mlU/L) |
CV death (ND) CV hospitalization (ND) |
5599 (51) |
75 |
<50 |
ND |
Increased risk of death in the highest TSH group (HR 1.54). TSH as an independent predictor of the combined endpoint |
[88] |
|
Total T3 ≤ 52.3 ng/dl |
CV death (38), non-CV death (16) |
144 (49) |
71 |
42 |
100% |
T3 as independent predictors for both all-cause and cardiac mortalities among critically ill patients with HF, and high NT-proBNP and low T3 levels predict a worse long-term outcome |
[89] |
|
SHY: TSH <0.35 µIU/mL SCH: TSH > 5.5 µIU/mL |
Overall death |
963 (26) |
52 |
32 |
72% |
SHY, SCH have higher all-cause mortality rates. However, only SHY (HR 1.793), not SCH, is an independent predictor for increased risk of overall death |
[90] |
|
SHY: TSH < 0.3 µIU/mL SCH: TSH > 5.0 µIU/mL |
CV death (1104), Hospitalization (1210), non-CV death (1402) |
4750 (22) |
73 |
31 |
63% |
SCH associated with an increased risk of the composite outcome of CV death or HF hospitalization (HR: 1.29), as well as all-cause death (HR: 1.36). When NT-proBNP was added to the predictive models, the association between SCH and all outcomes was eliminated |
[91] |
|
LT3: fT3 < 2.7 pmol/l SHY: TSH < 0.3 mIU/l SCH: TSH > 4.0 mIU/l HYPER: TSH < 0.1 mIU/l HYPO: TSH > 4.0 mIU/l |
CV death (153), non-CV death (111) |
758 (29) |
68 |
30 |
ND |
SCH, SHY; HYPO, HYPER are not relevant prognostic factors. LT3 is a significant indicator of poor prognosis |
[92] |
|
HYPO (>5.0 μU/mL), HYPER (<0.3 μU/mL) |
non-CV death (ND) |
2225 (48) |
59 |
24 |
ND |
HYPO and HYPER were associated with 58% and 85% increases in the risk for death (HR: 1.58; HR: 1.85) |
[93] |
|
LT3: fT3 < 2.1 ng/L |
CV death (64), non-CV death (46) |
442 (25) |
65 |
33 |
37% |
Pts with LT3 and higher BNP showed the highest risk of all-cause and cardiac death |
[94] |
|
SCH: TSH > 5.5 mlU/L |
Hospitalization (55), non-CV death (18), transplantation (6) |
338 (33) |
64 |
32 |
ND |
TSH levels, even slightly above normal range, are independently associated with a greater likelihood of HF progression |
[95] |
|
fT3/fT4 ratio ≤ 1.7 |
CV death (15), VF (1) |
111 (31) |
62 |
29 |
ND |
fT3/fT4 ratio ≤ 1.7 was associated with an increased risk of mortality, independent of other prognostic markers. Sensitivity, specificity, positive and negative predictivity of fT3/fT4 ratio ≤ 1.7 for cardiac mortality were 100%, 71%, 36%, and 100% |
[96] |
|
free T3, 3.2 to 6.5 pmol/L (2 to 4.2 pg/mL); |
CV death (47), non-CV death (17) |
281 (17) |
68 |
28 |
ND |
Low T3 levels are an independent predictor of mortality, adding prognostic information to conventional clinical (age) and functional cardiac parameters (LVEF) |
[97] |
|
fT3/reverseT3 ratio ≤ 4 |
Cardiac death (17), Transplantation (6) |
84 (16) |
50 |
18 |
100% |
A low fT3/reverse T3 ratio was a predictor of cardiac events, with a survival rate of 37%. The lowest ratio was associated with the poorest prognosis |
[98] |
SCH: Ssubclinical hypothyroidism; SCH: subclinical hypothyroidism; SHY: subclinical hyperthyroidism; HYPO: overt hypothyroidism; SHYPER: overt hyperthyroidism; HT: VAD: ventricular assist device; CV: cardiovascular; LT3: low T3 syndrome; fT3: free triiodothyronine; fT4: free thyroxine; TSH: thyrotropin; W: women; ND: no data.
However, the results on the prognostic role of subclinical TH abnormalities in HF are non-consensual, and the heterogeneity can depend on different factors, including type and severity of disease, sex, age, race, definition, modality of acquisition, and severity of TH dysfunction, type and number of events considered at follow-up, duration of follow-up. As shown in Table 1, the studies on HF included patients with different levels of left ventricular dysfunction, different ages, and with stable and unstable ischemic and non-ischemic HF. The ages ranged from 37 to 75 years, and the events included both hard and soft ones. Two examples may account for these discrepancies.
In the study of Mahal et al., the conclusion was that in hospitalized patients with HF and subclinical hypothyroidism (SCH), there was no increase in mortality and major morbidity, but there were only differences in the hospital characteristics [102]. Similarly, the study of Perez showed that hypothyroidism was associated with an increased risk of the composite outcome (cardiac death or HF hospitalization and all-cause death), but this association disappeared when NT-proBNP was included in the model [91]. However, in both studies, a major part of the population was old (>80 years, around 90% in the Mahal study, mean age 73 years old in the Perez study), and this may create trouble in the diagnosis of hypothyroidism. Accordingly, TSH normal reference range increases with age, and this may justify the use of different reference intervals in patients >60 years [103]. Another example is the enrolment of patients taking TH substitutive therapy, as in the study of Frey in which levothyroxine treatment was in 20% of patients with subclinical hyperthyroidism (SHY) (iatrogenic SHY) and in 27% of SCH patients [92]. However, a limitation of all the above-mentioned studies is that TH was assessed only one time, at the beginning of the enrolment without any follow-up. This limit is critical if researchers consider that TH disorders can develop in 27% of HF patients (LT3S 12.5%, SCH 10.4%, overt hypothyroidism 6.2%) during follow-up, highlighting the need to monitor TH metabolism longitudinally, also considering that these abnormalities were associated with additive worse prognosis factors [104]. Moreover, considering HF as a systemic disease, it is conceivable that TH metabolic abnormalities may further aggravate the function of other organs and systems, such as patients with renal insufficiency or those with anemia, inducing thus the development of a vicious circle favoring HF progression [105][106].
In AMI, LT3S has been more extensively assessed in terms of prognostic impact in comparison to the other TH abnormalities. This is in line with the evidence that in the acute phase of AMI, T3 circulating levels rapidly down-regulate with maximal changes 36 h after onset of symptoms [107]. This down-regulation is linked to an increase in the inactive T3 metabolite (reverse T3), which occurs rapidly within 12 h of the onset of symptoms. The reduction is strictly linked with the acute phase of the disease and its severity, as evidenced by the direct relationship among clinical status, myocardial necrosis extent, and LV dysfunction, as well as the intensity of the inflammatory and stress response and levels of T3 [108][109]. Also, changes in T3 levels were associated with early and late recovery of cardiac function after AMI [110]. Interestingly, the persistence of low TSH value in the acute phase of AMI and after 4 months was associated with post-ischemic LV remodeling, as well as the persistence of SCH was associated with more severe coronary artery stenosis and occurrence of cardiac events [111][112]. These data highlight the importance of maintaining TH homeostasis, and this should also be a key point of TH replacement therapy. In general, LT3S or low T3/T4 ratio were associated with a worse prognosis. The association with other predictive variables, such as NT-proBNP and GRACE risk score, increased the weight of prognostic stratification, identifying patients at higher risk of cardiac events [113][114]. Also, the negative prognostic impact of LT3S has been shown in patients with myocardial infarction with nonobstructive coronary arteries [115].
