Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mohammed Aufy | -- | 2662 | 2023-12-29 07:56:23 | | | |
| 2 | Fanny Huang | Meta information modification | 2662 | 2023-12-29 10:17:19 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Aufy, M.; Hussein, A.M.; Stojanovic, T.; Studenik, C.R.; Kotob, M.H. Proteolytic Activation of the Epithelial Sodium Channel. Encyclopedia. Available online: https://encyclopedia.pub/entry/53258 (accessed on 10 August 2026).
Aufy M, Hussein AM, Stojanovic T, Studenik CR, Kotob MH. Proteolytic Activation of the Epithelial Sodium Channel. Encyclopedia. Available at: https://encyclopedia.pub/entry/53258. Accessed August 10, 2026.
Aufy, Mohammed, Ahmed M. Hussein, Tamara Stojanovic, Christian R. Studenik, Mohamed H. Kotob. "Proteolytic Activation of the Epithelial Sodium Channel" Encyclopedia, https://encyclopedia.pub/entry/53258 (accessed August 10, 2026).
Aufy, M., Hussein, A.M., Stojanovic, T., Studenik, C.R., & Kotob, M.H. (2023, December 29). Proteolytic Activation of the Epithelial Sodium Channel. In Encyclopedia. https://encyclopedia.pub/entry/53258
Aufy, Mohammed, et al. "Proteolytic Activation of the Epithelial Sodium Channel." Encyclopedia. Web. 29 December, 2023.
Copy Citation
Epithelial sodium channel (ENaC) are integral to maintaining salt and water homeostasis in various biological tissues, including the kidney, lung, and colon. They enable the selective reabsorption of sodium ions, which is a process critical for controlling blood pressure, electrolyte balance, and overall fluid volume. ENaC activity is finely controlled through proteolytic activation, a process wherein specific enzymes, or proteases, cleave ENaC subunits, resulting in channel activation and increased sodium reabsorption. This regulatory mechanism plays a pivotal role in adapting sodium transport to different physiological conditions.
ENaC
epithelial sodium channel
ENaC activation
proteases
1. Introduction
Epithelial sodium channel (ENaC) are integral membrane proteins that play a crucial role in the regulation of sodium reabsorption in various epithelial tissues [1][2]. ENaC consist of the following three homologous subunits: α, β, and γ (Figure 1), which form a functional channel complex responsible for the selective transport of sodium ions across the epithelial cell membranes [3][4]. The activity of ENaC is tightly regulated to maintain sodium balance. In addition, certain hormones play an important role in ENaC activation. Angiotensin is another example of the hormonal activation of ENaC. Another example of the hormonal regulation of ENaC is AVP, also referred to as vasopressin or the antidiuretic hormone, which primarily plays a key role in regulating water balance and blood pressure within the body. An additional important regulatory mechanism involved in ENaC activity is proteolytic activation by proteases [5]. The significance of proteolytic activation in regulating ENaC activity is underscored by the fact that the dysregulation of this process can lead to various pathological conditions [6][7].
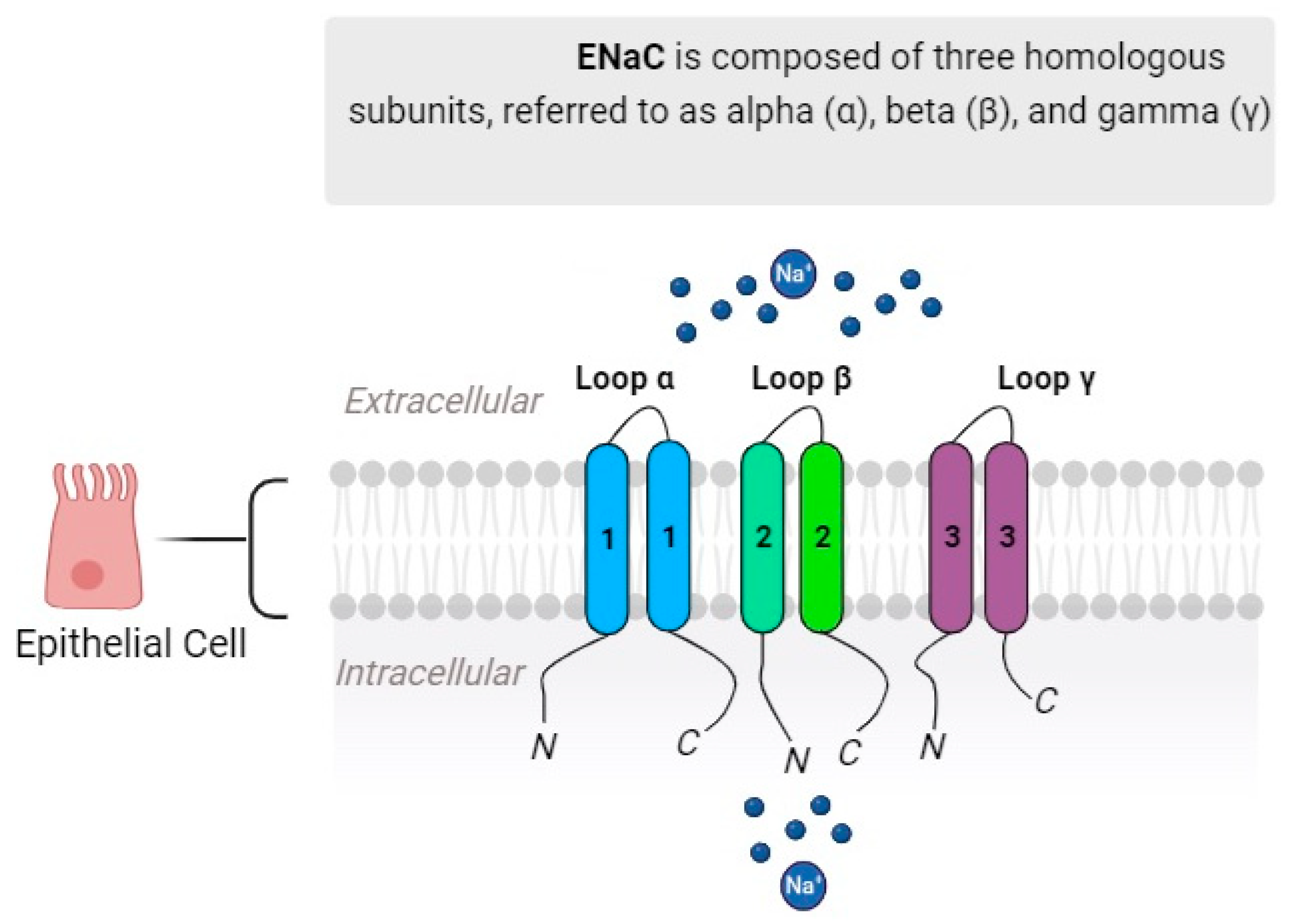
Figure 1. Canonical ENaC is composed of three homologous subunits referred to as alpha (α), beta (β), and gamma (γ). Together, these subunits combine to create a functional channel with multiple components, forming a heteromultimeric structure.
