 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Majid Mufaqam Syed-Abdul | -- | 3380 | 2023-12-28 06:30:20 | | | |
| 2 | Lindsay Dong | + 4 word(s) | 3384 | 2023-12-29 01:29:44 | | |
Video Upload Options
Metabolic-associated steatotic liver disease (MASLD) is a cluster of pathological conditions primarily developed due to the accumulation of ectopic fat in the hepatocytes. During the severe form of the disease, i.e., metabolic-associated steatohepatitis (MASH), accumulated lipids promote lipotoxicity, resulting in cellular inflammation, oxidative stress, and hepatocellular ballooning. If left untreated, the advanced form of the disease progresses to fibrosis of the tissue, resulting in irreversible hepatic cirrhosis or the development of hepatocellular carcinoma. Although numerous mechanisms have been identified as significant contributors to the development and advancement of MASLD, altered lipid metabolism continues to stand out as a major factor contributing to the disease.
1. Metabolic-Associated Steatotic Liver Disease
2. Pathophysiological Changes at the Molecular Level in MASLD
3. Lipid Synthesis in MASLD
3.1. Fatty Acids
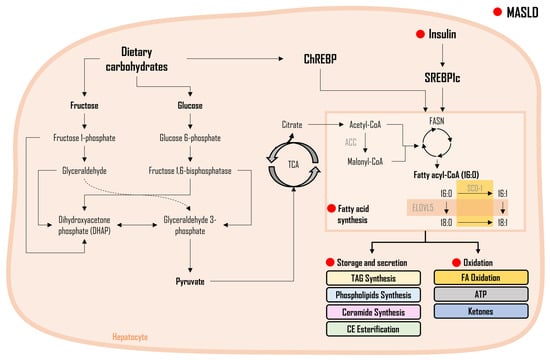
Saturated vs. Unsaturated FAs in MASLD
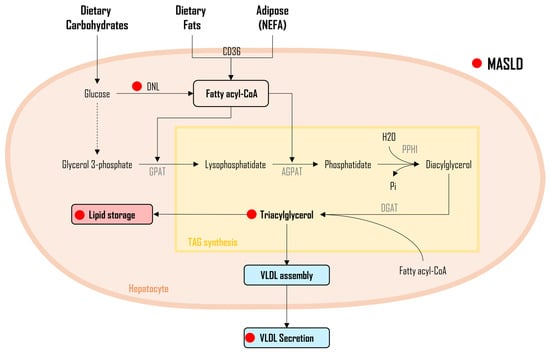
3.2. Triacylglycerol and Diacylglycerol
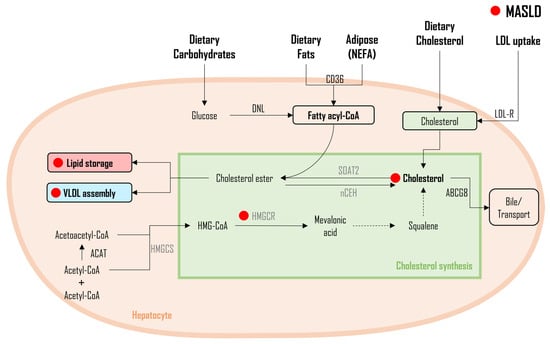
3.3. Cholesterol
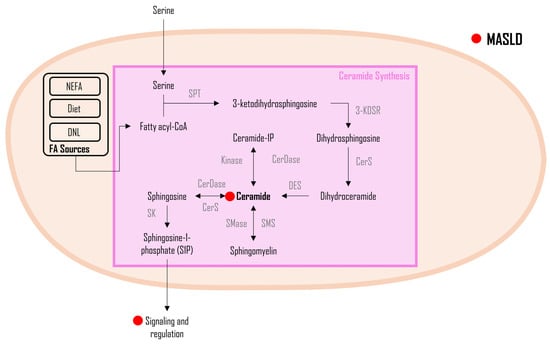
3.4. Ceramides
4. Lipid oxidation in MASLD
FA Oxidation
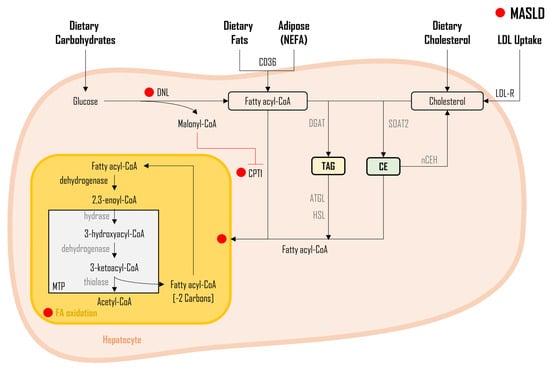
5. Lipid Flux and Storage in MASLD
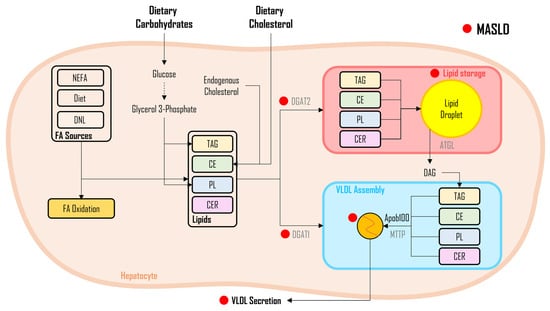
6. Interplay of Cholesterol Metabolism and DNL in MASLD
7. Conclusions
References
- Rinella, M.E.; Lazarus, J.V.; Ratziu, V.; Francque, S.M.; Sanyal, A.J.; Kanwal, F.; Romero, D.; Abdelmalek, M.F.; Anstee, Q.M.; Arab, J.P.; et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. Hepatology 2023, 78, 1966–1986.
- Sanyal, A.J.; American Gastroenterological, A. AGA technical review on nonalcoholic fatty liver disease. Gastroenterology 2002, 123, 1705–1725.
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20.
