Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sarfaraz K. Niazi | -- | 3161 | 2023-12-28 18:19:10 | | | |
| 2 | Sirius Huang | Meta information modification | 3161 | 2023-12-29 02:39:24 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Niazi, S.K.; Mariam, Z. Computer-Aided Drug Design and Drug Discovery. Encyclopedia. Available online: https://encyclopedia.pub/entry/53240 (accessed on 30 July 2026).
Niazi SK, Mariam Z. Computer-Aided Drug Design and Drug Discovery. Encyclopedia. Available at: https://encyclopedia.pub/entry/53240. Accessed July 30, 2026.
Niazi, Sarfaraz K., Zamara Mariam. "Computer-Aided Drug Design and Drug Discovery" Encyclopedia, https://encyclopedia.pub/entry/53240 (accessed July 30, 2026).
Niazi, S.K., & Mariam, Z. (2023, December 28). Computer-Aided Drug Design and Drug Discovery. In Encyclopedia. https://encyclopedia.pub/entry/53240
Niazi, Sarfaraz K. and Zamara Mariam. "Computer-Aided Drug Design and Drug Discovery." Encyclopedia. Web. 28 December, 2023.
Copy Citation
In the dynamic landscape of drug discovery, Computer-Aided Drug Design (CADD) emerges as a transformative force, bridging the realms of biology and technology. The core principle underpinning CADD are the utilization of computer algorithms on chemical and biological data to simulate and predict how a drug molecule will interact with its target—usually a protein or DNA sequence in the biological system.
Computer-Aided Drug Design (CADD)
Machine Learning and Artificial Intelligence (AI)
drug discovery
Chemoinformatics
molecular modeling
molecular docking
target identification
1. Introduction to Computer-Aided Drug Design (CADD)
Computer-Aided Drug Design (CADD): A Synthesis of Biology and Technology
Identifying and developing a novel therapeutic agent can be an exhaustive and expensive endeavor in the expansive realm of drug discovery, where biology converges with chemistry. Historically, this journey relied on serendipitous discoveries or traditional trial-and-error methodologies, often consuming decades and substantial resources without a guaranteed outcome. The late 20th century heralded a transformative epoch for this field with the introduction of Computer-Aided Drug Design (CADD), which blends the intricate complexities of biological systems with the predictive power of computational algorithms and the development of chemical as well as biological-data-curated databases [1]. The core principle underpinning CADD are the utilization of computer algorithms on chemical and biological data to simulate and predict how a drug molecule will interact with its target—usually a protein or DNA sequence in the biological system [2]. This can range from understanding the drug’s molecular structure or target and predicting how the drug will bind to forecasting the pharmacological effects and potential side effects.
CADDs birth was facilitated by two crucial advancements: the blossoming field of structural biology, which unveiled the three-dimensional architectures of biomolecules, and the exponential growth in computational power, which made it feasible to perform complex simulations in relatively shorter timeframes [3]. One of the earliest and most celebrated applications of CADD was in the design of the anti-influenza drug Zanamivir. This process showcased the potential of this approach to significantly truncate the drug discovery timeline [4]. At its core, CADD is subdivided into two main categories: structure-based drug design (SBDD) and ligand-based drug design (LBDD) [5]. SBDD leverages knowledge of the three-dimensional structure of the biological target, aiming to understand how potential drugs can fit and interact with it. In contrast, LBDD does not require knowledge of the target structure but instead focuses on known drug molecules and their pharmacological profiles to design new drug candidates (Figure 1) [6].
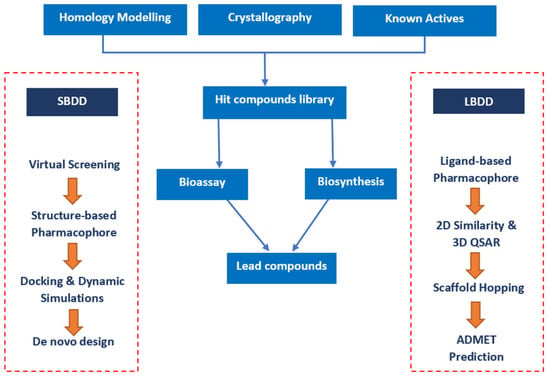
Figure 1. Conventional pathways in structure-based drug design (SBDD) and ligand-based drug design (LBDD) employ distinct methodologies. SBDD centers on target biomolecule structures, while LBDD relies on known ligand characteristics [7].