6. Thyroid and Heart Replacement Therapy in Heart Failure and Acute Myocardial Infarction
As researchers stated in a previous review, TH replacement therapy is still considered a pillar in patients with cardiac disorders. Actually, if researchers consider LT3S an acute physiological response to the acute disease phase, it is correct to avoid any treatment. However, according to the experimental evidence showing the key role of TH in cardioprotection, researchers also have to hypothesize that prolonged TH abnormalities could reduce or make cardioprotective mechanisms ineffective. Moreover, as already reported in the first paragraph of this research, the Coronary Drug Project (CDP) reinforced this dogma [116]. In HF and AMI patients, thyroxine and triiodothyronine were both used. Table 2 summarizes the main characteristics and findings of these studies in HF patients [117][118][119][120][121][122][123][124][125]. In general, the studies were carried out in a few of patients with different clinical characteristics and with different targets, mainly functional and morphological ones. Furthermore, the main questions regard the type, duration, administration modalities, and TH dosage. In the study, researchers administered T3 intravenously for three consecutive days in stable hospitalized HF patients having a severe LV dysfunction and LT3S [121]. The drug was well tolerated without any side effects, HR increase, or arrhythmias at Holter EKG monitoring. The results showed a significant increase in LV stroke volume, LVEF, and cardiac index. The improved cardiac function paralleled neuroendocrine deactivation, with a significant reduction in the vasoconstrictor/sodium-retaining norepinephrine and aldosterone, and in the plasma levels of their counterpart NT-proBNP. In contrast, Holmager et al. administered 3-month oral T3 therapy in stable chronic HF patients, without any clinical or functional benefit [118]. However, the differences between the two studies are consistent and could account for the contrasting results. First, patients of the study had lower LVEF and higher –NT-pro-BNP, indicating a more severe clinical and cardiac status than those in Holmager’s study. Furthermore, the type of administration and T3 dosage were definitely different, and this determined a higher increase in fT3 in the treated patients with respect to those of Holmager, from 1.74 pg/mL (range 1.62–1.93 pg/mL) to 3.43 (range 3.20–3.84 pg/mL) and from 1.4 (0.9–1.6 nmol/L) to 1.7 (1.3–3.4 nmol/L), respectively. Indeed, continuous intravenous administration of T3 guarantees the stability of circulating plasma T3 levels, which is unlikely with oral administration of two daily T3 doses. One suggestion coming from the discrepancies between these two studies is that patient selection is a key point for the effectiveness of TH replacement therapy. Thus, researchers can argue that the positive effect of TH replacement therapy in HF patients occurs in those with more severe clinical and cardiac status. A proof of this is the result of Malik’s study in which thyroxine (20 µg/h) was administered intravenously in patients with severe left ventricular dysfunction, evolving to cardiogenic shock, who were unresponsive to conventional inotropic therapy and intra-aortic balloon counterpulsation. All patients had improvements in cardiac index and hemodynamics at 24 and 36 h after beginning thyroxine. These effects were maintained to complete surgical treatment consisting of heart transplantation or LV device [123]. Another critical point is the modality of administration. If intravenous continuous infusion guarantees the maintenance of a stable TH circulating level, but it is not feasible in daily life, oral administration cannot guarantee this. One solution could be the use of slow-release T3 patches, but there are no studies on their application in HF patients. Importantly, two meta-analyses showed TH replacement therapy was well tolerated and confirmed the positive effects of TH replacement therapy in HF patients with a reduction in neuroendocrine and sympathetic activation, improvement in cardiac function, cardiac output, and diastolic function [126][127].
Table 2. TH replacement therapy in patients with ischemic and non-ischemic HF.
|
Patients (N) |
Study Design |
LVEF (%) |
TH Dose Treatment |
Main Findings |
Ref. |
|
39 ischemic and non-ischemic HF and LT3 |
Randomized, prospective, double-blind, placebo-controlled |
31 ± 6 |
T3 0.025 mg/day, OS |
Improvement in NYHA class, ↑ LVEF, ↓ LVESV, ↓ NT-proBNP, ↓ hs-C-reactive protein, ↑ 6-MWD |
[117] |
|
13 ischemic and non-ischemic HF and LT3 |
Randomized, double-blind, cross-over, placebo-controlled |
43 (37–52) |
Oral T3 twice daily for 3 months (tablet dose 20 µg) |
No clinical or functional benefit observed |
[118] |
|
163 ischemic, non-ischemic HF, SCH |
Uncontrolled |
N/A |
T4 dose necessary to normalize TSH |
↑ Physical performance at 6 min walking test |
[119] |
|
86 ischemic and non-ischemic HF |
Randomized (2:1) placebo-controlled |
28 ± 6 |
DTPA twice daily 90 mg increments (every 2 wks to maximum 360 mg) |
↑ CI ↓ SVR, lipoproteins, and cholesterol |
[120] |
|
20 ischemic and non-ischemic HF and LT3 |
Randomized, placebo-controlled |
25 (18–32) |
T3 3 days continuously infused (initial dose 20 g/m2) |
↑ LVSV, LVEDV, ↓ NT-proBNP, Aldosterone, NA |
[121] |
|
6 ischemic and non-ischemic HF |
Uncontrolled |
24 ± 3 |
T3 initial dose 20 mg/m2bs/d Continuous infusion (4 d) |
↓SVR ↑ CO and UO |
[122] |
|
10 cardiogenic shock |
Uncontrolled |
N/A |
T4 20 mg/h bolus + continuous infusion (36h) |
↑ CI, PCWP, and MAP |
[123] |
|
23 ischemic and non-ischemic HF |
Uncontrolled |
22 ± 1 |
T3 cumulative dose 0.15–2.7 mg/kg bolus + continuous infusion (6-12 h) |
↓ SVR ↑ CO |
[124] |
|
10 non-ischemic HF |
Randomized (1:1) placebo-controlled |
29 ± 6 |
T4 100 mg/d OS for 3 months |
Improvement in cardiovascular performance at rest, exercise, and dobutamine stress test |
[125] |
CI: cardiac index; CO: cardiac output; DITPA: 3,5-Diiodothyropropionic acid; HF: heart failure; LVEF: left ventricular ejection fraction; LVEDV: left ventricular end diastolic volume; LVSV: left ventricular stroke volume; MAP: mean arterial pressure; NT-proBNP: N-terminal pro-Brain natriuretic peptide; PCWP: pulmonary capillary wedge pressure; T3: triiodothyronine; T4: thyroxine; SVR: systemic vascular resistance; UO: urinary output, 6-MWD: 6 min walking distance; SCH: subclinical hypothyroidism; LT3: low T3 syndrome; NA: not applicable.