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84.
- Fazel, Y.; Koenig, A.B.; Sayiner, M.; Goodman, Z.D.; Younossi, Z.M. Epidemiology and natural history of non-alcoholic fatty liver disease. Metabolism 2016, 65, 1017–1025.
- Lopez-Velazquez, J.A.; Silva-Vidal, K.V.; Ponciano-Rodriguez, G.; Chavez-Tapia, N.C.; Arrese, M.; Uribe, M.; Mendez-Sanchez, N. The prevalence of nonalcoholic fatty liver disease in the Americas. Ann. Hepatol. 2014, 13, 166–178.
- Farrell, G.C.; Chitturi, S.; Lau, G.K.; Sollano, J.D.; Asia–Pacific Working Party on NAFLD. Guidelines for the assessment and management of non-alcoholic fatty liver disease in the Asia-Pacific region: Executive summary. J. Gastroenterol. Hepatol. 2007, 22, 775–777.
- Feijo, S.G.; Lima, J.M.; Oliveira, M.A.; Patrocinio, R.M.; Moura-Junior, L.G.; Campos, A.B.; Lima, J.W.; Braga, L.L. The spectrum of non alcoholic fatty liver disease in morbidly obese patients: Prevalence and associate risk factors. Acta Cir. Bras. 2013, 28, 788–793.
- Neuschwander-Tetri, B.A.; Caldwell, S.H. Nonalcoholic steatohepatitis: Summary of an AASLD Single Topic Conference. Hepatology 2003, 37, 1202–1219.
- Sherif, Z.A.; Saeed, A.; Ghavimi, S.; Nouraie, S.M.; Laiyemo, A.O.; Brim, H.; Ashktorab, H. Global epidemiology of nonalcoholic fatty liver disease and perspectives on US minority populations. Dig. Dis. Sci. 2016, 61, 1214–1225.
- Kabbany, M.N.; Conjeevaram Selvakumar, P.K.; Watt, K.; Lopez, R.; Akras, Z.; Zein, N.; Carey, W.; Alkhouri, N. Prevalence of nonalcoholic steatohepatitis-associated cirrhosis in the United States: An analysis of National Health and Nutrition Examination Survey data. Am. J. Gastroenterol. 2017, 112, 581–587.
- Ekstedt, M.; Franzen, L.E.; Mathiesen, U.L.; Thorelius, L.; Holmqvist, M.; Bodemar, G.; Kechagias, S. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology 2006, 44, 865–873.
- Musso, G.; Gambino, R.; Cassader, M.; Pagano, G. Meta-analysis: Natural history of non-alcoholic fatty liver disease (NAFLD) and diagnostic accuracy of non-invasive tests for liver disease severity. Ann. Med. 2011, 43, 617–649.