The rise of CADD is synonymous with the paradigm shift in drug discovery, where the process transitioned from being largely empirical to becoming more rational and targeted [8]. However, as with any scientific methodology, CADD has challenges. While predicting the behavior of biological systems solely based on computer simulations, it is important to acknowledge the inherent pitfalls. For instance, consider the hypothetical scenario where a computer simulation accurately models the biochemical interactions between a receptor and its target. However, if the simulation lacks crucial real-world data on external environmental factors or unexpected biological responses, the predictions may deviate significantly from the actual outcomes. These models, while sophisticated, often require experimental validation to ascertain their predictions [9][10]. In conclusion, CADD signifies the harmonious blend of biology and technology, aiming to expedite drug discovery. While it has already made significant strides in the field, its full potential is yet to be realized as newer computational methods and an increased understanding of biological systems come to the fore.
2. Key Techniques and Approaches in CADD
Delineating the Array of Techniques in Computer-Aided Drug Design
Computer-Aided Drug Design (CADD) is a powerful and interdisciplinary field that plays a pivotal role in modern drug discovery. It combines computational techniques with biological knowledge to identify and optimize potential drug candidates. This integration of diverse methodologies contributes to the versatility and effectiveness of CADD in the pharmaceutical industry. The vastness and versatility of CADD arise from the plethora of techniques and methodologies that underpin this field. This field’s effectiveness is rooted in its diverse methodologies, ranging from molecular modeling to predicting drug metabolism. Following Lipinski's rule is very important in CADD for getting the best oral drug likeliness. Compounds should have the fewest violations of criteria like molecular weight, lipophilicity, hydrogen bond donors and acceptors, and so on. This strategic integration of CADD principles and adherence to drug-likeness criteria collectively accelerates and refines the drug discovery process, showcasing the versatility and impactful role of CADD in various fields and the pharmaceutical industry (Figure 2) [11][12].
Figure 2. Elements of processing biomolecular data for CADD (image sourced from the European Research Council website [13]).
Molecular Modeling: At the heart of CADD lies molecular modeling, which encompasses a wide range of computational techniques used to model or mimic the behavior of molecules. This involves creating three-dimensional models of molecular structures, often proteins and ligands. This technique provides insights into molecules’ structural and functional attributes, facilitating a deeper understanding of how potential drugs might behave within the biological system [14]. It enables researchers to visualize and analyze the interactions between drug candidates and their target proteins, aiding in the design and optimization of potential drugs. Recently developed AI/ML-driven tools like AlphaFold2 [15], trTosetta [16][17], Robetta [18], RoseTTA Fold [19], ESMFold [20], and OmegaFold [21] have accelerated protein structure prediction by many folds [22]. Methods like molecular dynamics (MD) simulations can forecast the time-dependent behavior of molecules, capturing their motions and interactions over time through various tools like Gromacs [23], ACEMD [24], and OpenMM [25][26] (Table 1).
Table 1. Structure-predicting methods and tools.
