 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Nuno Saraiva | -- | 3501 | 2024-01-02 13:02:24 | | | |
| 2 | Lindsay Dong | + 2 word(s) | 3503 | 2024-01-03 02:12:26 | | |
Video Upload Options
The Golgi apparatus (GA) is a central hub in our cells, helping modify and move proteins and lipids. When the GA does not work correctly, it can affect cell processes linked to cancer. This dysregulation can impact how proteins are changed, where they go in and outside the cell, how cells use energy, or even the structure of the extracellular matrix and the environment. That is why targeting the GA could be an appealing approach to treat cancer. Surprisingly, there are no anticancer drugs approved that specifically target the GA.
1. Introduction
The Golgi apparatus (GA) is an ancient and fundamental eukaryotic organelle found in almost all eukaryotic organisms and cell types [1]. Situated at the vital intersection of the endocytic and exocytic pathways, the GA assumes a pivotal role as a crucial transport center for eukaryotic cells. This organelle serves as a dynamic site for protein and lipid modification and acts as the key orchestrator for lipid and protein sorting and as a store for several molecules and ions [2]. Given its central involvement in membrane and protein trafficking and secretion, and in the storage capacity of second messengers (such as Ca2+), it comes as no surprise that dysregulation of GA function is often associated with human pathologies, such as Batten’s disease [3] or more prevalent conditions like Alzheimer’s disease [4] or cancer [5].
The GA involvement in various cancer hallmarks is of paramount importance, shaping the malignant phenotype of cancer and tumor stromal cells (reviewed in [6]). From aberrant glycosylation to enhanced secretion, the GA apparatus orchestrates a series of intricate molecular events that can contribute to shape cancer progression. It acts as a central hub for the modification and trafficking of cell surface and secreted proteins, allowing cancer cells to evade apoptosis and sustain proliferative signaling. Additionally, it can promote angiogenesis and shape the tumor microenvironment, namely by controlling extracellular matrix (ECM) composition and stromal cell behavior. By regulating the processing and maturation of critical signaling molecules, the GA can have a profound impact on the activation of key oncogenic pathways, thus driving the uncontrolled growth of cancer cells. Moreover, the GA plays a crucial role in the acquisition of invasive and metastatic capabilities by cancer cells. It modulates the synthesis and trafficking of most ECM components and of ECM-modifying enzymes such as matrix metalloproteinases (MMPs), which are crucial enzymes involved in ECM remodeling [7]. By facilitating the secretion and activation of this type of protein, the GA enables cancer cells to breach the physical barriers of the surrounding tissues [5] and initiate the metastatic cascade. Furthermore, the GA is involved in the dysregulated post-translational protein modifications, such as the glycosylation patterns, observed in cancer cells [8]. Aberrant glycosylation of proteins and lipids, catalyzed by GA-resident enzymes, leads to the generation of unique carbohydrate structures on the cell surface. These altered glycan structures play a significant role in cancer cell adhesion, migration, and immune evasion, further fueling tumor progression and metastasis [8]. The GA acts as a key regulator of these glycosylation processes, thus having a profound influence on the functional properties of cancer cells. Some less explored facets of this enigmatic organelle also have the potential to play a significant role in influencing tumor cells by impacting signal transduction, redox regulation, and ion homeostasis.