- Anstee, Q.M.; Targher, G.; Day, C.P. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 330–344.
- Syed-Abdul, M.M. Expanding Pharmacists’ Role in the Management of Non-Alcoholic Fatty Liver Disease. Pharmacy 2023, 11, 151.
- Zhu, L.; Baker, S.S.; Liu, W.; Tao, M.H.; Patel, R.; Nowak, N.J.; Baker, R.D. Lipid in the livers of adolescents with nonalcoholic steatohepatitis: Combined effects of pathways on steatosis. Metabolism 2011, 60, 1001–1011.
- Mitsuyoshi, H.; Yasui, K.; Harano, Y.; Endo, M.; Tsuji, K.; Minami, M.; Itoh, Y.; Okanoue, T.; Yoshikawa, T. Analysis of hepatic genes involved in the metabolism of fatty acids and iron in nonalcoholic fatty liver disease. Hepatol. Res. 2009, 39, 366–373.
- Dorn, C.; Riener, M.O.; Kirovski, G.; Saugspier, M.; Steib, K.; Weiss, T.S.; Gabele, E.; Kristiansen, G.; Hartmann, A.; Hellerbrand, C. Expression of fatty acid synthase in nonalcoholic fatty liver disease. Int. J. Clin. Exp. Pathol. 2010, 3, 505–514.
- Nagaya, T.; Tanaka, N.; Kimura, T.; Kitabatake, H.; Fujimori, N.; Komatsu, M.; Horiuchi, A.; Yamaura, T.; Umemura, T.; Sano, K.; et al. Mechanism of the development of nonalcoholic steatohepatitis after pancreaticoduodenectomy. BBA Clin. 2015, 3, 168–174.
- Kohjima, M.; Enjoji, M.; Higuchi, N.; Kato, M.; Kotoh, K.; Yoshimoto, T.; Fujino, T.; Yada, M.; Yada, R.; Harada, N.; et al. Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int. J. Mol. Med. 2007, 20, 351–358.
- Greco, D.; Kotronen, A.; Westerbacka, J.; Puig, O.; Arkkila, P.; Kiviluoto, T.; Laitinen, S.; Kolak, M.; Fisher, R.M.; Hamsten, A.; et al. Gene expression in human NAFLD. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G1281–G1287.
- Ferre, P.; Foufelle, F. Hepatic steatosis: A role for de novo lipogenesis and the transcription factor SREBP-1c. Diabetes Obes. Metab. 2010, 12 (Suppl. 2), 83–92.
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131.
- Rong, S.; Cortes, V.A.; Rashid, S.; Anderson, N.N.; McDonald, J.G.; Liang, G.; Moon, Y.A.; Hammer, R.E.; Horton, J.D. Expression of SREBP-1c requires SREBP-2-mediated generation of a sterol ligand for LXR in livers of mice. Elife 2017, 6, e25015.
- Ahmed, M.H.; Byrne, C.D. Modulation of sterol regulatory element binding proteins (SREBPs) as potential treatments for non-alcoholic fatty liver disease (NAFLD). Drug Discov. Today 2007, 12, 740–747.
- Coassolo, L.; Liu, T.; Jung, Y.; Taylor, N.P.; Zhao, M.; Charville, G.W.; Nissen, S.B.; Yki-Jarvinen, H.; Altman, R.B.; Svensson, K.J. Mapping transcriptional heterogeneity and metabolic networks in fatty livers at single-cell resolution. iScience 2023, 26, 105802.
- Westerbacka, J.; Kolak, M.; Kiviluoto, T.; Arkkila, P.; Siren, J.; Hamsten, A.; Fisher, R.M.; Yki-Jarvinen, H. Genes involved in fatty acid partitioning and binding, lipolysis, monocyte/macrophage recruitment, and inflammation are overexpressed in the human fatty liver of insulin-resistant subjects. Diabetes 2007, 56, 2759–2765.
- Bechmann, L.P.; Gieseler, R.K.; Sowa, J.P.; Kahraman, A.; Erhard, J.; Wedemeyer, I.; Emons, B.; Jochum, C.; Feldkamp, T.; Gerken, G.; et al. Apoptosis is associated with CD36/fatty acid translocase upregulation in non-alcoholic steatohepatitis. Liver Int. 2010, 30, 850–859.