In the context of AMI, in a phase II, randomized, treated/untreated patients’ study (THYRST Study), researchers showed that T3 replacement therapy was safe and improved regional systolic function in patients with STEMI and LT3 [128]. Accordingly, the discharge/follow-up decrease in wall motion score index, a semiquantitative method to assess regional systolic wall function, was significantly greater in the T3-treated group, whereas there were no significant changes in systolic global function and in reduction in necrosis extent between the treated and untreated T3 groups. T3 was administered orally three times daily with a maximum dose of 15 mcg/m2/day and continued for 6 months starting after 72 h from hospital admission [128]. The effort was to maintain T3 circulating levels within normal ranges. Also, in the ThyRepair Study, the aim was the effects of acute administration of T3 in patients with anterior AMI, with the treatment starting after coronary revascularization intravenously at a bolus injection of 0.8 μg/kg followed by a constant infusion of 0.113 μg/kg/h i.v. for 48 h [129]. The results showed a reduced intra-hospital LV remodeling, but not at follow-up (lower LV end-diastolic and end-systolic volumes in treated patients) and a tendency to reduction in myocardial necrosis extent at follow-up in treated than in untreated patients. In contrast, the study of Jabbar et al. showed that levothyroxine treatment in AMI patients did not improve global systolic function [130]. The initial T4 dose was 25 μg per day, with the target to maintain TSH between 0.4 and 2.5 mU/L. The starting dose was within 21 days of AMI up to 52 weeks. In this research, the mean TSH value at the beginning of the treatment was 5.7 mU/L, and LVEF was quite normal (>50%). It is noteworthy that the enrolled patients of these three studies were clinically stable with a normal mean LVEF (>50%), and this, very likely, mitigated the effects of T3 therapy. Moreover, TH dose regimen was different, as well as the beginning and the lasting of treatment and the type of TH used. Thus, according to the results of the meta-analysis of Tharmapoopathy, there are still no indications of TH replacement therapy in patients with AMI [131]. Another critical point that could account for the discrepancies in the results on the efficacy of TH treatment in HF and AMI patients is that TH levels may not accurately reflect myocardial TH levels. In particular, cardiac diseases induce an increased expression of cardiac D3 deiodinase with the consequent conversion of T4 into reverse T3, that is, the inactive T3 form. Probably, patients with increased activation of this enzyme can benefit from the TH replacement therapy.
References
- Wicomb, W.; Cooper, D.K.; Hassoulas, J.; Rose, A.G.; Barnard, C.N. Orthotopic transplantation of the baboon heart after 20 to 24 hours’ preservation by continuous hypothermic perfusion with an oxygenated hyperosmolar solution. J. Thorac. Cardiovasc. Surg. 1982, 83, 133–140.
- Cooper, D.K.; Wicomb, W.N.; Rose, A.G.; Barnard, C.N. Orthotopic allotransplantation and autotransplantation of the baboon heart following twenty-four hours’ storage by a portable hypothermic perfusion system. Cryobiology 1983, 20, 385–394.
- Cushing, H. Some experimental and clinical observations concerning states of increased intracranial tension. Am. J. Med. Sci. 1902, 124, 373–400.
- Kocher, A. Ueber morbus Basedowi. Mitt. Grenzgeb. Med. Chir. 1901, 1, 1–13.
- Cooper, D.K.C.; Novitzky, D.; Wicomb, W.N. The pathophysiological effects of brain death on potential donor organs, with particular reference to the heart. Ann. R. Coll. Surg. Engl. 1989, 71, 261–266.
- Novitzky, D.; Rose, A.G.; Cooper, D.K.C.; Reichart, B. Interpretation of endomyocardial biopsy after heart transplantation. Potentially confusing factors. S Afr. Med. J. 1986, 70, 789–792.
- Novitzky, D.; Cooper, D.K.C.; Rose, A.G.; Reichart, B. Prevention of myocardial injury by pretreatment with verapamil hydrochloride prior to experimental brain death: Efficacy in a baboon model. Am. J. Emerg. Med. 1987, 5, 11–18.
- Novitzky, D.; Cooper, D.K.C.; Morrell, D.; Isaacs, S. Change from aerobic to anaerobic metabolism after brain death, and reversal following triiodothyronine (T3) therapy. Transplantation 1988, 45, 32–36.
- Hamilton, B.E. Clinical notes on hearts in hyperthyroidism. Boston Med. Surg. J. 1922, 186, 216–218.
- Pratschke, J.; Wilhelm, M.J.; Kusaka, M.; Basker, M.; Cooper, D.K.C.; Hancock, W.W.; Tilney, N.L. Brain death and its influence on donor organ quality and outcome after transplantation. Transplantation 1999, 67, 343–348.
- Goetsch, E. Newer methods in the diagnosis of thyroid disorders: Pathological and clinical: B. Adrenaline hypersensitiveness in clinical states of hyperthyroidism. NY State J. Med. 1918, 18, 259–267.
- McDonald, C.H.; Shepeard, W.L.; Green, M.F.; DeGroat, A.F. Response of the hyperthyroid heart to epinephrine. Am. J. Phys. 1935, 112, 227–230.
- Gaffney, T.E.; Braunwald, E.; Kahler, R.L. Effects of guanethidine on triiodothyronine-induced hyperthyroidism in man. New Eng. J. Med. 1961, 265, 16–20.
- Levey, G.S. The adrenergic nervous system in hyperthyroidism: Therapeutic role of beta adrenergic blocking drugs. Pharmacol. Ther. 1976, 1, 431–443.
- The coronary drug project. Findings leading to further modifi cations of its protocol with respect to dextrothyroxine. The coronary drug project research group. JAMA 1972, 220, 996–1008.
- Pilo, A.; Iervasi, G.; Vitek, F.; Ferdeghini, M.; Cazzuola, F.; Bianchi, R. Thyroidal and peripheral production of 3,5,3′—Triiodothyronine in humans by multicompartmental analysis. Am. J. Physiol. 1990, 258, E715–E726.
- Young, W.F., Jr.; Gorman, C.A.; Jiang, N.S.; Machacek, D.; Hay, I.D. L-thyroxine contamination of pharmaceutical D-thyroxine: Probable cause of therapeutic effect. Clin. Pharmacol. Ther. 1984, 36, 781–787.
- Papp, C. The heart in thyroid dysfunction. Postgrad. Med. J. 1945, 21, 45–51.
- Tata, J.R.; Ernster, L.; Lindberg, O.; Arrhenius, E.; Pedersen, S.; Hedman, R. The action of thyroid hormones at the cell level. Biochem. J. 1963, 86, 408–428.