2. Proteolytic Activation of ENaC
2.1. Furin-like Convertases
Furin-like convertases are a family of proteases involved in the proteolytic activation of ENaC [8]. These convertases cleave the extracellular domains of ENaC subunits, resulting in channel activation [3]. The α and γ subunits of ENaC contain two and one furin cleavage consensus sites, respectively (Figure 2), and the mutation or deletion of any of these sites impairs channel activation [9]. Furin-like convertases, including furin itself and PC5/6, have emerged as pivotal regulators of ENaC activity in various tissues, including the kidney and lung [10]. The furin inhibitor BOS-318, through the selective inhibition of furin, effectively and markedly suppresses ENaC-mediated sodium transport in differentiated human bronchial epithelial cells. This inhibition was observed in both short-term (IC50 = 263.0 nM) and long-term (IC50 = 17.4 nM) treatment conditions [11], corroborating with previous investigations that indicate reduced basal ENaC activity following furin deficiency or inhibition [12][13]. The attenuation of ENaC activity has been correlated with favorable outcomes related to airway hydration and mucociliary clearance. It has been demonstrated that BOS-318-mediated ENaC inhibition leads to a simultaneous elevation in the height of the airway surface liquid and increased rates of mucociliary clearance in cystic fibrosis airway epithelial cells [11]. These collective findings suggest that furin targeting has the promising potential to offer therapeutic advantages in the management of cystic fibrosis.
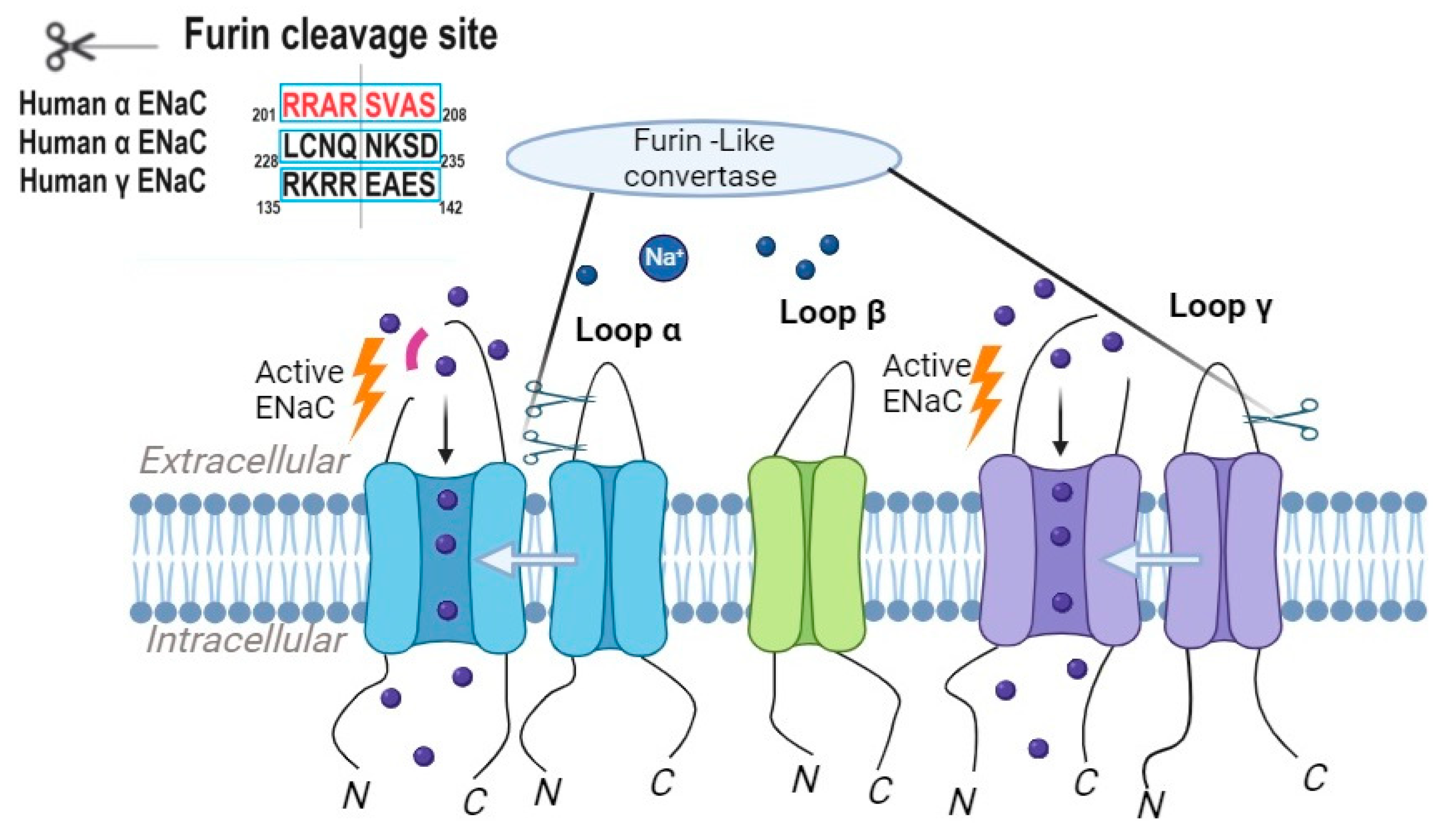
Figure 2. Proteolytic cleavage triggers the activation of ENaC, with furin initiating this process by cleaving the α subunit of ENaC twice and the γ subunit once.
2.2. Tissue Kallikreins
Other examples of key players in ENaC processing are tissue kallikreins, which are a family of serine proteases that are involved in the proteolytic activation of ENaC. Tissue kallikreins cleave the γ subunit of ENaC, leading to an increased channel open probability [14]. These proteases are expressed in various tissues, including the kidney and airway epithelia, and are implicated in the regulation of sodium reabsorption [15].