- Nakamuta, M.; Fujino, T.; Yada, R.; Yada, M.; Yasutake, K.; Yoshimoto, T.; Harada, N.; Higuchi, N.; Kato, M.; Kohjima, M.; et al. Impact of cholesterol metabolism and the LXRalpha-SREBP-1c pathway on nonalcoholic fatty liver disease. Int. J. Mol. Med. 2009, 23, 603–608.
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015, 21, 739–746.
- Moore, M.P.; Cunningham, R.P.; Meers, G.M.; Johnson, S.A.; Wheeler, A.A.; Ganga, R.R.; Spencer, N.M.; Pitt, J.B.; Diaz-Arias, A.; Swi, A.I.A.; et al. Compromised hepatic mitochondrial fatty acid oxidation and reduced markers of mitochondrial turnover in human NAFLD. Hepatology 2022, 76, 1452–1465.
- Fujita, K.; Nozaki, Y.; Wada, K.; Yoneda, M.; Fujimoto, Y.; Fujitake, M.; Endo, H.; Takahashi, H.; Inamori, M.; Kobayashi, N.; et al. Dysfunctional very-low-density lipoprotein synthesis and release is a key factor in nonalcoholic steatohepatitis pathogenesis. Hepatology 2009, 50, 772–780.
- Desterke, C.; Chiappini, F. Lipid related genes altered in NASH connect inflammation in liver pathogenesis progression to HCC: A canonical pathway. Int. J. Mol. Sci. 2019, 20, 5594.
- Miquilena-Colina, M.E.; Lima-Cabello, E.; Sanchez-Campos, S.; Garcia-Mediavilla, M.V.; Fernandez-Bermejo, M.; Lozano-Rodriguez, T.; Vargas-Castrillon, J.; Buque, X.; Ochoa, B.; Aspichueta, P.; et al. Hepatic fatty acid translocase CD36 upregulation is associated with insulin resistance, hyperinsulinaemia and increased steatosis in non-alcoholic steatohepatitis and chronic hepatitis C. Gut 2011, 60, 1394–1402.
- McDevitt, R.M.; Bott, S.J.; Harding, M.; Coward, W.A.; Bluck, L.J.; Prentice, A.M. De novo lipogenesis during controlled overfeeding with sucrose or glucose in lean and obese women. Am. J. Clin. Nutr. 2001, 74, 737–746.
- Softic, S.; Cohen, D.E.; Kahn, C.R. Role of dietary fructose and hepatic de novo lipogenesis in fatty liver disease. Dig. Dis. Sci. 2016, 61, 1282–1293.
- Syed-Abdul, M.M.; Parks, E.J.; Ramos-Roman, M.A. Maternal Metabolism of Glucose and Lipids in Lactating and Non-lactating Women at 6-Weeks Postpartum. Diabetes 2017, 66, LB56.
- Baykal, A.P.; Parks, E.J.; Shamburek, R.; Syed-Abdul, M.M.; Chacko, S.; Cochran, E.; Startzell, M.; Gharib, A.M.; Ouwerkerk, R.; Abd-Elmoniem, K.Z.; et al. Leptin decreases de novo lipogenesis in patients with lipodystrophy. JCI Insight 2020, 5, e137180.
- Ramos-Roman, M.A.; Syed-Abdul, M.M.; Casey, B.M.; Alger, J.R.; Liu, Y.L.; Parks, E.J. Lactation alters the relationship between liver lipid synthesis and hepatic fat stores in the postpartum period. J. Lipid Res. 2022, 63, 100288.
- Syed-Abdul, M.M.; Jacome-Sosa, M.; Hu, Q.; Gaballah, A.H.; Winn, N.C.; Lee, N.T.; Mucinski, J.M.; Manrique-Acevedo, C.; Lastra, G.; Anderson, J.M.; et al. The Tailgate Study: Differing metabolic effects of a bout of excessive eating and drinking. Alcohol 2021, 90, 45–55.
- Ramos-Roman, M.A.; Syed-Abdul, M.M.; Adams-Huet, B.; Casey, B.M.; Parks, E.J. Lactation Versus Formula Feeding: Insulin, Glucose, and Fatty Acid Metabolism During the Postpartum Period. Diabetes 2020, 69, 1624–1635.