- Dillmann, W.H. Hormonal influences on cardiac myosin ATPase activity and myosin isoenzyme distribution. Mol. Cell Endocrinol. 1984, 34, 169–181.
- Oppenheimer, J.H.; Koerner, D.; Schwartz, H.L.; Surks, M.I. Specific nuclear triiodothyronine binding sites in rat liver and kidney. J. Clin. Endocrinol. Metab. 1972, 35, 330–333.
- Fazio, S.; Palmieri, E.A.; Lombardi, G.; Biondi, B. Effects of thyroid hormone on the cardiovascular system. Recent Prog. Horm. Res. 2004, 59, 31–50.
- Razvi, S.; Jabbar, A.; Pingitore, A.; Danzi, S.; Biondi, B.; Klein, I.; Peeters, R.; Zaman, A.; Iervasi, G. Thyroid hormones and cardiovascular function and diseases. J. Am. Coll. Cardiol. 2018, 24, 1781–1796.
- Klein, I. Thyroid hormone and the cardiovascular system. Am. J. Med. 1990, 88, 631–637.
- Jabbar, A.; Pingitore, A.; Pearce, S.H.; Zaman, A.; Iervasi, G.; Razvi, S. Thyroid hormones and cardiovascular disease. Nat. Rev. Cardiol. 2017, 14, 39–55.
- Hartong, R.; Wang, N.; Kurokawa, R.; Lazar, M.A.; Glass, C.K.; Apriletti Dillmann, W.H. Delineation of three different thyroid hormone-response elements in promoter of rat sarcoplasmic reticulum Ca2_-ATPase gene. J. Biol. Chem. 1994, 269, 13021–13029.
- Klein, I. Chapter 81: Endocrine disorders and cardiovascular disease. In Braunwald’s Heart Disease, 10th ed.; Elsevier: Philadelphia, PA, USA, 2014; pp. 1793–1808.
- Mastorci, F.; Sabatino, L.; Vassalle, C.; Pingitore, A. Cardioprotection and Thyroid Hormones in the Clinical Setting of Heart Failure. Front. Endocrinol. 2020, 10, 927.
- Sun, Z.; Ojamaa, K.; Coetzee, W.A.; Artman, M.; Klein, I. Effects of thyroid hormone on action potential and repolarization currents in rat ventricular myocytes. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E302–E307.
- Pachucki, J.; Burmeister, L.A.; Larsen, P.R. Thyroid hormone regulates hyperpolarization-activated cyclic nucleotide-gated channel (HCN2) mRNA in the rat heart. Circ. Res. 1999, 85, 498–503.
- Danzi, S.; Klein, I. Thyroid hormone and blood pressure regulation. Curr. Hypertens. Rep. 2003, 5, 513–520.
- Klein, I.; Ojamaa, K. Thyroid hormone and the cardiovascular system. N. Engl. J. Med. 2001, 344, 501–509.
- Biondi, B.; Palmieri, E.A.; Lombardi, G.; Fazio, S. Effects of thyroid hormone on cardiac function: The relative importance of heart rate, loading conditions, and myocardial contractility in the regulation of cardiac performance in human hyperthyroidism. J. Clin. Endocrinol. Metab. 2002, 87, 968–974.
- Volpe, R. Immunoregulation in autoimmune thyroid disease. Thyroid 1994, 4, 373–377.
- Cooper, D.S.; Biondi, B. Subclinical thyroid disease. Lancet 2012, 379, 1142–1154.
- Kahaly, G.J.; Dillmann, W.H. Thyroid hormone action in the heart. Endocr. Rev. 2005, 26, 704–728.
- Mohr-Kahaly, S.; Kahaly, G.; Meyer, J. . Z. Kardiol. 1996, 85 (Suppl. S6), 219–231.
- Weltman, N.Y.; Wang, D.; Redetzke, R.A.; Gerdes, A.M. Longstanding hyperthyroidism is associated with normal or enhanced intrinsic cardiomyocyte function despite decline in global cardiac function. PLoS ONE 2012, 7, e46655.
- Klein, I. Endocrine disorders and cardiovascular disease. In Braunwald’s Heart Disease: A Textbook of Cardiovascular Medicine, 7th ed.; Zipes, D.P., Libby, P., Bonow, R., Braunwald, E., Eds.; Saunders: Philadelphia, PA, USA, 2005; pp. 2051–2065.
- Fredlund, B.O.; Olsson, S.B. Long QT interval and ventricular tachycardia of “torsade de pointe” type in hypothyroidism. Acta Med. Scand. 1983, 213, 231–235.
- Sawin, C.T.; Geller, A.; Wolf, P.A.; Belanger, A.J.; Baker, E.; Bacharach, P.; Wilson, P.W.; Benjamin, E.J.; D’Agostino, R.B. Low serum thyrotropin concentrations as a risk factor for atrial fibrillation in older persons. N. Engl. J. Med. 1994, 331, 1249–1252.
- Parle, J.V.; Maisonneuve, P.; Sheppard, M.C.; Boyle, P.; Franklyn, J.A. Prediction of all-cause and cardiovascular mortality in elderly people from one low serum thyrotropin result: A 10-year cohort study. Lancet 2001, 358, 861–865.
- Iervasi, G.; Molinaro, S.; Landi, P.; Taddei, M.C.; Galli, E.; Mariani, F.; L’abbate, A.; Pingitore, A. Association between increased mortality and mild thyroid dysfunction in cardiac patients. Arch. Intern. Med. 2007, 167, 1526–1532.
- Cappola, A.R.; Fried, L.P.; Arnold, A.M.; Danese, M.D.; Kuller, L.H.; Burke, G.L.; Tracy, R.P.; Ladenson, P.W. Thyroid status, cardiovascular risk, and mortality in older adults. JAMA 2006, 295, 1033–1041.
- Rodondi, N.; Bauer, D.C.; Cappola, A.R.; Cornuz, J.; Robbins, J.; Fried, L.P.; Ladenson, P.W.; Vittinghoff, E.; Gottdiener, J.S.; Newman, A.B. Subclinical thyroid dysfunction, cardiac function, and the risk of heart failure. The Cardiovascular Health study. J. Am. Coll. Cardiol. 2008, 52, 1152–1159.
- Rodondi, N.; Aujesky, D.; Vittinghoff, E.; Cornuz, J.; Bauer, D.C. Subclinical hypothyroidism and the risk of coronary heart disease: A meta-analysis. Am. J. Med. 2006, 119, 541–551.
- Monzani, F.; Di Bello, V.; Caraccio, N.; Bertini, A.; Giorgi, D.; Giusti, C.; Ferrannini, E. Effect of levothyroxine on cardiac function and structure in subclinical hypothyroidism: A double blind, placebo-controlled study. J. Clin. Endocrinol. Metab. 2001, 86, 1110–1115.