| Methods | Programs |
|---|---|
| Homology Modeling/Comparative Modeling: Create a 3D model of the target protein using a homologous protein’s empirically confirmed structure as a guide. | MODELLER, SWISS-MODEL, Phyre2, RaptorX, I-TASSER |
| Ab Initio Modeling: Build a 3D model of the target protein by sampling the protein’s conformational space without using experimental data. | Rosetta, QUARK, AlphaFold, ESMFold, PCONS5 |
| Threading: Build a 3D model of the target protein by aligning the protein sequence with the sequences of proteins of known structure. | MUSTER, 3D-PSSM, LOMETS, HHpred |
| Hybrid Modeling: Combine two or more modeling approaches to improve the accuracy of the predicted structure. | CABS-flex, PrimeX, GalaxyHomomer |
| Molecular Dynamics: Simulate the behavior of the protein over time using classical or quantum mechanics. | GROMACS, NAMD, CHARMM |
| Knowledge-based methods: Use existing knowledge about protein structure and function to predict the structure of the target protein. | ProSMoS, ProQ3D, I-TASSER-2GO |
| Template-free methods: Build a 3D model of the target protein without using templates or homologous proteins. | CONFOLD2, MetaPSICOV, TrRosetta |
| Fragment-assembly methods: Build a 3D model of the target protein by assembling fragments of known protein structures. | PEP-FOLD3, Robetta, QUARK |
Docking and Virtual Screening: Docking involves predicting the orientation and position of a drug molecule when it binds to its target protein. It estimates the binding affinity between the drug and its target, which is crucial in drug design [27]. Utilizing advanced tools such as AutoDock Vina [28], AutoDock GOLD [29], Glide [30], DOCK [31], LigandFit [32], and SwissDock [33], researchers can predict binding affinities and orientations with precision (Table 2). Conversely, virtual screening, a complementary approach, involves sifting through vast compound libraries to identify potential drug candidates [34]. Tools like DOCK [26], LigandFit [27], and ChemBioServer [35] facilitate this process, rapidly evaluating interactions and identifying compounds with high binding affinities. DOCK is renowned for structure-based drug design; LigandFit integrates into the Schrödinger suite; and ChemBioServer is an online platform for efficient virtual screening. The synergy of these docking and virtual screening tools enhances the accuracy of predictions, contributing significantly to the identification of promising drug candidates in the complex landscape of computational drug design. Many researchers, like Pinzi and Sohoo, have extensively discussed using these tools, showcasing their implementation in advancing the field of computational drug design [36][37].
Table 2. Docking Tools, their advantages, and disadvantages.
| Tool | Application | Advantages | Disadvantages |
|---|---|---|---|
| AutoDock Vina | Predicting the binding affinities and orientations of ligands. | Fast, accurate, and easy to use. | May not be as accurate for complex systems. |
| AutoDock GOLD | Predicting the binding affinities and orientations of ligands, especially for flexible ligands. | Accurate for flexible ligands. | Requires a license and can be expensive. |
| Glide | Predicting the binding affinities and orientations of ligands. | Accurate and integrated with other Schrödinger tools. | Requires the Schrödinger suite, which can be expensive. |
| DOCK | Predicting the binding affinities and orientations of ligands and performing virtual screening. | It is versatile and can be used for both docking and virtual screening. | Can be slower than other tools. |
| LigandFit | Predicting the binding affinities and orientations of ligands. | Easy to use and integrated with other Schrödinger tools. | May not be as accurate for complex systems. |
| SwissDock | Predicting the binding affinities and orientations of ligands. | Easy to use and accessible online. | May not be as accurate for complex systems. |
Quantitative Structure-Activity Relationship (QSAR): QSAR modeling explores the relationship between the chemical structure of molecules and their biological activities. Through statistical methods, QSAR models can predict the pharmacological activity of new compounds based on their structural attributes, enabling chemists to make informed modifications to enhance a drug’s potency or reduce its side effects [38][39]. A study by Luo et al. used the Similarity Ensemble Approach (SEA) as a key tool to check how accurate k-nearest neighbors (kNN) Quantitative Structure-Activity Relationship (QSAR) models were. These models were systematically constructed for known ligands associated with individual G Protein-Coupled Receptor (GPCR) targets to reveal active and inactive molecules [40]. In the meantime, Raj et al. did a different study using the molecular field analysis method to make QSAR models for 50 compounds that showed anti-HIV activity. The findings underscored the critical role of electrostatic and steric interactions in influencing the anti-HIV activity of the compounds [41]. Along with this, Nigsch et al. used a new method in another study that combined a deep neural network with QSAR models to look at a group of 1000 chemicals that are known to have anti-cancer properties. According to this study, integrating the QSAR approach with deep learning techniques proved advantageous, enabling the identification of critical structural characteristics that significantly contributed to the compounds’ anti-cancer efficacy [42].
Pharmacophore Modeling: A pharmacophore is a spatial arrangement of essential features in a molecule necessary for its pharmacological activity. Pharmacophore modeling is an important part of modern drug discovery. It involves figuring out how important molecular features are arranged in space to affect a molecule's pharmacological activity. This approach is potent for medicinal chemists, enabling the rational design of novel compounds with optimized pharmacological properties [43].