- Kim, C.W.; Addy, C.; Kusunoki, J.; Anderson, N.N.; Deja, S.; Fu, X.; Burgess, S.C.; Li, C.; Ruddy, M.; Chakravarthy, M.; et al. Acetyl CoA carboxylase inhibition reduces hepatic steatosis but elevates plasma triglycerides in mice and humans: A bedside to bench investigation. Cell Metab. 2017, 26, 394–406.e396.
- Mao, J.; DeMayo, F.J.; Li, H.; Abu-Elheiga, L.; Gu, Z.; Shaikenov, T.E.; Kordari, P.; Chirala, S.S.; Heird, W.C.; Wakil, S.J. Liver-specific deletion of acetyl-CoA carboxylase 1 reduces hepatic triglyceride accumulation without affecting glucose homeostasis. Proc. Natl. Acad. Sci. USA 2006, 103, 8552–8557.
- Savage, D.B.; Choi, C.S.; Samuel, V.T.; Liu, Z.X.; Zhang, D.; Wang, A.; Zhang, X.M.; Cline, G.W.; Yu, X.X.; Geisler, J.G.; et al. Reversal of diet-induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl-CoA carboxylases 1 and 2. J. Clin. Investig. 2006, 116, 817–824.
- Lee, A.K.; Kyriakou, T.; Weston, A.J.; O’Dell, S.D. Functional single-nucleotide polymorphism in acetyl-CoA carboxylase ACACB gene promoter. DNA Cell Biol. 2010, 29, 703–712.
- Chakravarthy, M.V.; Pan, Z.; Zhu, Y.; Tordjman, K.; Schneider, J.G.; Coleman, T.; Turk, J.; Semenkovich, C.F. “New” hepatic fat activates PPARalpha to maintain glucose, lipid, and cholesterol homeostasis. Cell Metab. 2005, 1, 309–322.
- Wu, M.; Singh, S.B.; Wang, J.; Chung, C.C.; Salituro, G.; Karanam, B.V.; Lee, S.H.; Powles, M.; Ellsworth, K.P.; Lassman, M.E.; et al. Antidiabetic and antisteatotic effects of the selective fatty acid synthase (FAS) inhibitor platensimycin in mouse models of diabetes. Proc. Natl. Acad. Sci. USA 2011, 108, 5378–5383.
- Ntambi, J.M.; Miyazaki, M.; Stoehr, J.P.; Lan, H.; Kendziorski, C.M.; Yandell, B.S.; Song, Y.; Cohen, P.; Friedman, J.M.; Attie, A.D. Loss of stearoyl-CoA desaturase-1 function protects mice against adiposity. Proc. Natl. Acad. Sci. USA 2002, 99, 11482–11486.
- Cohen, P.; Miyazaki, M.; Socci, N.D.; Hagge-Greenberg, A.; Liedtke, W.; Soukas, A.A.; Sharma, R.; Hudgins, L.C.; Ntambi, J.M.; Friedman, J.M. Role for stearoyl-CoA desaturase-1 in leptin-mediated weight loss. Science 2002, 297, 240–243.
- Jiang, G.; Li, Z.; Liu, F.; Ellsworth, K.; Dallas-Yang, Q.; Wu, M.; Ronan, J.; Esau, C.; Murphy, C.; Szalkowski, D.; et al. Prevention of obesity in mice by antisense oligonucleotide inhibitors of stearoyl-CoA desaturase-1. J. Clin. Investig. 2005, 115, 1030–1038.
- Dif, N.; Euthine, V.; Gonnet, E.; Laville, M.; Vidal, H.; Lefai, E. Insulin activates human sterol-regulatory-element-binding protein-1c (SREBP-1c) promoter through SRE motifs. Biochem. J. 2006, 400, 179–188.
- Matsuzaka, T.; Shimano, H. Insulin-dependent and -independent regulation of sterol regulatory element-binding protein-1c. J. Diabetes Investig. 2013, 4, 411–412.
- Postic, C.; Girard, J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: Lessons from genetically engineered mice. J. Clin. Investig. 2008, 118, 829–838.
- Abdul-Wahed, A.; Guilmeau, S.; Postic, C. Sweet sixteenth for ChREBP: Established roles and future goals. Cell Metab. 2017, 26, 324–341.