Tissue kallikrein knockout mice (TK−/−) are genetically modified mice where the TK gene is disrupted or deleted, resulting in reduced or absent TK activity and impaired γ-ENaC processing. When membrane proteins from the renal cortex of TK−/− mice are incubated with TK in an in vitro setting, a 70 kDa band of gamma-ENaC appears [14]. This finding indicates that TK can promote γ-ENaC cleavage in vitro. In other words, the introduction of TK in an in vitro environment partially restores the processing of gamma-ENaC [14].
Among the tissue kallikreins, Kallikrein 1 (KLK1) has emerged as a key protease involved in the proteolytic activation of ENaC. KLK1 has been demonstrated to enhance the cleavage of γ-ENaC in the native collecting duct (CD), suggesting the potential direct involvement of this endogenous protease in the regulation of sodium (Na+) balance. This finding hints at the role of KLK1 in modulating γ-ENaC activity within the CD, which could impact sodium reabsorption and, subsequently, sodium homeostasis in the body [16]. This discovery may have implications for understanding and potentially managing the conditions related to sodium regulation, such as hypertension and electrolyte imbalances, although further research is necessary to confirm and fully comprehend these implications. In the lung, Kallikrein is predominantly produced by bronchial epithelial cells and has been implicated in the regulation of ENaC activity [14].
Regulation of KLK-Mediated ENaC Activation by Hormonal and Signaling Pathways
The activity of KLK7 in mediating ENaC proteolytic processing is regulated by hormonal and signaling pathways. One important regulator of KLK activity is the serine protease inhibitor LEKTI (lympho-epithelial Kazal-type-related inhibitor) [17]. LEKTI binds to and inhibits KLK, preventing its proteolytic activity on ENaC [17]. This regulatory interaction ensures precise control over ENaC activation via KLK.
Furthermore, several hormonal and signaling pathways have been shown to modulate KLK expression and activity, thereby influencing ENaC activation. For instance, the glucocorticoid hormone cortisol has been found to upregulate KLK expression in airway epithelial cells, leading to increased ENaC proteolytic processing and sodium transport [18]. This hormonal regulation of KLK provides a mechanism for the fine-tuning of ENaC activity in response to physiological and pathological conditions.
Overall, the proteolytic activation of ENaC by tissue kallikreins is subjected to regulation by various hormonal and signaling pathways. The presence of serine protease inhibitors, such as LEKTI, helps to fine-tune the activity of KLK and ensure precise control over ENaC activation. Hormones like cortisol have been shown to influence KLK expression and activity, thereby modulating ENaC proteolytic processing and sodium transport.
In conclusion, tissue kallikreins, particularly KLK, play a crucial role in the proteolytic activation of ENaC. Their ability to cleave the γ subunit of ENaC enhances the channel’s open probability and facilitates sodium reabsorption in various tissues. KLK activity is regulated by serine protease inhibitors and influenced by hormonal and signaling pathways. The dysregulation of KLK-mediated ENaC activation can contribute to the development of electrolyte imbalances and respiratory disorders. Further research is warranted to elucidate the intricate regulatory mechanisms involved in exploring the therapeutic potential of targeting KLK and related pathways for the treatment of ENaC-related disorders.
2.3. Prostasin