2. Pharmacological Approaches
2.1. Golgi Apparatus Trafficking Disrupting Agents
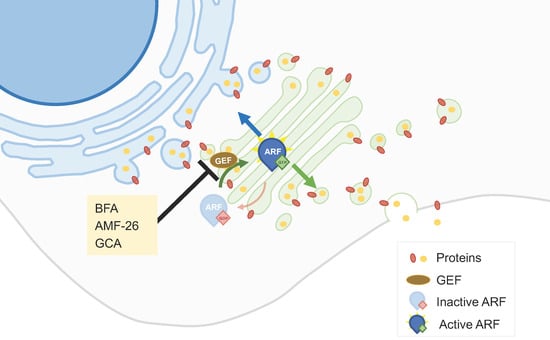
2.2. Golgi-Milieu-Disrupting Agents
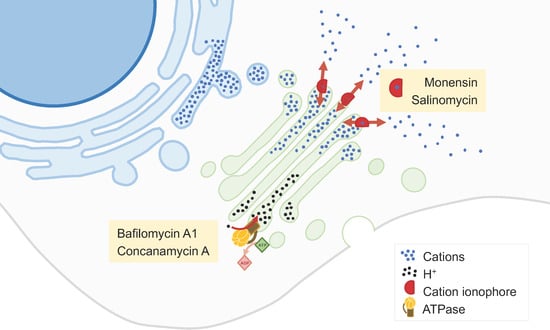
2.3. Inhibitors of the Synthetic Glycosilation Machinery
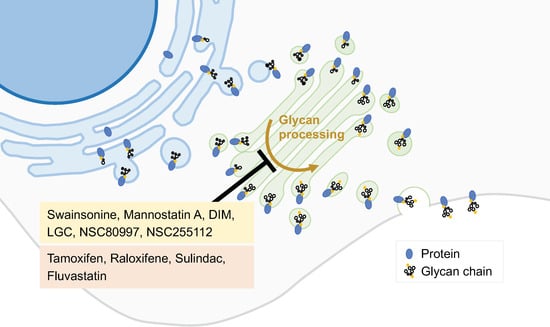
2.4. Inhibitors of Peripheral Golgi-Associated Proteins
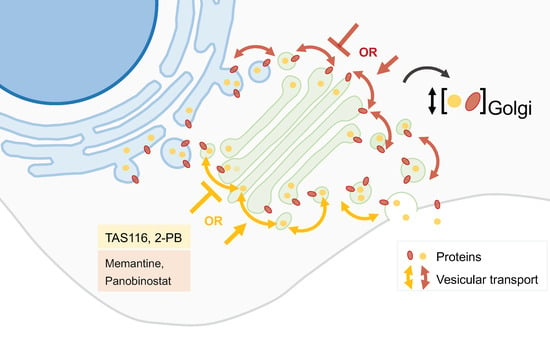
2.5. Drug Repurposing
3. Nanotechnological Approaches
3.1. Passive Targeting
3.2. Active Targeting
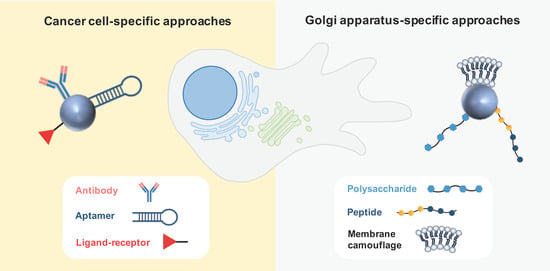
3.2.1. Cancer Cell-Specific Approaches
3.2.2. Golgi-Apparatus-Specific Approaches
3.3. Physical Targeting
4. Conclusions
Various nanotechnological approaches have been proposed for the delivery of GA-process-modulating drugs to cancer cells, making use of passive, active, and physical targeting strategies with successful outcomes in cellular and animal studies. In the last 4 years, elegant targeting approaches, making use of ligand–receptor interactions, transformable peptides, and membrane camouflage, have been suggested for the specific delivery of anticancer compounds to the GA. This may have only been the beginning of the field of organelle-specific targeting, which may be an asset to specifically modulate the GA functions with benefits in terms of anticancer efficacy, particularly related to the control of metastasis and chemoresistance. Thus, it is expected that some of these approaches may reach clinical trials in the near-future. Despite the results of the foreseen trials, questions remain regarding the potential translation of these nanoplatforms into clinical practice as these advanced delivery systems are associated with laborious and costly production methods. Nevertheless, the journey to develop a cost-effective, clinical-translatable, GA-targeting nanoplatform with remarkable anticancer efficacy and proper safety profiles is ongoing with promising initial breakthroughs.