- Benhamed, F.; Denechaud, P.D.; Lemoine, M.; Robichon, C.; Moldes, M.; Bertrand-Michel, J.; Ratziu, V.; Serfaty, L.; Housset, C.; Capeau, J.; et al. The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J. Clin. Investig. 2012, 122, 2176–2194.
- Samuel, V.T.; Shulman, G.I. Nonalcoholic Fatty Liver Disease as a Nexus of Metabolic and Hepatic Diseases. Cell Metab. 2018, 27, 22–41.
- Dentin, R.; Tomas-Cobos, L.; Foufelle, F.; Leopold, J.; Girard, J.; Postic, C.; Ferre, P. Glucose 6-phosphate, rather than xylulose 5-phosphate, is required for the activation of ChREBP in response to glucose in the liver. J. Hepatol. 2012, 56, 199–209.
- Ortega-Prieto, P.; Postic, C. Carbohydrate Sensing Through the Transcription Factor ChREBP. Front. Genet. 2019, 10, 472.
- Chambers, K.T.; Chen, Z.; Lai, L.; Leone, T.C.; Towle, H.C.; Kralli, A.; Crawford, P.A.; Finck, B.N. PGC-1beta and ChREBP partner to cooperatively regulate hepatic lipogenesis in a glucose concentration-dependent manner. Mol. Metab. 2013, 2, 194–204.
- Uyeda, K.; Repa, J.J. Carbohydrate response element binding protein, ChREBP, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell Metab. 2006, 4, 107–110.
- Iroz, A.; Montagner, A.; Benhamed, F.; Levavasseur, F.; Polizzi, A.; Anthony, E.; Regnier, M.; Fouche, E.; Lukowicz, C.; Cauzac, M.; et al. A specific ChREBP and PPARalpha cross-talk is required for the glucose-mediated FGF21 response. Cell Rep. 2017, 21, 403–416.
- Baraille, F.; Planchais, J.; Dentin, R.; Guilmeau, S.; Postic, C. Integration of ChREBP-mediated glucose sensing into whole body metabolism. Physiology 2015, 30, 428–437.
- Jois, T.; Chen, W.; Howard, V.; Harvey, R.; Youngs, K.; Thalmann, C.; Saha, P.; Chan, L.; Cowley, M.A.; Sleeman, M.W. Deletion of hepatic carbohydrate response element binding protein (ChREBP) impairs glucose homeostasis and hepatic insulin sensitivity in mice. Mol. Metab. 2017, 6, 1381–1394.
- Denechaud, P.D.; Dentin, R.; Girard, J.; Postic, C. Role of ChREBP in hepatic steatosis and insulin resistance. FEBS Lett. 2008, 582, 68–73.
- Erion, D.M.; Popov, V.; Hsiao, J.J.; Vatner, D.; Mitchell, K.; Yonemitsu, S.; Nagai, Y.; Kahn, M.; Gillum, M.P.; Dong, J.; et al. The role of the carbohydrate response element-binding protein in male fructose-fed rats. Endocrinology 2013, 154, 36–44.
- Oh, A.R.; Sohn, S.; Lee, J.; Park, J.M.; Nam, K.T.; Hahm, K.B.; Kim, Y.B.; Lee, H.J.; Cha, J.Y. ChREBP deficiency leads to diarrhea-predominant irritable bowel syndrome. Metabolism 2018, 85, 286–297.
- Hall, A.M.; Finck, B.N. ChREBP refines the hepatic response to fructose to protect the liver from injury. J. Clin. Investig. 2017, 127, 2533–2535.
- Zhang, D.; Tong, X.; VanDommelen, K.; Gupta, N.; Stamper, K.; Brady, G.F.; Meng, Z.; Lin, J.; Rui, L.; Omary, M.B.; et al. Lipogenic transcription factor ChREBP mediates fructose-induced metabolic adaptations to prevent hepatotoxicity. J. Clin. Investig. 2017, 127, 2855–2867.
- Yan, J.H.; Guan, B.J.; Gao, H.Y.; Peng, X.E. Omega-3 polyunsaturated fatty acid supplementation and non-alcoholic fatty liver disease: A meta-analysis of randomized controlled trials. Medicine 2018, 97, e12271.