For example, pharmacophore modeling has proven instrumental in kinase inhibitors. Zhang et al. used pharmacophore modeling to find important parts of active kinase inhibitors, such as regions that donate and accept hydrogen bonds and regions that do not attract water. The identified pharmacophore elements provided valuable guidance for designing novel compounds, resulting in improved selectivity and potency against specific kinases implicated in disease pathways [44]. Similarly, pharmacophore modeling has been applied to design ligands targeting G protein-coupled receptors (GPCRs). Fidom et al. exemplified this application by elucidating the spatial arrangement of features crucial for GPCR binding, such as aromatic interactions and hydrogen bonding [45][46]. The pharmacophore models that were made made it easier to make ligands that were more specific and had higher affinity for certain GPCRs that are involved in many therapeutic areas. In summary, the strategic use of pharmacophore modeling enables systematic analysis of the essential features contributing to a molecule’s pharmacological activity. This knowledge enhances understanding of ligand-receptor interactions and empowers researchers to rationally design compounds with enhanced efficacy and reduced side effects, shaping the future landscape of pharmaceutical research.
Prediction of Drug Metabolism and Pharmacokinetics (DMPK): The ultimate success of a drug is not solely determined by its ability to bind to its target. Its metabolic stability, solubility, and how it is distributed in the body (pharmacokinetics) play pivotal roles. CADD offers tools that can predict the DMPK properties of compounds, allowing researchers to anticipate and address potential issues related to drug metabolism, bioavailability, and potential drug-drug interactions [47].
Novo Drug Design: Unlike other methods that modify existing molecules, de novo drug design creates new drug molecules from scratch. This technique leverages computational algorithms to generate new molecular structures that fit specific criteria, opening the door to many novel drug candidates [48].
In summary, the techniques embedded within CADD provide an integrated, multi-faceted approach to drug discovery. CADD makes sure that drug candidates are strong and selective, and have the best pharmacokinetic and safety profiles, by providing a set of tools that includes molecular modeling and drug metabolism prediction.
3. Integration of Machine Learning and AI in CADD
3.1. Machine Learning and AI: The New Vanguard in Drug Discovery
The technological renaissance that defines the 21st century has borne witness to the meteoric rise of Machine Learning (ML) and Artificial Intelligence (AI). These computational realms, known for their data-driven decision-making capabilities, have begun to significantly influence the sphere of Computer-Aided Drug Design (CADD), reshaping the contours of drug discovery [49]. Machine Learning, a subset of AI, hinges on algorithms that can learn patterns from vast data sets without being explicitly programmed for specific tasks (Figure 3) [50]. In drug discovery, ML has been instrumental in predicting molecular properties, understanding drug-receptor interactions, and forecasting biological responses based on chemical structures. Techniques such as deep learning, which uses neural networks modeled after the human brain, show immense potential for predicting complex drug-related outcomes with remarkable accuracy [51].
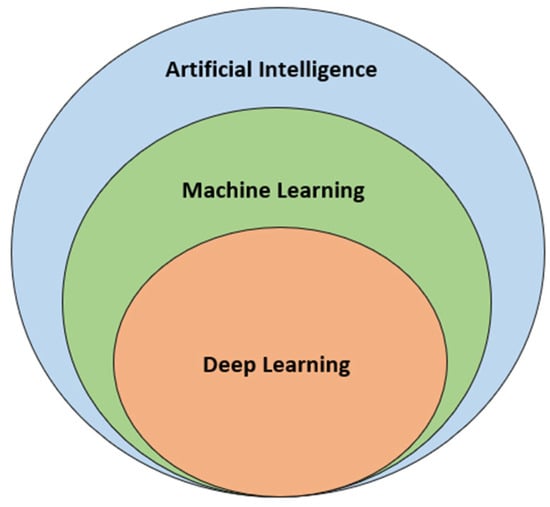
Figure 3. Subsets of Artificial Intelligence: Machine Learning and Deep Learning.
3.2. Implications of ML in CADD
Predicting Drug-Drug Interactions: One of the challenges in drug discovery is understanding how a new drug might interact with other medications a patient might be taking. ML algorithms can process large databases of known drug-drug interactions to predict potential harmful combinations for novel compounds [52].
Drug Repurposing: Drug repurposing involves finding new therapeutic applications for existing drugs. By analyzing vast datasets, Machine Learning can identify potential new targets for existing medications, thus saving both time and costs associated with traditional drug discovery [53].
Generative Adversarial Networks (GANs) in Drug Design: GANs are a form of AI where two neural networks (a generator and a discriminator) are trained in tandem. The generator creates molecular structures while the discriminator evaluates them. Over time, the generator becomes adept at creating feasible and potentially bioactive molecular structures, which can be synthesized and tested in the lab [54].