Prostasin, also known as channel-activating protease-1 (CAP1), a glycosylphosphatidylinositol-anchored serine protease, is present in the prostate gland, kidney, bronchi, colon, liver, lung, pancreas, and salivary glands. Its substrate specificity is trypsin-like [19]. It has been identified in nuclear and membrane fractions [20]. Prostasin’s activation of ENaC occurs through the cleavage of the γ-subunit, resulting in the release of an inhibitory peptide from the extracellular domain. The serine protease prostasin enhances ENaC activation by initiating the cleavage of the γ- subunit at a site located away from the furin cleavage site (Figure 3). Furin cleaves gamma ENaC at site 43, initiating a key molecular event. Subsequently, prostasin further refines this process by cleaving γ ENaC at site 186. ENaC channels that lack this specific furin and/or prostasin cleavage site exhibit increased activity, resulting in a higher open probability. Additionally, a synthetic peptide is designed to match the fragment cleaved from the gamma subunit functions as a reversible inhibitor of native ENaC in mouse cortical-collecting duct cells and primary cultures of human airway epithelial cells [19].
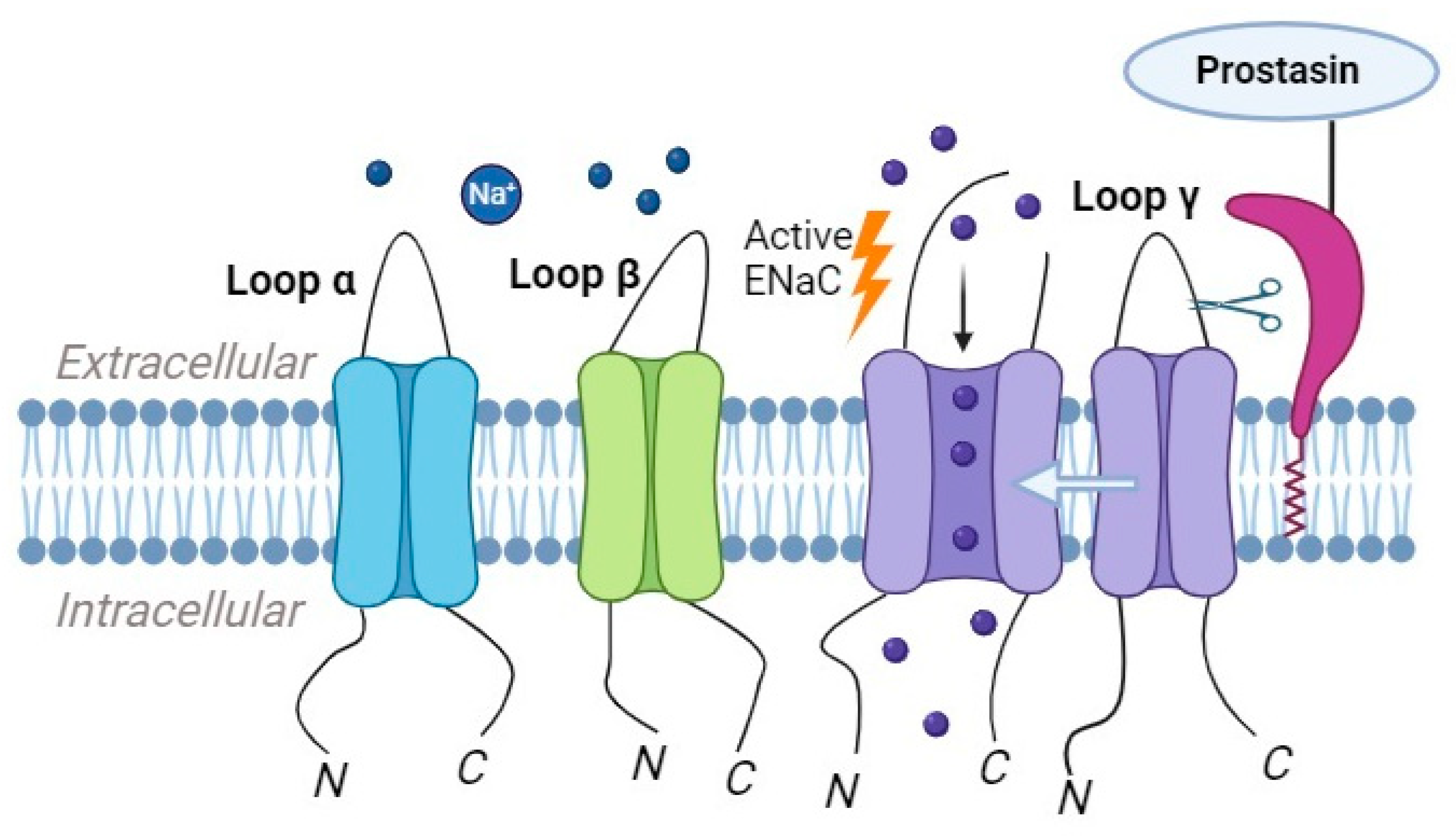
Figure 3. Prostasin has the capability to cleave ENaC, leading to the subsequent cleavage of the γ-subunit, which results in the release of an inhibitory peptide.
The activation of prostasin and its subsequent effect on ENaC activity are tightly regulated. Prostasin is initially synthesized as an inactive zymogen, and its activation involves the proteolytic cleavage of the propeptide domain. This cleavage can be mediated by other proteases, such as matriptase, which activates prostasin by cleaving to the propeptide domain [21].
Once activated, prostasin interacts with ENaC, leading to the proteolytic processing of the γ subunit RKRK186 site [22]. This processing enhances the channel’s open probability and promotes sodium transport. The exact mechanisms by which prostasin activates ENaC are not fully understood, but it is believed to involve changes in channel gating properties and interactions with other regulatory proteins [18].
The targeted deletion of prostasin in specific tissues, such as the lung and colon, in adult epithelial phenotypes has provided clear evidence implicating prostasin in ENaC-mediated sodium transport [23][24][25]. However, this association was not observed in the skin [25][26][27][28]. Notably, mice with an alveolar-specific prostasin knockout exhibited a 40% reduction in the ENaC-mediated Na+ current, leading to impaired alveolar fluid clearance. Nevertheless, unchallenged mice did not show alveolar edema, alterations in lung morphology, or changes in their tight junction protein abundance; these effects only manifested under increased hydrostatic pressure [23]. Similarly, a decrease in ENaC activity was noted in colon-specific prostasin knockout mice.
2.4. Plasmin as a Regulator of ENaC Activity
Plasmin, as a highly potent and reactive serine protease, exhibits key roles in several physiological processes, such as thrombolysis, embryogenesis, cancer progression, and wound healing [29]. It is another serine protease that is implicated in the regulation of ENaC activity. This enzyme is derived from plasminogen through the action of plasminogen activators, such as the tissue-type plasminogen activator (tPA) or urokinase-type plasminogen activator (uPA) [21]. Plasmin can directly cleave the γ subunit of ENaC, leading to an increase in the channel’s open probability and enhanced sodium reabsorption (Figure 4) [30].
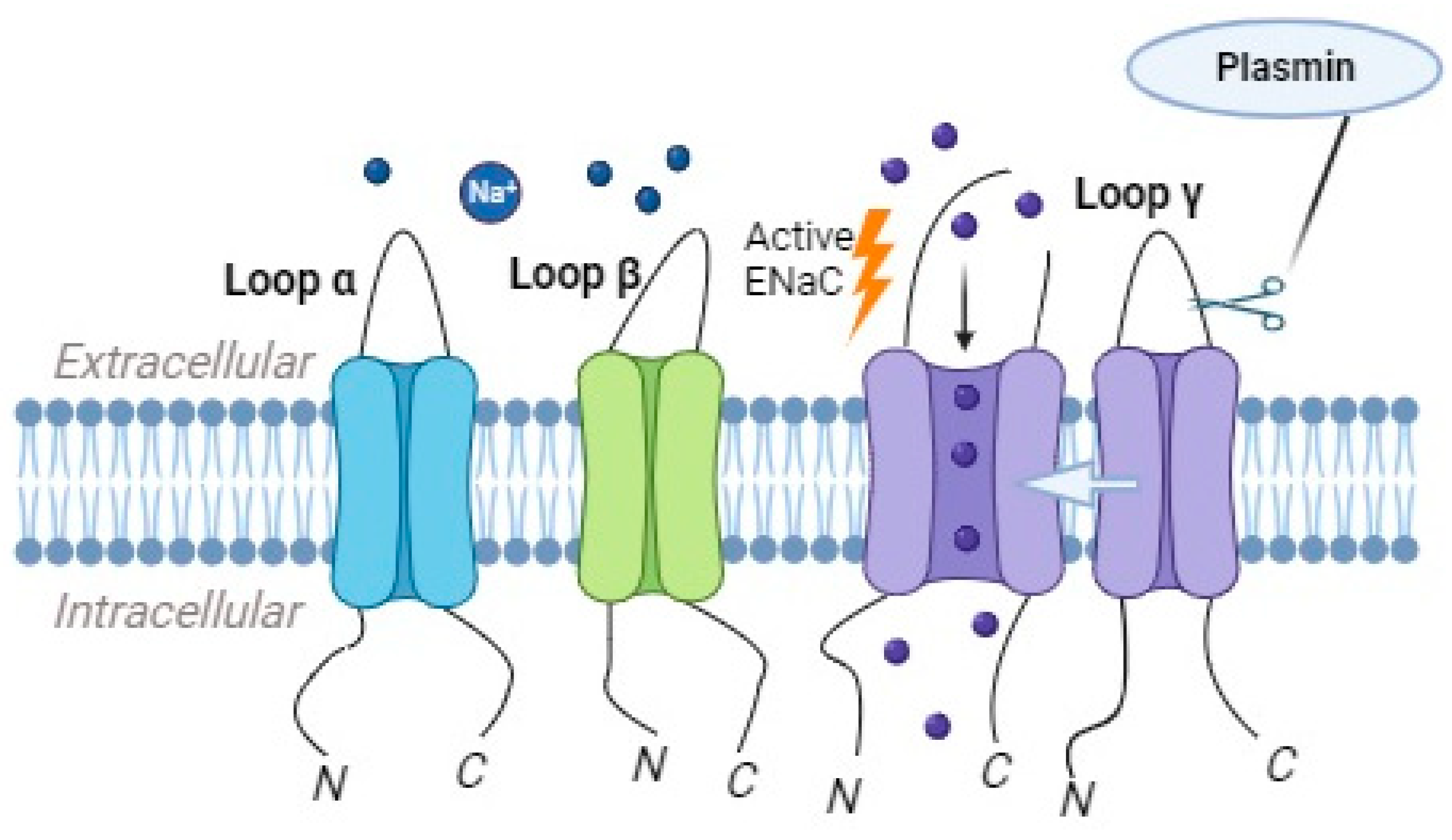
Figure 4. Plasmin has the ability to directly cleave the γ subunit of the epithelial sodium channel (ENaC), which results in the increased likelihood of the channel being open and consequently enhances the reabsorption of sodium ions.