- Chiappini, F.; Coilly, A.; Kadar, H.; Gual, P.; Tran, A.; Desterke, C.; Samuel, D.; Duclos-Vallee, J.C.; Touboul, D.; Bertrand-Michel, J.; et al. Metabolism dysregulation induces a specific lipid signature of nonalcoholic steatohepatitis in patients. Sci. Rep. 2017, 7, 46658.
- Araya, J.; Rodrigo, R.; Videla, L.A.; Thielemann, L.; Orellana, M.; Pettinelli, P.; Poniachik, J. Increase in long-chain polyunsaturated fatty acid n-6/n-3 ratio in relation to hepatic steatosis in patients with non-alcoholic fatty liver disease. Clin. Sci. 2004, 106, 635–643.
- Chong, M.F.; Hodson, L.; Bickerton, A.S.; Roberts, R.; Neville, M.; Karpe, F.; Frayn, K.N.; Fielding, B.A. Parallel activation of de novo lipogenesis and stearoyl-CoA desaturase activity after 3d of high-carbohydrate feeding. Am. J. Clin. Nutr. 2008, 87, 817–823.
- Knebel, B.; Fahlbusch, P.; Dille, M.; Wahlers, N.; Hartwig, S.; Jacob, S.; Kettel, U.; Schiller, M.; Herebian, D.; Koellmer, C.; et al. Fatty liver due to increased de novo lipogenesis: Alterations in the hepatic peroxisomal proteome. Front. Cell Dev. Biol. 2019, 7, 248.
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922.
- Petersen, M.C.; Shulman, G.I. Roles of Diacylglycerols and Ceramides in Hepatic Insulin Resistance. Trends Pharmacol. Sci. 2017, 38, 649–665.
- Birkenfeld, A.L.; Shulman, G.I. Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 diabetes. Hepatology 2014, 59, 713–723.
- Morino, K.; Petersen, K.F.; Shulman, G.I. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes 2006, 55 (Suppl. 2), S9–S15.
- Jornayvaz, F.R.; Shulman, G.I. Diacylglycerol activation of protein kinase Cepsilon and hepatic insulin resistance. Cell Metab. 2012, 15, 574–584.
- Samuel, V.T.; Liu, Z.X.; Qu, X.; Elder, B.D.; Bilz, S.; Befroy, D.; Romanelli, A.J.; Shulman, G.I. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J. Biol. Chem. 2004, 279, 32345–32353.
- Amin, N.B.; Darekar, A.; Anstee, Q.M.; Wong, V.W.; Tacke, F.; Vourvahis, M.; Lee, D.S.; Charlton, M.; Alkhouri, N.; Nakajima, A.; et al. Efficacy and safety of an orally administered DGAT2 inhibitor alone or coadministered with a liver-targeted ACC inhibitor in adults with non-alcoholic steatohepatitis (NASH): Rationale and design of the phase II, dose-ranging, dose-finding, randomised, placebo-controlled MIRNA (Metabolic Interventions to Resolve NASH with fibrosis) study. BMJ Open 2022, 12, e056159.
- Feldstein, A.E.; Charatcharoenwitthaya, P.; Treeprasertsuk, S.; Benson, J.T.; Enders, F.B.; Angulo, P. The natural history of non-alcoholic fatty liver disease in children: A follow-up study for up to 20 years. Gut 2009, 58, 1538–1544.
- Browning, J.D.; Szczepaniak, L.S.; Dobbins, R.; Nuremberg, P.; Horton, J.D.; Cohen, J.C.; Grundy, S.M.; Hobbs, H.H. Prevalence of hepatic steatosis in an urban population in the United States: Impact of ethnicity. Hepatology 2004, 40, 1387–1395.
- Conus, F.; Rabasa-Lhoret, R.; Peronnet, F. Characteristics of metabolically obese normal-weight (MONW) subjects. Appl. Physiol. Nutr. Metab. 2007, 32, 4–12.
- Bugianesi, E.; Gastaldelli, A.; Vanni, E.; Gambino, R.; Cassader, M.; Baldi, S.; Ponti, V.; Pagano, G.; Ferrannini, E.; Rizzetto, M. Insulin resistance in non-diabetic patients with non-alcoholic fatty liver disease: Sites and mechanisms. Diabetologia 2005, 48, 634–642.