Predictive Toxicology: One of the primary reasons drug candidates fail in clinical trials is unforeseen toxicity. By looking at past data on drug-induced toxicities, ML models can help predict possible bad effects. This helps get rid of potentially toxic compounds early on in the discovery process. QSAR models also predict toxicological effects by connecting the structure of a molecule with its possible toxicity using properties like molecular weight, lipophilicity, and electronic properties. More details, like solubility, metabolic stability, and the identification of toxicophores, give us a full picture. This helps us find hazards early on and choose which compounds to test in experiments in computational toxicology.
The integration of AI and ML into CADD signifies more than just the adoption of new technologies. It represents a paradigm shift from traditional hypothesis-driven research to data-driven discovery, leveraging the power of big data and computational prowess to inform decision-making at every step of drug discovery [55]. However, while these technologies promise a revolution in drug discovery, challenges persist. Issues such as data quality, interpretability of AI models, and the need for experimental validation continue to be focal areas of attention in this integration [56]. In essence, the synergy of ML, AI, and CADD sets the stage for a new era in drug discovery. An era characterized by increased efficiency, reduced costs, and the rapid delivery of effective therapeutics to patients in need.
4. Challenges and Limitations in CADD
Understanding the Obstacles: The Roadblocks in Computer-Aided Drug Design
While CADD offers unparalleled advantages in expediting and refining drug discovery, it is crucial to recognize its inherent challenges. A notable obstacle is the scarcity of experts proficient in AI/ML within CADD. Initiatives like specialized training programs and targeted recruitment are crucial; for example, organizations like in-silico Medicine are pioneering efforts to bridge this gap, fostering a skilled workforce capable of harnessing advanced computational techniques for drug discovery. Addressing these limitations can lead to better strategies and pave the way for more effective drug discovery workflows [57].
Accuracy of Predictive Models: In CADD, a major challenge lies in ensuring the accuracy of computational models, given that molecular dynamics simulations, docking scores, and machine learning predictions all rely on theoretical models. These models may not fully capture the intricate nuances of biological systems. To enhance accuracy, it is essential to delve into the intricacies of scoring algorithms [58]. Scoring algorithms in drug discovery are pivotal for predicting the binding affinity between molecules and their targets. To ensure their accuracy, it is imperative to actively mitigate the risk of false positives and negatives. This involves meticulous calibration of scoring parameters, the incorporation of diverse molecular descriptors, and continuous validation against experimental data. For instance, refining docking scores through rigorous validation against known binding affinities can enhance the reliability of predictions. By optimizing the balance between sensitivity and specificity, researchers can bolster confidence in scoring algorithms, reducing the likelihood of inaccuracies in drug discovery predictions [59][60][61].
Data Quality and Quantity: The predictions made by CADD tools are only as good as the data they are trained on. The predictions are likely inaccurate if the underlying data are of poor quality or insufficient. The lack of curated, high-quality datasets, especially in the context of machine learning in drug discovery, is a recurring challenge [62]. Removing outliers and ensuring consistent data formatting can refine molecular interaction datasets, minimizing inaccuracies and bolstering the reliability of computational models. Standardized experimental protocols, like using the same assay conditions and endpoint measurements every time, also help improve the quality of data in CADD, making sure that the results are strong and reliable.
Over-reliance on Computational Predictions: While CADD is a powerful tool, over-reliance on its predictions without subsequent experimental validation can lead to misguided efforts. Balancing computational predictions with experimental evidence is essential for a successful drug discovery [63].
Time and Computational Cost: Some advanced CADD techniques, especially those involving extensive molecular dynamics simulations or intricate machine learning models, require vast computational resources. The associated costs, both in terms of time and infrastructure, can be prohibitive for some research groups [64].
Representing Molecular Flexibility: Most biological molecules, including potential drug compounds and their target proteins, are highly flexible. Accurately representing this flexibility, especially in techniques like molecular docking, is challenging and can significantly impact the results of CADD studies [65].
Interpretability of AI Models: As AI and machine learning models become more complex, their predictions become more challenging to interpret. This ‘black-box’ nature of AI models can make it challenging to understand why a particular compound is predicted to be active or how its structure might be optimized [66].