References
- Klute, M.J.; Melancon, P.; Dacks, J.B. Evolution and Diversity of the Golgi. Cold Spring Harb. Perspect. Biol. 2011, 3, a007849.
- Ma, W.; Mayr, C. A Membraneless Organelle Associated with the Endoplasmic Reticulum Enables 3′UTR-Mediated Protein-Protein Interactions. Cell 2018, 175, 1492–1506.e19.
- Kremmidiotis, G. The Batten disease gene product (CLN3p) is a Golgi integral membrane protein. Hum. Mol. Genet. 1999, 8, 523–531.
- Crawford, K.; Leonenko, G.; Baker, E.; Grozeva, D.; Lan-Leung, B.; Holmans, P.; Williams, J.; O’Donovan, M.C.; Escott-Price, V.; Ivanov, D.K. Golgi apparatus, endoplasmic reticulum and mitochondrial function implicated in Alzheimer’s disease through polygenic risk and RNA sequencing. Mol. Psychiatry 2023, 28, 1327–1336.
- Almeida, N.; Carrara, G.; Palmeira, C.M.; Fernandes, A.S.; Parsons, M.; Smith, G.L.; Saraiva, N. Stimulation of cell invasion by the Golgi Ion Channel GAAP/TMBIM4 via an H2O2-Dependent Mechanism. Redox Biol. 2020, 28, 101361.
- Martins, M.; Fernandes, A.S.; Saraiva, N. GOLGI: Cancer cell fate control. Int. J. Biochem. Cell Biol. 2022, 145, 106174.
- Kajiho, H.; Kajiho, Y.; Frittoli, E.; Confalonieri, S.; Bertalot, G.; Viale, G.; Di Fiore, P.P.; Oldani, A.; Garre, M.; Beznoussenko, G.V.; et al. RAB2A controls MT1-MMP endocytic and E-cadherin polarized Golgi trafficking to promote invasive breast cancer programs. EMBO Rep. 2016, 17, 1061–1080.
- Magalhães, A.; Duarte, H.O.; Reis, C.A. Aberrant Glycosylation in Cancer: A Novel Molecular Mechanism Controlling Metastasis. Cancer Cell 2017, 31, 733–735.
- Donaldson, J.G.; Jackson, C.L. ARF family G proteins and their regulators: Roles in membrane transport, development and disease. Nat. Rev. Mol. Cell Biol. 2011, 12, 362–375.
- Mishev, K.; Dejonghe, W.; Russinova, E. Small Molecules for Dissecting Endomembrane Trafficking: A Cross-Systems View. Chem. Biol. 2013, 20, 475–486.
- Ohashi, Y.; Iijima, H.; Yamaotsu, N.; Yamazaki, K.; Sato, S.; Okamura, M.; Sugimoto, K.; Dan, S.; Hirono, S.; Yamori, T. AMF-26, a Novel Inhibitor of the Golgi System, Targeting ADP-ribosylation Factor 1 (Arf1) with Potential for Cancer Therapy. J. Biol. Chem. 2012, 287, 3885–3897.
- Shao, R.-G.; Shimizu, T.; Pommier, Y. Brefeldin A Is a Potent Inducer of Apoptosis in Human Cancer Cells Independently of p53. Exp. Cell Res. 1996, 227, 190–196.
- Satoh, M.; Ito, A.; Nojiri, H.; Handa, K.; Numahata, K.; Ohyama, C.; Saito, S.; Hoshi, S.; Hakomori, S.-I. Enhanced GM3 expression, associated with decreased invasiveness, is induced by brefeldin A in bladder cancer cells. Int. J. Oncol. 2001, 19, 723–731.
- Fu, D.; Bebawy, M.; Kable, E.P.W.; Roufogalis, B.D. Dynamic and intracellular trafficking of P-glycoprotein-EGFP fusion protein: Implications in multidrug resistance in cancer. Int. J. Cancer 2004, 109, 174–181.
- Zhao, Q.; Wu, M.; Zheng, X.; Yang, L.; Zhang, Z.; Li, X.; Chen, J. ERGIC3 Silencing Additively Enhances the Growth Inhibition of BFA on Lung Adenocarcinoma Cells. Curr. Cancer Drug Targets 2020, 20, 67–75.