The activation of plasminogen and the subsequent generation of plasmin are tightly regulated by the presence of plasminogen activator inhibitors (PAIs) and other regulatory factors. The balance between plasminogen activators and their inhibitors determines the level of plasmin activity and its impact on the ENaC’s function.
In nephrotic syndrome, the involvement of plasmin in activating ENaC serves as a notable illustration. The suppression of plasma aldosterone in nephrotic syndrome has prompted investigations into the alternative pathway of epithelial sodium channel (ENaC) activation through extracellular proteolysis [31][32]. Urine samples from nephrotic syndrome patients have been found to contain soluble proteolytic activity, with plasmin identified as the major protease aberrantly filtrated from the plasma as a plasminogen [33][34]. In vitro studies have shown that exposure to nephrotic urine containing plasmin or purified plasmin enhances an inward amiloride-sensitive current in collecting duct cells [33][35]. Plasmin has been demonstrated to release an inhibitory peptide tract from the exodomain of the γENaC subunit (Figure 4), either through direct cleavage at high concentrations or via Glycosylphophatidylinositol-anchored prostasin at low concentrations [35][36]. The addition of α2-antiplasmin and aprotinin has been shown to inhibit the ability of nephrotic urine and plasmin to evoke a current [35][36]. Furthermore, the in vitro cleavage of γENaC corresponds to a distinct shift in the migratory pattern of renal tissue γENaC to lower molecular weight isoforms on SDS-PAGE gels in proteinuric conditions [37][38]. The presence of plasminogen immunoreactivity in the urine of preeclampsia (PE) patients with preeclampsia, compared to uncomplicated pregnancies, has prompted the hypothesis that manifest preeclampsia may be linked to plasmin-dependent protease activity in urine [39]. This activity is believed to have the potential to enhance epithelial ENaC activity. The plasmin catalytic activity in PE patients was elevated by about 40-fold compared to normal individuals [39].
Plasmin has been involved in various physiological and pathological processes, including inflammation and tissue remodeling. Its role in ENaC activation suggests that it may contribute to sodium and fluid balance in certain tissues, such as the kidney and lungs.
2.5. The Role of Different Cathepsins in ENaC Processing and Activation
Cathepsins are a family of lysosomal proteases involved in the degradation and processing of various proteins [40]. Emerging evidence suggests that specific cathepsins play a role in the processing and activation of ENaC, contributing to the regulation of sodium reabsorption and electrolyte balance [41].
Cathepsins B and S have been identified as key players in ENaC processing and activation. These cathepsins belong to the cysteine protease family and are predominantly localized in endosomes and lysosomes within the cells. Their proteolytic activity can modulate the function and activity of ENaC [42][43][44].
Cathepsin B, in particular, has been implicated in the proteolytic cleavage of the α subunit of ENaC (Figure 5). The cleavage of the α subunit via cathepsin B leads to increased ENaC activity and enhanced sodium transport. Studies have shown that inhibiting cathepsin B’s activity results in reduced ENaC activity and impaired sodium reabsorption [43].