- Ripoli, A.; Pingitore, A.; Favilli, B.; Bottoni, A.; Turchi, S.; Osman, N.F.; De Marchi, D.; Lombardi, M.; L’Abbate, A.; Iervasi, G. Does subclinical hypothyroidism affect cardiac pump performance? Evidence from a magnetic resonance imaging study. J. Am. Coll. Cardiol. 2005, 45, 439–445.
- Hak, A.E.; Pols, H.A.; Visser, T.J.; Drexhage, H.A.; Hofman, A.; Witteman, J.C. Subclinical hypothyroidism is an independent risk factor for atherosclerosis and myocardial infarction in elderly women: The Rotterdam Study. Ann. Intern. Med. 2000, 132, 270–278.
- Kübler, W.; Haass, M. Cardioprotection: Definition, classification, and fundamental principles. Heart 1996, 75, 330–333.
- Pingitore, A.; Nicolini, G.; Kusmic, C.; Iervasi, G.; Grigolini, P.; Forini, F. Cardioprotection and thyroid hormones. Heart Fail. Rev. 2016, 21, 391–399.
- Forini, F.; Pitto, L.; Nicolini, G. Thyroid hormone, mitochondrial function and cardioprotection. In Thyroid and Heart; Iervasi, G., Pingitore, A., Gerdes, A.M., Razvi, A., Eds.; Springer Nature: Cham, Switzerland, 2020; Chapter 9.
- Goldenthal, M.J.; Ananthakrishnan, R.; Marín-García, J. Nuclear-mitochondrial cross-talk in cardiomyocyte T3 signaling: A time-course analysis. J. Mol. Cell Cardiol. 2005, 39, 319–326.
- Galli, E.; Pingitore, A.; Iervasi, G. The role of thyroid hormone in the pathophysiology of heart failure: Clinical evidence. Heart Fail. Rev. 2010, 15, 155–169.
- Garber, J.R.; Cobin, R.H.; Gharib, H.; Hennessey, J.V.; Klein, I.; Mechanick, J.I.; Pessah-Pollack, R.; Singer, P.A.; Woeber for the American Association of Clinical Endocrinologists; American Association of Clinical Endocrinologists; et al. Clinical practice guidelines for hypothyroidism in adults: Cosponsored by the American Association of Clinical Endocrinologists and the American Thyroid Association. Thyroid 2012, 22, 1200–1235.
- Pan, Y.; Wang, Y.; Shi, W.; Liu, Y.; Cao, S.; Yu, T. Mitochondrial proteomics alterations in rat hearts following ischemia/reperfusion and diazoxide post conditioning. Mol. Med. Rep. 2021, 23, 161.
- Canale, P.; Nicolini, G.; Pitto, L.; Kusmic, C.; Rizzo, M.; Balzan, S.; Iervasi, G.; Forini, F. Role of miR-133/Dio3 Axis in the T3-Dependent Modulation of Cardiac mitoK-ATP Expression. Int. J. Mol. Sci. 2022, 23, 6549.
- Sabatino, L. Nrf2-Mediated Antioxidant Defense and Thyroid Hormone Signaling: A Focus on Cardioprotective Effects. Antioxidants 2023, 12, 1177.
- Della Nera, G.; Sabatino, L.; Gaggini, M.; Gorini, F.; Vassalle, C. Vitamin D Determinants, Status, and Antioxidant/Anti-inflammatory-Related Effects in Cardiovascular Risk and Disease: Not the Last Word in the Controversy. Antioxidants 2023, 18, 948.
- Al-Oanzi, Z.H.; Alenazy, F.O.; Alhassan, H.H.; Alruwaili, Y.; Alessa, A.I.; Alfarm, N.B.; Alanazi, M.O.; Alghofaili, S.I. The Role of Vitamin D in Reducing the Risk of Metabolic Disturbances That Cause Cardiovascular Diseases. J. Cardiovasc. Dev. Dis. 2023, 10, 209.
- Alrefaie, Z.; Awad, H. Effect of vitamin D3 on thyroid function and deiodinase 2 expression in diabetic rats. Arch. Physiol. Biochem. 2015, 121, 206–209.
- Miura, M.; Tanaka, K.; Komatsu, Y.; Suda, M.; Yasoda, A.; Sakuma, Y.; Ozasa, A.; Nakao, K. A novel interaction between thyroid hormones and 1,25(OH)(2)D(3) in osteoclast formation. Biochem. Biophys. Res. Commun. 2002, 291, 987–994.
- Gouveia, C.H.; Christoffolete, M.A.; Zaitune, C.R.; Dora, J.M.; Harney, J.W.; Maia, A.L.; Bianco, A.C. Type 2 iodothyronine selenodeiodinase is expressed throughout the mouse skeleton and in the MC3T3-E1 mouse osteoblastic cell line during differentiation. Endocrinology 2005, 146, 195–200.
- Berg, J.P.; Liane, K.M.; Bjørhovde, S.B.; Bjøro, T.; Torjesen, P.A.; Haug, E. Vitamin D receptor binding and biological effects of cholecalciferol analogues in rat thyroid cells. J. Steroid Biochem. Mol. Biol. 1994, 50, 145–150.
- D’Emden, M.C.; Wark, J.D. 1,25-Dihydroxyvitamin D3 enhances thyrotropin releasing hormone induced thyrotropin secretion in normal pituitary cells. Endocrinology 1987, 121, 1192–1194.
- Wang, Y.; Liu, Q.; Dong, H.; Feng, Y.; Raguthu, C.; Liang, X.; Liu, C.; Zhang, Z.; Yao, X. The protective effect of iodide intake adjustment and 1,25(OH)2D3 supplementation in rat offspring following excess iodide intake. Ther. Adv. Endocrinol. Metab. 2020, 11, 2042018820958295.
- Vassalle, C.; Parlanti, A.; Pingitore, A.; Berti, S.; Iervasi, G.; Sabatino, L. Vitamin D, Thyroid Hormones and Cardiovascular Risk: Exploring the Components of This Novel Disease Triangle. Front. Physiol. 2021, 12, 722912.
- Babić Leko, M.; Jureško, I.; Rozić, I.; Pleić, N.; Gunjača, I.; Zemunik, T. Vitamin D and the Thyroid: A Critical Review of the Current Evidence. Int. J. Mol. Sci. 2023, 24, 3586.
- Pingitore, A.; Mastorci, F.; Berti, S.; Sabatino, L.; Palmieri, C.; Iervasi, G.; Vassalle, C. Hypovitaminosis D and Low T3 Syndrome: A Link for Therapeutic Challenges in Patients with Acute Myocardial Infarction. J. Clin. Med. 2021, 10, 5267.
- Mirhosseini, N.; Brunel, L.; Muscogiuri, G.; Kimball, S. Physiological serum 25-hydroxyvitamin D concentrations are associated with improved thyroid function-observations from a community-based program. Endocrine 2017, 58, 563–573.