- Shen, S.-M.; Ji, Y.; Zhang, C.; Dong, S.-S.; Yang, S.; Xiong, Z.; Ge, M.-K.; Yu, Y.; Xia, L.; Guo, M.; et al. Nuclear PTEN safeguards pre-mRNA splicing to link Golgi apparatus for its tumor suppressive role. Nat. Commun. 2018, 9, 2392.
- Sáenz, J.B.; Sun, W.J.; Chang, J.W.; Li, J.; Bursulaya, B.; Gray, N.S.; Haslam, D.B. Golgicide A reveals essential roles for GBF1 in Golgi assembly and function. Nat. Chem. Biol. 2009, 5, 157–165.
- Yang, X.-Z.; Cui, S.-Z.; Zeng, L.-S.; Cheng, T.-T.; Li, X.-X.; Chi, J.; Wang, R.; Zheng, X.F.S.; Wang, H.-Y. Overexpression of Rab1B and MMP9 predicts poor survival and good response to chemotherapy in patients with colorectal cancer. Aging 2017, 9, 914–931.
- Yao, M.; Wang, L.; Leung, P.S.C.; Li, Y.; Liu, S.; Wang, L.; Guo, X.; Zhou, G.; Yan, Y.; Guan, G.; et al. The Clinical Significance of GP73 in Immunologically Mediated Chronic Liver Diseases: Experimental Data and Literature Review. Clin. Rev. Allergy Immunol. 2018, 54, 282–294.
- Shiina, I.; Umezaki, Y.; Ohashi, Y.; Yamazaki, Y.; Dan, S.; Yamori, T. Total Synthesis of AMF-26, an Antitumor Agent for Inhibition of the Golgi System, Targeting ADP-Ribosylation Factor 1. J. Med. Chem. 2013, 56, 150–159.
- Del Giudice, S.; De Luca, V.; Parizadeh, S.; Russo, D.; Luini, A.; Di Martino, R. Endogenous and Exogenous Regulatory Signaling in the Secretory Pathway: Role of Golgi Signaling Molecules in Cancer. Front. Cell Dev. Biol. 2022, 10, 611.
- Tan, X.; Banerjee, P.; Pham, E.A.; Rutaganira, F.U.N.; Basu, K.; Bota-Rabassedas, N.; Guo, H.-F.; Grzeskowiak, C.L.; Liu, X.; Yu, J.; et al. PI4KIIIβ is a therapeutic target in chromosome 1q–amplified lung adenocarcinoma. Sci. Transl. Med. 2020, 12, eaax3772.
- Janardhanan, P.; Somasundaran, A.K.; Balakrishnan, A.J.; Pilankatta, R. Sensitization of cancer cells towards Cisplatin and Carboplatin by protein kinase D inhibitors through modulation of ATP7A/B (copper transport ATPases). Cancer Treat. Res. Commun. 2022, 32, 100613.
- Li, Q.Q.; Hsu, I.; Sanford, T.; Railkar, R.; Balaji, N.; Sourbier, C.; Vocke, C.; Balaji, K.C.; Agarwal, P.K. Protein kinase D inhibitor CRT0066101 suppresses bladder cancer growth in vitro and xenografts via blockade of the cell cycle at G2/M. Cell. Mol. Life Sci. 2018, 75, 939–963.
- Zhang, M.; Xu, N.; Xu, W.; Ling, G.; Zhang, P. Potential therapies and diagnosis based on Golgi-targeted nano drug delivery systems. Pharmacol. Res. 2022, 175, 105861.
- Vanneste, M.; Huang, Q.; Li, M.; Moose, D.; Zhao, L.; Stamnes, M.A.; Schultz, M.; Wu, M.; Henry, M.D. High content screening identifies monensin as an EMT-selective cytotoxic compound. Sci. Rep. 2019, 9, 1200.