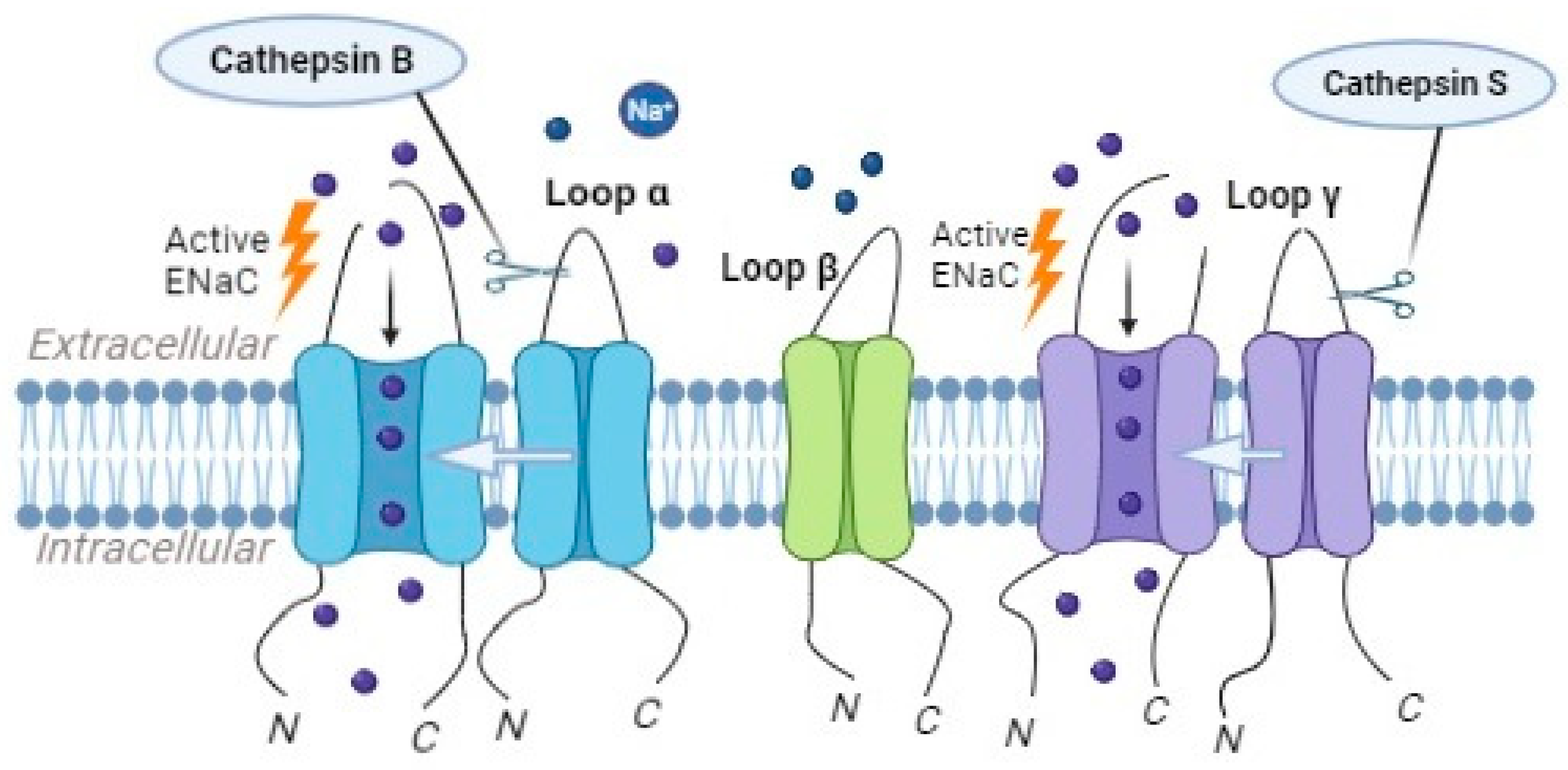
Figure 5. Cathepsin B can activate ENaC, causing the α-subunit to be cleaved, while cathepsin S triggers ENaC activation by cleaving the γ-subunit.
Individuals with nephrotic syndrome frequently exhibit signs of fluid accumulation, such as the development of edema or hypertension. The fundamental disruption in sodium regulation revolves around the improper stimulation of ENaC [41].
In the initial stages of sodium retention in NS, a comprehensive examination revealed the heightened expression of full-length ENaC subunits and the cleaved product of αENaC, accompanied by an increase in ENaC activity, as demonstrated through amiloride application. There was also an upregulation in the expression of Na+/K+-ATPase in the collecting duct. Urinary proteolytic activity showed an elevation, and mass spectrometry identified several proteases, including cathepsin B, which was determined to be involved in the processing of αENaC. The renal levels of both precursor and active cathepsin B were elevated, with localization observed in glomeruli and intercalated cells. Hypertension was prevented by inhibiting cathepsin B. In conclusion, the identified mechanism involves the cathepsin B-induced processing of αENaC, resulting in heightened channel activity and the development of hypertension [41].
Cystic fibrosis is another disease that shows the combination of ENaC functioning and cathepsin B. In both normal and cystic fibrosis human bronchial epithelial cultures, cathepsin B was found in the apical plasma membrane and the airway surface liquid (ASL). Cathepsin B activity was notably higher in acidic ASL, correlating with an increased presence of ENaC in the plasma membrane and a decrease in the ASL volume. The activation of ENaC, influenced by acid/CTSB, was mitigated by the cell-impermeable, cathepsin B-selective inhibitor CA074, suggesting that inhibiting CTSB might be therapeutically beneficial.
Given that cathepsins primarily reside in lysosomal/endosomal compartments, the site of ENaC processing by cathepsin B poses a challenge. One investigation proposed multiple mechanisms to elucidate the location of ENaC processing by cathepsin B [41]. Cathepsin B undergoes post-translational processing in the rough endoplasmic reticulum, Golgi, endosomal, and lysosomal compartments. Additionally, cathepsin B is secreted into the extracellular space by various tumor epithelial cells. Considering these aspects and in light of the findings that cathepsin B is secreted on the apical side of some epithelial cells, the following several possibilities were considered: cathepsin B cleaving ENaC within the Golgi during post-translational modifications of cathepsin B and ENaC assembly, extracellular cleavage where ENaC is in the apical membrane, or cleavage within endosomes during ENaC recycling [41].
Cathepsin S, another member of the cathepsin family, has been suggested to play a role in the regulation of ENaC activity. Studies have demonstrated that cathepsin S can cleave the γ subunits of ENaC (Figure 5), leading to an increase in the channel’s opening probability and enhanced sodium reabsorption [42]. Mutations in the critical region of γENaC processing by cathepsin S, specifically at valine residues V182 and V193, prevented the proteolytic activation of ENaC by Cathepsin S [42].
The precise regulation and specific roles of cathepsins in ENaC processing and activation are not yet fully understood. However, it is clear that cathepsins, in addition to other proteases, contribute to the proteolytic regulation of ENaC, influencing sodium transport and electrolyte balance.
The activity of cathepsins and their impact on ENaC processing can be modulated by various factors. For example, changes in the pH within the endosomal–lysosomal compartments can influence cathepsin activity and subsequent ENaC activation. Additionally, the presence of endogenous inhibitors, such as cystatins, can regulate cathepsin activity and prevent excessive proteolysis [45].
References
- Blass, G.; Klemens, C.A.; Brands, M.W.; Palygin, O.; Staruschenko, A. Postprandial Effects on ENaC-Mediated Sodium Absorption. Sci. Rep. 2019, 9, 4296.
- Shabbir, W.; Topcagic, N.; Aufy, M. Activation of autosomal recessive Pseudohypoaldosteronism1 ENaC with aldosterone. Eur. J. Pharmacol. 2021, 901, 174090.
- Frindt, G.; Meyerson, J.R.; Satty, A.; Scandura, J.M.; Palmer, L.G. Expression of ENaC subunits in epithelia. J. Gen. Physiol. 2022, 154, e202213124.