Despite these challenges, the potential benefits of CADD in drug discovery are immense. By acknowledging these limitations and continually striving to address them through innovation and research, CADD will remain at the forefront of modern drug discovery, shaping the future of therapeutics.
References
- Johnson, A.M.; Smith, B.C. Historical perspectives in drug discovery: The advent of computational tools. J. Drug Discov. 1995, 12, 5–15.
- Patel, Y.; Chalmers, D.K. Modeling drug-receptor interactions: Advances and challenges. J. Med. Chem. 2003, 46, 2543–2554.
- Green, P.L.; Edwards, P.H. Structural biology and computational chemistry: A symbiotic relationship. Chem. Rev. 2010, 110, 5678–5698.
- Walker, N.T.; Williams, J.P. Zanamivir: The making of a drug. Nat. Biotechnol. 1997, 15, 232–235.
- Martinez, A. Computational strategies in drug design. Drug Discov. Today 2006, 11, 149–155.
- CC BY 4.0 Deed/Attribution 4.0 International/Creative Commons (n.d.). Available online: https://creativecommons.org/licenses/by/4.0/ (accessed on 10 November 2023).
- Lu, W.; Zhang, R.; Jiang, H.; Zhang, H.; Luo, C. Computer-Aided drug design in epigenetics. Front. Chem. 2018b, 6, 57.
- Kapoor, L.; Oprea, T.I. From empirical to rational drug discovery: The importance of CADD. Drug Des. Rev. 2018, 15, 345–356.
- Fu, C.; Xiang, M.A.; Chen, S.; Dong, G.; Liu, Z.; Chen, C.; Liang, J.; Cao, Y.; Zhang, M.; Liu, Q. Molecular drug simulation and experimental validation of the CD36 receptor competitively binding to Long-Chain fatty acids by 7-Ketocholesteryl-9-carboxynonanoate. ACS Omega 2023, 8, 28277–28289.
- Schaduangrat, N.; Lampa, S.; Simeon, S.; Gleeson, M.P.; Spjuth, O.; Nantasenamat, C. Towards reproducible computational drug discovery. J. Cheminform. 2020, 12, 9.
- Thompson, M.A. Techniques in Computer-Aided Drug Design. Bioorganic Med. Chem. 2004, 12, 3101–3110.
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25.
- ERC. Computational Biology: Spotlight on ERC projects. Available online: https://erc.europa.eu/projects-statistics/science-stories/computational-biology-spotlight-erc-projects (accessed on 27 November 2023).
- Leach, A.R.; Gillet, V.J. Molecular modeling: Principles and applications. J. Chem. Inf. Model. 2007, 47, 5–27.
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589.
- Yang, J.; Anishchenko, I.; Park, H.; Peng, Z.; Ovchinnikov, S.; Baker, D. Improved protein structure prediction using predicted interresidue orientations. Proc. Natl. Acad. Sci. USA 2020, 117, 1496–1503.
- Du, Z.; Su, H.; Wang, W.; Ye, L.; Wei, H.; Peng, Z.; Anishchenko, I.; Baker, D.; Yang, J. The trRosetta server for fast and accurate protein structure prediction. Nat. Protoc. 2021, 16, 5634–5651.
- Kim, D.E.; Chivian, D.; Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004, 32, W526–W531.
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876.
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y.; et al. Language models of protein sequences at the scale of evolution enable accurate structure prediction. bioRxiv 2022, 2020, 500902.
- Wu, R.; Ding, F.; Wang, R.; Shen, R.; Zhang, X.; Luo, S.; Su, C.; Wu, Z.; Xie, Q.; Berger, B.; et al. High-resolution de novo structure prediction from primary sequence. bioRxiv 2022.
- Niazi, S.K.; Mariam, Z. Reinventing Therapeutic Proteins: Mining a treasure of new therapies. Biologics 2023, 3, 72–94.
- Berendsen, H.J.C.; Van Der Spoel, D.; Van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56.
- Harvey, M.; Giupponi, G.; De Fabritiis, G. ACEMD: Accelerating Biomolecular Dynamics in the Microsecond Time Scale. J. Chem. Theory Comput. 2009, 5, 1632–1639.