- Iervasi, G.; Pingitore, A.; Landi, P.; Raciti, M.; Ripoli, A.; Scarlattini, M.; L’Abbate, A.; Donato, L. Low-T3 syndrome: A strong prognostic predictor of death in patients with heart disease. Circulation 2003, 107, 708–713.
- Pingitore, A.; Iervasi, G.; Barison, A.; Prontera, C.; Pratali, L.; Emdin, M.; Giannessi, D.; Neglia, D. Early activation of an altered thyroid hormone profile in asymptomatic or mildly symptomatic idiopathic left ventricular dysfunction. J. Card. Fail. 2006, 12, 520–526.
- Molinaro, S.; Iervasi, G.; Lorenzoni, V.; Coceani, M.; Landi, P.; Srebot, V.; Mariani, F.; L’Abbate, A.; Pingitore, A. Persistence of mortality risk in patients with acute cardiac diseases and mild thyroid dysfunction. Am. J. Med. Sci. 2012, 343, 65–70.
- De Matteis, G.; Covino, M.; Burzo, M.L.; Della Polla, D.A.; Petti, A.; Bruno, C.; Franceschi, F.; Mancini, A.; Gambassi, G. Prognostic role of hypothyroidism and low free-triiodothyronine levels in patients hospitalized with acute heart failure. Intern. Emerg. Med. 2021, 16, 1477–1486.
- Zhou, P.; Huang, L.Y.; Zhai, M.; Huang, Y.; Zhuang, X.F.; Liu, H.H.; Zhang, Y.H.; Zhang, J. The prognostic value of free triiodothyronine/free thyroxine ratio in patients hospitalized with heart failure. Zhonghua Yi Xue Za Zhi. 2023, 103, 1679–1684. (In Chinese)
- Wang, C.; Han, S.; Li, Y.; Tong, F.; Li, Z.; Sun, Z. Value of FT3/FT4 Ratio in Prognosis of Patients with Heart Failure: A Propensity-Matched Study. Front. Cardiovasc. Med. 2022, 9, 859608.
- Samuel, N.A.; Cuthbert, J.J.; Brown, O.I.; Kazmi, S.; Cleland, J.G.F.; Rigby, A.S.; Clark, A.L. Relation Between Thyroid Function and Mortality in Patients with Chronic Heart Failure. Am. J. Cardiol. 2021, 139, 57–63.
- Iacoviello, M.; Parisi, G.; Gioia, M.I.; Grande, D.; Rizzo, C.; Guida, P.; Lisi, F.; Giagulli, V.A.; Licchelli, B.; Di Serio, F.; et al. Thyroid Disorders and Prognosis in Chronic Heart Failure: A Long-Term Follow-Up Study. Endocr. Metab. Immune Disord. Drug Targets. 2020, 20, 437–445.
- Li, X.; Yao, Y.; Chen, Z.; Fan, S.; Hua, W.; Zhang, S.; Fan, X. Thyroid-stimulating hormone within the normal range and risk of major adverse cardiovascular events in nonischemic dilated cardiomyopathy patients with severe left ventricular dysfunction. Clin. Cardiol. 2019, 42, 120–128.
- Sato, Y.; Yoshihisa, A.; Kimishima, Y.; Kiko, T.; Kanno, Y.; Yokokawa, T.; Abe, S.; Misaka, T.; Sato, T.; Oikawa, M.; et al. Low T3 Syndrome Is Associated with High Mortality in Hospitalized Patients With Heart Failure. J. Card. Fail. 2019, 25, 195–203.
- Kannan, L.; Shaw, P.A.; Morley, M.P.; Brandimarto, J.; Fang, J.C.; Sweitzer, N.K.; Cappola, T.P.; Cappola, A.R. Thyroid dysfunction in heart failure and cardiovascular outcomes. Circ. Heart Fail. 2018, 11, e005266.
- Sato, Y.; Yoshihisa, A.; Kimishima, Y.; Kiko, T.; Watanabe, S.; Kanno, Y.; Abe, S.; Miyata, M.; Sato, T.; Suzuki, S.; et al. Subclinical hypothyroidism is associated with adverse prognosis in heart failure patients. Can. J. Cardiol. 2018, 34, 80–87.
- Chen, Y.Y.; Shu, X.R.; Su, Z.Z.; Lin, R.J.; Zhang, H.F.; Yuan, W.L.; Wang, J.F.; Xie, S.L. A low-normal free triiodothyronine level is associated with adverse prognosis in euthyroid patients with heart failure receiving cardiac resynchronization therapy. Int. Heart J. 2017, 58, 908–914.
- Hayashi, T.; Hasegawa, T.; Kanzaki, H.; Funada, A.; Amaki, M.; Takahama, H.; Ohara, T.; Sugano, Y.; Yasuda, S.; Ogawa, H.; et al. Subclinical hypothyroidism is an independent predictor of adverse cardiovascular outcomes in patients with acute decompensated heart failure. ESC Heart Fail. 2016, 3, 168–176.
- Wang, W.; Guan, H.; Fang, W.; Zhang, K.; Gerdes, A.M.; Iervasi, G.; Tang, Y.-D. Free triiodothyronine level correlates with myocardial injury and prognosis in idiopathic dilated cardiomyopathy: Evidence from cardiac MRI and SPECT/PET imaging. Sci. Rep. 2016, 6, 39811.
- Okayama, D.; Minami, Y.; Kataoka, S.; Shiga, T.; Hagiwara, N. Thyroid function on admission and outcome in patients hospitalized for acute decompensated heart failure. J. Cardiol. 2015, 66, 205–211.
- Wang, W.; Guan, H.; Gerdes, M.; Iervasi, G.; Yang, Y.; Tang, Y. Thyroid status, cardiac function and mortality in patients with idiopathic dilated cardiomyopathy. J. Clin. Endocrinol. Metab. 2015, 100, 3210–3218.
- Chen, S.; Shauer, A.; Zwas, D.R.; Lotan, C.; Keren, A.; Gotsman, I. The effect of thyroid function on clinical outcome in patients with heart failure. Eur. J. Heart Fail. 2014, 16, 217–226.
- Chuang, C.P.; Jong, Y.S.; Wu, C.Y.; Lo, H.M. Impact of triiodothyronine and N-terminal pro-B-type natriuretic peptide on the long term survival of critically ill patients with acute heart failure. Am. J. Cardiol. 2014, 113, 845–850.
- Li, X.; Yang, X.; Wang, Y.; Ding, L.; Wang, J.; Hua, W. The prevalence and prognostic effects of subclinical thyroid dysfunction in dilated cardiomyopathy patients: A single-center cohort study. J. Card. Fail. 2014, 20, 506–512.
- Perez, A.C.; Jhund, P.S.; Stott, D.J.; Gullestad, L.; Cleland, J.G.; van Veldhuisen, D.J.; Wikstrand, J.; Kjekshus, J.; McMurray, J.J. Thyroid-stimulating hormone and clinical outcomes: The CORONA trial (controlled rosuvastatin multinational study in heart failure). JACC Heart Fail. 2014, 2, 35–40.