- Hanukoglu, I.; Hanukoglu, A. Epithelial sodium channel (ENaC) family: Phylogeny, structure-function, tissue distribution, and associated inherited diseases. Gene 2016, 579, 95–132.
- Vallet, V.; Chraibi, A.; Gaeggeler, H.P.; Horisberger, J.D.; Rossier, B.C. An epithelial serine protease activates the amiloride-sensitive sodium channel. Nature 1997, 389, 607–610.
- Anand, D.; Hummler, E.; Rickman, O.J. ENaC activation by proteases. Acta Physiol. 2022, 235, e13811.
- Kleyman, T.R.; Myerburg, M.M.; Hughey, R.P. Regulation of ENaCs by proteases: An increasingly complex story. Kidney Int. 2006, 70, 1391–1392.
- Kota, P.; Gentzsch, M.; Dang, Y.L.; Boucher, R.C.; Stutts, M.J. The N terminus of α-ENaC mediates ENaC cleavage and activation by furin. J. Gen. Physiol. 2018, 150, 1179–1187.
- Sheng, S.; Carattino, M.D.; Bruns, J.B.; Hughey, R.P.; Kleyman, T.R. Furin cleavage activates the epithelial Na+ channel by relieving Na+ self-inhibition. Am. J. Physiol. Renal Physiol. 2006, 290, F1488–F1496.
- Harris, M.; Garcia-Caballero, A.; Stutts, M.J.; Firsov, D.; Rossier, B.C. Preferential assembly of epithelial sodium channel (ENaC) subunits in Xenopus oocytes: Role of furin-mediated endogenous proteolysis. J. Biol. Chem. 2008, 283, 7455–7463.
- Douglas, L.E.J.; Reihill, J.A.; Ho, M.W.Y.; Axten, J.M.; Campobasso, N.; Schneck, J.L.; Rendina, A.R.; Wilcoxen, K.M.; Martin, S.L. A highly selective, cell-permeable furin inhibitor BOS-318 rescues key features of cystic fibrosis airway disease. Cell Chem. Biol. 2022, 29, 947–957.e8.
- Douglas, L.E.J.; Reihill, J.A.; Montgomery, B.M.; Martin, S.L. Furin as a therapeutic target in cystic fibrosis airways disease. Eur. Respir. Rev. 2023, 32, 220256.
- Reihill, J.A.; Walker, B.; Hamilton, R.A.; Ferguson, T.E.; Elborn, J.S.; Stutts, M.J.; Harvey, B.J.; Saint-Criq, V.; Hendrick, S.M.; Martin, S.L. Inhibition of protease-epithelial sodium channel signaling improves mucociliary function in cystic fibrosis airways. Am. J. Respir. Crit. Care Med. 2016, 194, 701710.
- Picard, N.; Eladari, D.; El Moghrabi, S.; Planès, C.; Bourgeois, S.; Houillier, P.; Wang, Q.; Burnier, M.; Deschenes, G.; Knepper, M.A.; et al. Defective ENaC processing and function in tissue kallikrein-deficient mice. J. Biol. Chem. 2008, 283, 4602–4611.
- Patel, A.B.; Chao, J.; Palmer, L.G. Tissue kallikrein activation of the epithelial Na channel. Am. J. Physiol. Renal Physiol. 2012, 303, F540–F550.
- Gondzik, V.; Weber, W.M.; Awayda, M.S. Coupling of epithelial Na+ and Cl- channels by direct and indirect activation by serine proteases. Am. J. Physiol. Cell Physiol. 2012, 303, C936–C946.
- Antalis, T.M.; Buzza, M.S. Extracellular: Plasma Membrane Proteases—Serine Proteases. Encycl. Cell Biol. 2016, 650–660.
- Miller, G.S.; List, K. The matriptase-prostasin proteolytic cascade in epithelial development and pathology. Cell Tissue Res. 2013, 351, 245–253.
- Aggarwal, S.; Dabla, P.K.; Arora, S. Prostasin: An Epithelial Sodium Channel Regulator. J. Biomark. 2013, 2013, 179864.
- Chen, L.M.; Skinner, M.L.; Kauffman, S.W.; Chao, J.; Chao, L.; Thaler, C.D.; Chai, K.X. Prostasin is a glycosylphosphatidylinositol-anchored active serine protease. J. Biol. Chem. 2001, 276, 21434–21442.
- Keragala, C.B.; Medcalf, R.L. Plasminogen: An enigmatic zymogen. Blood 2021, 137, 2881–2889.
- Carattino, M.D.; Mueller, G.M.; Palmer, L.G.; Frindt, G.; Rued, A.C.; Hughey, R.P.; Kleyman, T.R. Prostasin interacts with the epithelial Na+ channel and facilitates cleavage of the γ-subunit by a second protease. Am. J. Physiol. Renal Physiol. 2014, 307, F1080–F1087.
- Planès, C.; Randrianarison, N.H.; Charles, R.; Frateschi, S.; Cluzeaud, F.; Vuagniaux, G.; Soler, P.; Clerici, C.; Rossier, B.C.; Hummler, E. ENaC-mediated alveolar fluid clearance and lung fluid balance depend on the channel-activating protease 1. EMBO Mol. Med. 2010, 2, 26–37.
- Malsure, S.; Wang, Q.; Charles, R.P.; Sergi, C.; Perrier, R.; Christensen, B.M.; Maillard, M.; Rossier, B.C.; Hummler, E. Colon-specific deletion of epithelial sodium channel causes sodium loss and aldosterone resistance. J. Am. Soc. Nephrol. 2014, 25, 1453–1464.
- Frateschi, S.; Keppner, A.; Malsure, S.; Iwaszkiewicz, J.; Sergi, C.; Merillat, A.M.; Fowler-Jaeger, N.; Randrianarison, N.; Planès, C.; Hummler, E. Mutations of the serine protease CAP1/Prss8 lead to reduced embryonic viability, skin defects, and decreased ENaC activity. Am. J. Pathol. 2012, 181, 605–615.
- Leyvraz, C.; Charles, R.P.; Rubera, I.; Guitard, M.; Rotman, S.; Breiden, B.; Sandhoff, K.; Hummler, E. The epidermal barrier function is dependent on the serine protease CAP1/Prss8. J. Cell Biol. 2005, 170, 487–496.