- Eastman, P.; Swails, J.; Chodera, J.D.; McGibbon, R.T.; Zhao, Y.; Beauchamp, K.A.; Wang, L.; Simmonett, A.C.; Harrigan, M.P.; Stern, C.; et al. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol. 2017, 13, e1005659.
- Adcock, S.A.; McCammon, J.A. Molecular dynamics: Survey of methods for simulating the activity of proteins. Chem. Rev. 2006, 106, 1589–1615.
- Morris, G.M.; Lim-Wilby, M. Molecular docking. Methods Mol. Biol. 2008, 443, 365–382.
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461.
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791.
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Shenkin, P.S. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749.
- Ewing, T.J.A.; Makino, S.; Skillman, A.G.; Kuntz, I.D. DOCK 4.0: Search strategies for automated molecular docking of flexible molecule databases. J. Comput. Aided Mol. Des. 2001, 15, 411–428.
- Venkatachalam, C.M.; Jiang, X.; Oldfield, T.; Waldman, M. LigandFit: A novel method for the shape-directed rapid docking of ligands to protein active sites. J. Mol. Graph. Model. 2003, 21, 289–307.
- Bitencourt-Ferreira, G.; De Azevedo, W.F. Docking with SwissDock. Methods Mol. Biol. 2019, 2053, 189–202.
- Willett, P. Virtual screening using molecular docking. Drug Discov. Today Technol. 2006, 3, 229–234.
- Karatzas, E.; Zamora, J.E.; Athanasiadis, E.; Dellis, D.; Cournia, Z.; Spyrou, G.M. ChemBioServer 2.0: An advanced web server for filtering, clustering and networking of chemical compounds facilitating both drug discovery and repurposing. Bioinformatics 2020, 36, 2602–2604.
- Pinzi, L.; Rastelli, G. Molecular docking: Shifting paradigms in drug discovery. Int. J. Mol. Sci. 2019, 20, 4331.
- Sahoo, R.N.; Pattanaik, S.; Pattnaik, G.; Mallick, S.; Mohapatra, R. Review on the use of Molecular Docking as the First Line Tool in Drug Discovery and Development. Indian J. Pharm. Sci. 2022, 84, 1334–1337.
- Hansch, C.; Leo, A. Exploring QSAR: Hydrophobic, Electronic, and Steric Constants; ACS Professional Reference Book: Washington, DC, USA, 1995.
- Niazi, S.K.; Mariam, Z. Recent Advances in Machine-Learning-Based Chemoinformatics: A Comprehensive Review. Int. J. Mol. Sci. 2023, 24, 11488.
- Luo, M.; Wang, X.S.; Tropsha, A. Comparative Analysis of QSAR-based vs. Chemical Similarity Based Predictors of GPCRs Binding Affinity. Mol. Inform. 2016, 35, 36–41.
- Raj, N.; Jain, S.K. 3d QSAR studies in conjunction with k-nearest neighbor molecular field analysis (k-NN-MFA) on a series of substituted 2-phenyl-benzimidazole derivatives as an anti allergic agents. Dig. J. Nanomater. Biostructures 2011, 6, 1811–1821.
- Nigsch, F.; Bender, A.; van Buuren, B.; Tissen, J.; Nigsch, E.; Mitchell, J.B. Melting point prediction employing k-nearest neighbor algorithms and genetic parameter optimization. J. Chem. Inf. Model. 2006, 46, 2412–2422.
- Güner, O.F. Pharmacophore perception, development, and use in drug design. J. Med. Chem. 2002, 45, 5–12.
- Zhang, Y.; Yang, S.; Jiao, Y.; Liu, H.; Yuan, H.; Lü, S.; Ran, T.; Yao, S.; Ke, Z.; Xu, J.; et al. An integrated virtual screening approach for VEGFR-2 inhibitors. J. Chem. Inf. Model. 2013, 53, 3163–3177.
- Fidom, K.; Ísberg, V.; Hauser, A.S.; Mordalski, S.; Lehto, T.; Bojarski, A.J.; Gloriam, D.E. A new crystal structure fragment-based pharmacophore method for G protein-coupled receptors. Methods 2015, 71, 104–112.
- Munk, C.; Harpsøe, K.; Hauser, A.S.; Isberg, V.; Gloriam, D.E. Integrating structural and mutagenesis data to elucidate GPCR ligand binding. Curr. Opin. Pharmacol. 2016, 30, 51–58.