- Frey, A.; Kroiss, M.; Berliner, D.; Seifert, M.; Allolio, B.; Güder, G.; Ertl, G.; Angermann, C.E.; Störk, S.; Fassnacht, M. Prognostic impact of subclinical thyroid dysfunction in heart failure. Int. J. Cardiol. 2013, 168, 300–305.
- Mitchell, J.E.; Hellkamp, A.S.; Mark, D.B.; Anderson, J.; Johnson, G.W.; Poole, J.E.; Lee, K.L.; Bardy, G.H. Thyroid function in heart failure and impact on mortality. JACC Heart Fail. 2013, 1, 48–55.
- Passino, C.; Pingitore, A.; Landi, P.; Fontana, M.; Zyw, L.; Clerico, A.; Emdin, M.; Iervasi, G. Prognostic value of combined measurement of brain natriuretic peptide and triiodothyronine in heart failure. J. Card. Fail. 2009, 15, 35–40.
- Iacoviello, M.; Guida, P.; Guastamacchia, E.; Triggiani, V.; Forleo, C.; Catanzaro, R.; Cicala, M.; Basile, M.; Sorrentino, S.; Favale, S. Prognostic role of sub-clinical hypothyroidism in chronic heart failure outpatients. Curr. Pharm. Des. 2008, 14, 2686–2692.
- Kozdag, G.; Ural, D.; Vural, A.; Agacdiken, A.; Kahraman, G.; Sahin, T.; Ural, E.; Komsuoglu, B. Relation between free triiodothyronine/free thyroxine ratio, echocardiographic parameters and mortality in dilated cardiomyopathy. Eur. J. Heart Fail. 2005, 7, 113–118.
- Pingitore, A.; Landi, P.; Taddei, M.C.; Ripoli, A.; L’Abbate, A.; Iervasi, G. Triiodothyronine levels for risk stratification of patients with chronic heart failure. Am. J. Med. 2005, 118, 132136.
- Hamilton, M.A.; Stevenson, L.W.; Luu, M.; Walden, J.A. Altered thyroid hormone metabolism in advanced heart failure. J. Am. Coll. Cardiol. 1990, 16, 91–95.
- Lubrano, V.; Pingitore, A.; Carpi, A.; Iervasi, G. Relationship between triiodothyronine and proinflammatory cytokines in chronic heart failure. Biomed. Pharmacother. 2010, 64, 165–169.
- Shen, Y.; Chen, G.; Su, S.; Zhao, C.; Ma, H.; Xiang, M. Independent Association of Thyroid Dysfunction and Inflammation Predicts Adverse Events in Patients with Heart Failure via Promoting Cell Death. J. Cardiovasc. Dev. Dis. 2022, 9, 290.
- Cittadini, A.; Salzano, A.; Iacoviello, M.; Triggiani, V.; Rengo, G.; Cacciatore, F.; Maiello, C.; Limongelli, G.; Masarone, D.; Perticone, F.; et al. Multiple hormonal and metabolic deficiency syndrome predicts outcome in heart failure: The T.O.S.CA. Registry. Eur. J. Prev. Cardiol. 2021, 28, 1691–1700.
- Mahal, S.; Datta, S.; Ravat, V.; Patel, P.; Saroha, B.; Patel, R.S. Does subclinical hypothyroidism affect hospitalization outcomes and mortality in congestive cardiac failure patients? Cureus 2018, 10, e2766.
- Fontes, R.; Coeli, C.R.; Aguiar, F.; Vaisman, M. Reference interval of thyroid stimulating hormone and free thyroxine in a reference population over 60 years old and in very old subjects (over 80 years): Comparison to young subjects. Thyroid. Res. 2013, 6, 13.
- Silva-Tinoco, R.; Castillo-Martínez, L.; Orea-Tejeda, A.; Orozco-Gutiérrez, J.J.; Vázquez-Díaz, O.; Montaño-Hernández, P.; Flores-Rebollar, A.; Reza-Albarrán, A. Developing thyroid disorders is associated with poor prognosis factors in patient with stable chronic heart failure. Int. J. Cardiol. 2011, 147, e24–e25.
- Merla, R.; Martinez, J.D.; Martinez, M.A.; Khalife, W.; Bionat, S.; Bionat, J.; Barbagelata, A. Hypothyroidism and renal function in patients with systolic heart failure. Tex. Heart Inst. J. 2010, 37, 66–69.
- Drechsler, C.; Schneider, A.; Gutjahr-Lengsfeld, L.; Kroiss, M.; Carrero, J.J.; Krane, V.; Allolio, B.; Wanner, C.; Fassnacht, M. Thyroid function, cardiovascular events, and mortality in diabetic hemodialysis patients. Am. J. Kidney Dis. 2014, 63, 988–996.
- Friberg, L.; Werner, S.; Eggertsen, G.; Ahnve, S. Rapid down-regulation of thyroid hormones in acute myocardial infarction: Is it cardioprotective in patients with angina? Arch. Intern. Med. 2002, 162, 1388–1394.
- Wang, W.Y.; Tang, Y.D.; Yang, M.; Cui, C.; Mu, M.; Qian, J.; Yang, Y.J. Free triiodothyronine level indicates the degree of myocardial injury in patients with acute ST-elevation myocardial infarction. Chin. Med. J. 2013, 126, 3926–3929.
- Ceremuzyński, L.; Górecki, A.; Czerwosz, L.; Chamiec, T.; Bartoszewicz, Z. Herbaczyńska-Cedro Low serum triiodothyronine in acute myocardial infarction indicates major heart injury. K. Kardiol. Pol. 2004, 60, 468–480.
- Lymvaios, I.; Mourouzis, I.; Cokkinos, D.V.; Dimopoulos, M.A.; Toumanidis, S.T.; Pantos, C. Thyroid hormone and recovery of cardiac function in patients with acute myocardial infarction: A strong association? Eur. J. Endocrinol. 2011, 165, 107–114.
- Reindl, M.; Feistritzer, H.J.; Reinstadler, S.J.; Mueller, L.; Tiller, C.; Brenner, C.; Mayr, A.; Henninger, B.; Mair, J.; Klug, G.; et al. Thyroid-stimulating hormone and adverse left ventricular remodeling following ST-segment elevation myocardial infarction. Eur. Heart J. Acute Cardiovasc. Care. 2019, 8, 717–726.
- Han, C.; Xu, K.; Wang, L.; Zhang, Y.; Zhang, R.; Wei, A.; Dong, L.; Hu, Y.; Xu, J.; Li, W.; et al. Impact of persistent subclinical hypothyroidism on clinical outcomes in non-ST-segment elevation acute coronary syndrome undergoing percutaneous coronary intervention. Clin. Endocrinol. 2022, 96, 70–81.
- Brozaitiene, J.; Mickuviene, N.; Podlipskyte, A.; Burkauskas, J.; Bunevicius, R. Relationship and prognostic importance of thyroid hormone and N-terminal pro-B-Type natriuretic peptide for patients after acute coronary syndromes: A longitudinal observational study. Cardiovasc. Disord. 2016, 16, 45.