- Wang, H.; Zhang, H.; Zhu, Y.; Wu, Z.; Cui, C.; Cai, F. Anticancer Mechanisms of Salinomycin in Breast Cancer and Its Clinical Applications. Front. Oncol. 2021, 11, 654428.
- Marjanović, M.; Mikecin Dražić, A.-M.; Mioč, M.; Paradžik, M.; Kliček, F.; Novokmet, M.; Lauc, G.; Kralj, M. Salinomycin disturbs Golgi function and specifically affects cells in epithelial-to-mesenchymal transition. J. Cell Sci. 2023, 136, jcs260934.
- Nguyen, S.L.T.; Nguyen, T.C.; Do, T.T.; Vu, T.L.; Nguyen, T.T.; Do, T.T.; Nguyen, T.H.T.; Le, T.H.; Trinh, D.K.; Nguyen, T.A.T. Study on the Anticancer Activity of Prodigiosin from Variants of Serratia Marcescens QBN VTCC 910026. Biomed Res. Int. 2022, 2022, 4053074.
- Chen, F.; Kang, R.; Liu, J.; Tang, D. The V-ATPases in cancer and cell death. Cancer Gene Ther. 2022, 29, 1529–1541.
- Lu, X.; Chen, L.; Chen, Y.; Shao, Q.; Qin, W. Bafilomycin A1 inhibits the growth and metastatic potential of the BEL-7402 liver cancer and HO-8910 ovarian cancer cell lines and induces alterations in their microRNA expression. Exp. Ther. Med. 2015, 10, 1829–1834.
- Lee, Z.Y.; Loo, J.S.E.; Wibowo, A.; Mohammat, M.F.; Foo, J.B. Targeting cancer via Golgi α-mannosidase II inhibition: How far have we come in developing effective inhibitors? Carbohydr. Res. 2021, 508, 108395.
- Obata, Y.; Horikawa, K.; Takahashi, T.; Akieda, Y.; Tsujimoto, M.; Fletcher, J.A.; Esumi, H.; Nishida, T.; Abe, R. Oncogenic signaling by Kit tyrosine kinase occurs selectively on the Golgi apparatus in gastrointestinal stromal tumors. Oncogene 2017, 36, 3661–3672.
- Saito, Y.; Takahashi, T.; Obata, Y.; Nishida, T.; Ohkubo, S.; Nakagawa, F.; Serada, S.; Fujimoto, M.; Ohkawara, T.; Nishigaki, T.; et al. TAS-116 inhibits oncogenic KIT signalling on the Golgi in both imatinib-naïve and imatinib-resistant gastrointestinal stromal tumours. Br. J. Cancer 2020, 122, 658–667.
- Lio, S.C.; Johnson, J.; Chatterjee, A.; Ludwig, J.W.; Millis, D.; Banie, H.; Sircar, J.C.; Sinha, A.; Richards, M.L. Disruption of Golgi processing by 2-phenyl benzimidazole analogs blocks cell proliferation and slows tumor growth. Cancer Chemother. Pharmacol. 2008, 61, 1045–1058.
- Koyama, R.; Hakamata, W.; Hirano, T.; Nishio, T. Identification of Small-Molecule Inhibitors of Human Golgi Mannosidase via a Drug Repositioning Screen. Chem. Pharm. Bull. 2018, 66, 678–681.
- Yu, R.; Longo, J.; van Leeuwen, J.E.; Zhang, C.; Branchard, E.; Elbaz, M.; Cescon, D.W.; Drake, R.R.; Dennis, J.W.; Penn, L.Z. Mevalonate Pathway Inhibition Slows Breast Cancer Metastasis via Reduced N -glycosylation Abundance and Branching. Cancer Res. 2021, 81, 2625–2635.
- Tiwari, H.; Rai, N.; Singh, S.; Gupta, P.; Verma, A.; Singh, A.K.; Kajal; Salvi, P.; Singh, S.K.; Gautam, V. Recent Advances in Nanomaterials-Based Targeted Drug Delivery for Preclinical Cancer Diagnosis and Therapeutics. Bioengineering 2023, 10, 760.