- Ekins, S.; Williams, A.J. In silico pharmacokinetics: ADME in drug discovery. Drug Discov. World 2007, 8, 17–24.
- Schneider, G.; Fechner, U. Computer-based de novo design of drug-like molecules. Nat. Rev. Drug Discov. 2005, 4, 649–663.
- Zhang, L.; Tan, J. AI and its role in drug discovery. J. Drug Discov. Des. 2019, 5, 1–10.
- Bishop, C.M. Pattern Recognition and Machine Learning; Springer: Berlin/Heidelberg, Germany, 2006.
- Goh, G.B.; Hodas, N.O.; Vishnu, A. Deep learning for computational chemistry. J. Comput. Chem. 2017, 38, 1291–1307.
- Vilar, S.; Uriarte, E.; Santana, L.; Lorberbaum, T.; Hripcsak, G. Predicting drug-drug interactions through drug structural similarities and interaction networks incorporating pharmacokinetics and pharmacodynamics knowledge. J. Cheminformatics 2016, 8, 12.
- Aliper, A.; Plis, S.; Artemov, A.; Ulloa, A.; Mamoshina, P.; Zhavoronkov, A. Deep learning applications for predicting pharmacological properties of drugs and drug repurposing using transcriptomic data. Mol. Pharm. 2016, 13, 2524–2530.
- Kadurin, A.; Aliper, A.; Kazennov, A.; Mamoshina, P.; Vanhaelen, Q.; Khrabrov, K.; Zhavoronkov, A. The cornucopia of meaningful leads: Applying deep adversarial autoencoders for new molecule development in oncology. Oncotarget 2017, 8, 10883–10890.
- Mayr, A.; Klambauer, G.; Unterthiner, T.; Hochreiter, S. DeepTox: Toxicity prediction using deep learning. Front. Environ. Sci. 2016, 3, 80.
- Chen, H.; Engkvist, O.; Wang, Y.; Olivecrona, M.; Blaschke, T. The rise of deep learning in drug discovery. Drug Discov. Today 2018, 23, 1241–1250.
- Jorgensen, W.L. The many roles of computation in drug discovery. Science 2004, 303, 1813–1818.
- Warren, G.L.; Andrews, C.W.; Capelli, A.M.; Clarke, B.; LaLonde, J.; Lambert, M.H.; Murray, C.W. A critical assessment of docking programs and scoring functions. J. Med. Chem. 2006, 49, 5912–5931.
- Aleksandrova, A. Machine-Learning Scoring Functions to Improve Structure-Based Binding Affinity Prediction and Virtual Screening. 2016. Available online: https://www.academia.edu/28830051/Machine_learning_scoring_functions_to_improve_structure_based_binding_affinity_prediction_and_virtual_screening (accessed on 10 November 2023).
- Fujimoto, K.; Minami, S.; Yanai, T. Machine-Learning- and Knowledge-Based scoring functions incorporating ligand and protein fingerprints. ACS Omega 2022, 7, 19030–19039.
- Guedes, I.A.; Barreto, A.; Marinho, D.; Krempser, E.; Kuenemann, M.A.; Spérandio, O.; Dardenne, L.E.; Miteva, M.A. New machine learning and physics-based scoring functions for drug discovery. Sci. Rep. 2021, 11, 3198.
- Walters, W.P.; Murcko, M.A. Prediction of ‘drug-likeness’. Adv. Drug Deliv. Rev. 2002, 54, 255–271.
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949.
- Dror, R.O.; Dirks, R.M.; Grossman, J.P.; Xu, H.; Shaw, D.E. Biomolecular simulation: A computational microscope for molecular biology. Annu. Rev. Biophys. 2012, 41, 429–452.
- Teague, S.J. Implications of protein flexibility for drug discovery. Nat. Rev. Drug Discov. 2003, 2, 527–541.
- Ching, T.; Himmelstein, D.S.; Beaulieu-Jones, B.K.; Kalinin, A.A.; Do, B.T.; Way, G.P.; Greene, C.S. Opportunities and obstacles for deep learning in biology and medicine. J. R. Soc. Interface 2018, 15, 20170387.
More
Information
Subjects:
Medicine, Research & Experimental
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.6K
Entry Collection:
Biopharmaceuticals Technology
Revisions:
2 times
(View History)
Update Date:
29 Dec 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No