- Crisante, G.; Battista, L.; Iwaszkiewicz, J.; Nesca, V.; Mérillat, A.M.; Sergi, C.; Zoete, V.; Frateschi, S.; Hummler, E. The CAP1/Prss8 catalytic triad is not involved in PAR2 activation and protease nexin-1 (PN-1) inhibition. FASEB J. 2014, 28, 4792–4805.
- Frateschi, S.; Camerer, E.; Crisante, G.; Rieser, S.; Membrez, M.; Charles, R.P.; Beermann, F.; Stehle, J.C.; Breiden, B.; Sandhoff, K.; et al. PAR2 absence completely rescues inflammation and ichthyosis caused by altered CAP1/Prss8 expression in mouse skin. Nat. Commun. 2011, 2, 161.
- Deryugina, E.I.; Quigley, J.P. Cell surface remodeling by plasmin: A new function for an old enzyme. J. Biomed. Biotechnol. 2012, 2012, 564259.
- Deng, Q.; Kakizoe, Y.; Iwata, Y.; Nakagawa, T.; Miyasato, Y.; Nakagawa, M.; Nishiguchi, K.; Nagayoshi, Y.; Adachi, M.; Narita, Y.; et al. The serine protease plasmin plays detrimental roles in epithelial sodium channel activation and podocyte injury in Dahl salt-sensitive rats. Hypertens. Res. 2023, 46, 50–62.
- Deschênes, G.; Wittner, M.; Stefano, A.; Jounier, S.; Doucet, A. Collecting duct is a site of sodium retention in PAN nephrosis: A rationale for amiloride therapy. J. Am. Soc. Nephrol. 2001, 12, 598–601.
- Deschênes, G.; Guigonis, V.; Doucet, A. Molecular mechanism of edema formation in nephrotic syndrome . Arch Pediatr. 2004, 11, 1084–1094.
- Svenningsen, P.; Bistrup, C.; Friis, U.G.; Bertog, M.; Haerteis, S.; Krueger, B.; Stubbe, J.; Jensen, O.N.; Thiesson, H.C.; Uhrenholt, T.R.; et al. Plasmin in nephrotic urine activates the epithelial sodium channel. J. Am. Soc. Nephrol. 2009, 20, 299–310.
- Vaziri, N.D.; Gonzales, E.C.; Shayestehfar, B.; Barton, C.H. Plasma levels and urinary excretion of fibrinolytic and protease inhibitory proteins in nephrotic syndrome. J. Lab. Clin. Med. 1994, 124, 118–124.
- Passero, C.J.; Mueller, G.M.; Rondon-Berrios, H.; Tofovic, S.P.; Hughey, R.P.; Kleyman, T.R. Plasmin activates epithelial Na+ channels by cleaving the gamma subunit. J. Biol. Chem. 2008, 283, 36586–36591.
- Svenningsen, P.; Uhrenholt, T.R.; Palarasah, Y.; Skjødt, K.; Jensen, B.L.; Skøtt, O. Prostasin-dependent activation of epithelial Na+ channels by low plasmin concentrations. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R1733–R1741.
- Gadau, J.; Peters, H.; Kastner, C.; Kühn, H.; Nieminen-Kelhä, M.; Khadzhynov, D.; Krämer, S.; Castrop, H.; Bachmann, S.; Theilig, F. Mechanisms of tubular volume retention in immune-mediated glomerulonephritis. Kidney Int. 2009, 75, 699–710.
- Kim, S.W.; Wang, W.; Nielsen, J.; Praetorius, J.; Kwon, T.H.; Knepper, M.A.; Frøkiaer, J.; Nielsen, S. Increased expression and apical targeting of renal ENaC subunits in puromycin aminonucleoside-induced nephrotic syndrome in rats. Am. J. Physiol. Renal Physiol. 2004, 286, F922–F935.
- Buhl, K.B.; Friis, U.G.; Svenningsen, P.; Gulaveerasingam, A.; Ovesen, P.; Frederiksen-Møller, B.; Jespersen, B.; Bistrup, C.; Jensen, B.L. Urinary plasmin activates collecting duct ENaC current in preeclampsia. Hypertension 2012, 60, 1346–1351.
- Aufy, M.; Abdelaziz, R.F.; Hussein, A.M.; Topcagic, N.; Shamroukh, H.; Abdel-Maksoud, M.A.; Salem, T.Z.; Studenik, C.R. Impact of Enniatin B and Beauvericin on Lysosomal Cathepsin B Secretion and Apoptosis Induction. Int. J. Mol. Sci. 2023, 24, 2030.
- Larionov, A.; Dahlke, E.; Kunke, M.; Zanon Rodriguez, L.; Schiessl, I.M.; Magnin, J.L.; Kern, U.; Alli, A.A.; Mollet, G.; Schilling, O.; et al. Cathepsin B increases ENaC activity leading to hypertension early in nephrotic syndrome. J. Cell Mol. Med. 2019, 23, 6543–6553.
- Haerteis, S.; Krappitz, M.; Bertog, M.; Krappitz, A.; Baraznenok, V.; Henderson, I.; Lindström, E.; Murphy, J.E.; Bunnett, N.W.; Korbmacher, C. Proteolytic activation of the epithelial sodium channel (ENaC) by the cysteine protease cathepsin-S. Pflugers Arch. 2012, 464, 353–365.
- Alli, A.A.; Song, J.Z.; Al-Khalili, O.; Bao, H.F.; Ma, H.P.; Alli, A.A.; Eaton, D.C. Cathepsin B is secreted apically from Xenopus 2F3 cells and cleaves the epithelial sodium channel (ENaC) to increase its activity. J. Biol. Chem. 2012, 287, 30073–30083.
- Tan, C.D.; Hobbs, C.; Sameni, M.; Sloane, B.F.; Stutts, M.J.; Tarran, R. Cathepsin B contributes to Na+ hyperabsorption in cystic fibrosis airway epithelial cultures. J. Physiol. 2014, 592, 5251–5268.
- Evans, T.I.; Joo, N.S.; Keiser, N.W.; Yan, Z.; Tyler, S.R.; Xie, W.; Zhang, Y.; Hsiao, J.J.; Cho, H.J.; Wright, M.E.; et al. Glandular Proteome Identifies Antiprotease Cystatin C as a Critical Modulator of Airway Hydration and Clearance. Am. J. Respir. Cell Mol. Biol. 2016, 54, 469–481.
More
Information
Subjects:
Pharmacology & Pharmacy
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
2 times
(View History)
Update Date:
29 Dec 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No