- Yu, R.-Y.; Xing, L.; Cui, P.-F.; Qiao, J.-B.; He, Y.-J.; Chang, X.; Zhou, T.-J.; Jin, Q.-R.; Jiang, H.-L.; Xiao, Y. Regulating the Golgi apparatus by co-delivery of a COX-2 inhibitor and Brefeldin A for suppression of tumor metastasis. Biomater. Sci. 2018, 6, 2144–2155.
- Liu, W.; Wei, J.; Huo, P.; Lu, Y.; Chen, Y.; Wei, Y. Controlled release of brefeldin A from electrospun PEG–PLLA nanofibers and their in vitro antitumor activity against HepG2 cells. Mater. Sci. Eng. C 2013, 33, 2513–2518.
- Basu, S.M.; Yadava, S.K.; Singh, R.; Giri, J. Lipid nanocapsules co-encapsulating paclitaxel and salinomycin for eradicating breast cancer and cancer stem cells. Colloids Surfaces B Biointerfaces 2021, 204, 111775.
- Gao, J.; Liu, J.; Xie, F.; Lu, Y.; Yin, C.; Shen, X. Co-Delivery of Docetaxel and Salinomycin to Target Both Breast Cancer Cells and Stem Cells by PLGA/TPGS Nanoparticles. Int. J. Nanomed. 2019, 14, 9199–9216.
- Anees, M.; Mehrotra, N.; Tiwari, S.; Kumar, D.; Kharbanda, S.; Singh, H. Polylactic acid based biodegradable hybrid block copolymeric nanoparticle mediated co-delivery of salinomycin and doxorubicin for cancer therapy. Int. J. Pharm. 2023, 635, 122779.
- Norouzi, M.; Abdali, Z.; Liu, S.; Miller, D.W. Salinomycin-loaded Nanofibers for Glioblastoma Therapy. Sci. Rep. 2018, 8, 9377.
- Zhang, Y.; Zhang, H.; Wang, X.; Wang, J.; Zhang, X.; Zhang, Q. The eradication of breast cancer and cancer stem cells using octreotide modified paclitaxel active targeting micelles and salinomycin passive targeting micelles. Biomaterials 2012, 33, 679–691.
- Tefas, L.R.; Toma, I.; Sesarman, A.; Banciu, M.; Jurj, A.; Berindan-Neagoe, I.; Rus, L.; Stiufiuc, R.; Tomuta, I. Co-delivery of gemcitabine and salinomycin in PEGylated liposomes for enhanced anticancer efficacy against colorectal cancer. J. Liposome Res. 2023, 33, 234–250.
- Singh, M.; Griffin, T.; Salimi, A.; Micetich, R.G.; Atwal, H. Potentiation of ricin A immunotoxin by monoclonal antibody targeted monensin containing small unilamellar vesicles. Cancer Lett. 1994, 84, 15–21.
- Li, H.; Zhang, P.; Luo, J.; Hu, D.; Huang, Y.; Zhang, Z.-R.; Fu, Y.; Gong, T. Chondroitin Sulfate-Linked Prodrug Nanoparticles Target the Golgi Apparatus for Cancer Metastasis Treatment. ACS Nano 2019, 13, 9386–9396.
- Luo, J.; Gong, T.; Ma, L. Chondroitin-modified lipid nanoparticles target the Golgi to degrade extracellular matrix for liver cancer management. Carbohydr. Polym. 2020, 249, 116887.
- Zhang, C.; Yan, L.; Wang, X.; Zhu, S.; Chen, C.; Gu, Z.; Zhao, Y. Progress, challenges, and future of nanomedicine. Nano Today 2020, 35, 101008.
- Xue, F.; Wen, Y.; Wei, P.; Gao, Y.; Zhou, Z.; Xiao, S.; Yi, T. A smart drug: A pH-responsive photothermal ablation agent for Golgi apparatus activated cancer therapy. Chem. Commun. 2017, 53, 6424–6427.