- Yu, T.; Tian, C.; Song, J.; He, D.; Wu, J.; Wen, Z.; Sun, Z.; Sun, Z. Value of the fT3/fT4 ratio and its combination with the GRACE risk score in predicting the prognosis in euthyroid patients with acute myocardial infarction undergoing percutaneous coronary intervention: A prospective cohort study. BMC Cardiovasc. Disord. 2018, 18, 181.
- Gao, S.; Ma, W.; Huang, S.; Lin, X.; Yu, M. Impact of low triiodothyronine syndrome on long-term outcomes in patients with myocardial infarction with nonobstructive coronary arteries. Ann. Med. 2021, 53, 741–749.
- Pingitore, A.; Chen, Y.; Gerdes, A.M.; Iervasi, G. Acute myocardial infarction and thyroid function: New pathophysiological and therapeutic perspectives. Ann. Medicine. Ann. Med. 2012, 44, 745–757.
- Amin, A.; Chitsazan, M.; Taghavi, S.; Ardeshiri, M. Effects of triiodothyronine replacement therapy in patietns with chronic stable heart failure and low-triidotrhyronine syndrome: A randomized, double-blind, placebo-controlled study. ESC Heart Fail. 2015, 2, 5–11.
- Holmager, P.; Schmidt, U.; Mark, P.; Andersen, U.; Dominguez, H.; Raymond, I.; Zerahn, B.; Nygaard, B.; Kistorp, C.; Faber, J. Long-term L-Triiodothyronine (T3) treatment in stable systolic heart failure patients: A randomised, double-blind, cross-over, placebo-controlled intervention study. Clin. Endocrinol. 2015, 83, 931–937.
- Curotto Grasiosi, J.C.; Peressotti, B.; Machado, R.A.; Filipini, E.C.; Angel, A.; Delgado, J.; Quiroga, G.A.C.; Mansilla, C.R.; Quesada, M.D.M.M.; Degregorio, A.; et al. Improvement in functional capacity after levothyroxine treatment in patients with chronic heart failure and sublinical hypothyroidism. Endocrinol. Nutr. 2013, 60, 427–432.
- Goldman, S.; McCarren, M.; Morkin, E.; Ladenson, P.W.; Edson, R.; Warren, S.; Ohm, J.; Thai, H.; Churby, L.; Barnhill, J.; et al. DITPA (3,5-Diiodothyropropionic Acid), a thyroid hormone analog to treat heart failure: Phase II trial veterans affairs cooperative study. Circulation 2009, 119, 3093–3100.
- Pingitore, A.; Galli, E.; Barison, A.; Iervasi, A.; Scarlattini, M.; Nucci, D.; L’Abbate, A.; Mariotti, R.; Iervasi, G. Acute effects of triiodothyronine (T3) replacement therapy in patients with chronic heart failure and low-T3 syndrome: A randomized, placebo-controlled study. J. Clin. Endocrinol. Metab. 2008, 93, 1351–1358.
- Iervasi, G.; Emdin, M.; Colzani, R.M.P.; Placidi, S.; Sabatino, L.; Scarlattini, M.; Formichi, B. Beneficial effects of long-term triiodothyronine (T3) infusion in patients with advanced heart failure and low T3 syndrome. In Proceedings of the 2nd International Congress on Heart Disease—New Trends in Research, Diagnosis and Treatment, Washington, DC, USA, 21–24 July 2001; Kimchi, A., Ed.; Medimond Medical Publications: Englewood, NJ, USA, 2001; pp. 549–553.
- Malik, F.S.; Mehra, M.R.; Uber, P.A.; Park, M.H.; Scott, R.L.; Van Meter, C.H. Intravenous thyroid hormone supplementation in heart failure with cardiogenic shock. J. Card. Fail. 1999, 5, 31–37.
- Hamilton, M.A.; Stevenson, L.W.; Fonarow, G.C.; Steimle, A.; Goldhaber, J.I.; Child, J.S.; Chopra, I.J.; Moriguchi, J.D.; Hage, A. Safety and hemodynamic effects of intravenous triiodothyronine in advanced congestive heart failure. Am. J. Cardiol. 1998, 81, 443–447.
- Moruzzi, P.; Doria, E.; Agostoni, P.G. Medium-term effectiveness of L-thyroxine treatment in idiopathic dilated cardiomyopathy. Am. J. Med. 1996, 101, 461–467.
- Shi, C.; Bao, Y.; Chen, X.; Tian, L. The Effectiveness of Thyroid Hormone Replacement Therapy on Heart Failure and Low-Triiodothyronine Syndrome: An Updated Systematic Review and Meta-analysis of Randomized Controlled Trials. Endocr. Pract. 2022, 28, 1178–1186.
- Chen, X.; Bao, Y.; Shi, C.; Tian, L. Effectiveness and Safety of Thyroid Hormone Therapy in Patients with Dilated Cardiomyopathy: A Systematic Review and Meta-analysis of RCTs. Am. J. Cardiovasc. Drugs. 2022, 22, 647–656.
- Pingitore, A.; Mastorci, F.; Piaggi, P.; Aquaro, G.D.; Molinaro, S.; Ravani, M.; De Caterina, A.; Trianni, G.; Ndreu, R.; Berti, S.; et al. Usefulness of Triiodothyronine Replacement Therapy in Patients With ST Elevation Myocardial Infarction and Borderline/Reduced Triiodothyronine Levels (from the THIRST Study). Am. J. Cardiol. 2018, 123, 905–912.
- Pantos, C.I.; Trikas, A.G.; Pissimisis, E.G.; Grigoriou, K.P.; Stougiannos, P.N.; Dimopoulos, A.K.; Linardakis, S.I.; Alexopoulos, N.A.; Evdoridis, C.G.; Gavrielatos, G.D.; et al. Effects of Acute Triiodothyronine Treatment in Patients with Anterior Myocardial Infarction Undergoing Primary Angioplasty: Evidence from a Pilot Randomized Clinical Trial (ThyRepair Study). Thyroid 2022, 32, 714–724.
- Jabbar, A.; Ingoe, L.; Junejo, S.; Carey, P.; Addison, C.; Thomas, H.; Parikh, J.D.; Austin, D.; Hollingsworth, K.G.; Stocken, D.D.; et al. Effect of Levothyroxine on Left Ventricular Ejection Fraction in Patients With Subclinical Hypothyroidism and Acute Myocardial Infarction: A Randomized Clinical Trial. JAMA 2020, 324, 249–258.
- Tharmapoopathy, M.; Thavarajah, A.; Kenny, R.P.W.; Pingitore, A.; Iervasi, G.; Dark, J.; Bano, A.; Razvi, S. Efficacy and Safety of Triiodothyronine Treatment in Cardiac Surgery or Cardiovascular Diseases: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Thyroid 2022, 32, 879–896.