- Luo, J.; Yang, H.; Song, B.L. Mechanisms and regulation of cholesterol homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 225–245.
- Farrell, G. Should we lower lipids in nonalcoholic fatty liver disease? Clin. Gastroenterol. Hepatol. 2014, 12, 152–155.
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An overview of sphingolipid metabolism: From synthesis to breakdown. In Sphingolipids as Signaling and Regulatory Molecules; Springer: New York, NY, USA, 2010; Volume 688, pp. 1–23.
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150.
- Merrill, A.H., Jr.; Schmelz, E.M.; Dillehay, D.L.; Spiegel, S.; Shayman, J.A.; Schroeder, J.J.; Riley, R.T.; Voss, K.A.; Wang, E. Sphingolipids--the enigmatic lipid class: Biochemistry, physiology, and pathophysiology. Toxicol. Appl. Pharmacol. 1997, 142, 208–225.
- Uchida, Y. Ceramide signaling in mammalian epidermis. Biochim. Biophys. Acta 2014, 1841, 453–462.
- Wakil, S.J.; Abu-Elheiga, L.A. Fatty acid metabolism: Target for metabolic syndrome. J. Lipid Res. 2009, 50, S138–S143.
- Fritz, I. The effect of muscle extracts on the oxidation of palmitic acid by liver slices and homogenates. Acta Physiol. Scand. 1955, 34, 367–385.
- Fritz, I.B.; Yue, K.T. Long-chain carnitine acyltransferase and the role of acylcarnitine derivatives in the catalytic increase of fatty acid oxidation induced by carnitine. J. Lipid Res. 1963, 4, 279–288.
- Bremer, J. Carnitine in intermediary metabolism. The biosynthesis of palmitylcarnitine by cell subfractions. J. Biol. Chem. 1963, 238, 2774–2779.
- McGarry, J.D.; Brown, N.F. The mitochondrial carnitine palmitoyltransferase system. From concept to molecular analysis. Eur. J. Biochem. 1997, 244, 1–14.
- Bartlett, K.; Eaton, S. Mitochondrial beta-oxidation. Eur. J. Biochem. 2004, 271, 462–469.
- Eaton, S.; Bartlett, K.; Pourfarzam, M. Mammalian mitochondrial beta-oxidation. Biochem. J. 1996, 320 Pt 2, 345–357.
- Rector, R.S.; Payne, R.M.; Ibdah, J.A. Mitochondrial trifunctional protein defects: Clinical implications and therapeutic approaches. Adv. Drug Deliv. Rev. 2008, 60, 1488–1496.
- Angdisen, J.; Moore, V.D.; Cline, J.M.; Payne, R.M.; Ibdah, J.A. Mitochondrial trifunctional protein defects: Molecular basis and novel therapeutic approaches. Curr. Drug Targets Immune Endocr. Metabol. Disord. 2005, 5, 27–40.
- Eaton, S.; Bursby, T.; Middleton, B.; Pourfarzam, M.; Mills, K.; Johnson, A.W.; Bartlett, K. The mitochondrial trifunctional protein: Centre of a beta-oxidation metabolon? Biochem. Soc. Trans. 2000, 28, 177–182.
- Grevengoed, T.J.; Klett, E.L.; Coleman, R.A. Acyl-CoA metabolism and partitioning. Annu. Rev. Nutr. 2014, 34, 1–30.
- Alves-Bezerra, M.; Cohen, D.E. Triglyceride metabolism in the liver. Compr. Physiol. 2017, 8, 1–8.
- Kerr, T.A.; Davidson, N.O. Cholesterol and nonalcoholic fatty liver disease: Renewed focus on an old villain. Hepatology 2012, 56, 1995–1998.
- Wurie, H.R.; Buckett, L.; Zammit, V.A. Diacylglycerol acyltransferase 2 acts upstream of diacylglycerol acyltransferase 1 and utilizes nascent diglycerides and de novo synthesized fatty acids in HepG2 cells. FEBS J. 2012, 279, 3033–3047.
- Yen, C.L.; Stone, S.J.; Koliwad, S.; Harris, C.; Farese, R.V., Jr. Thematic review series: Glycerolipids. DGAT enzymes and triacylglycerol biosynthesis. J. Lipid Res. 2008, 49, 2283–2301.
- Lewis, G.F.; Uffelman, K.D.; Szeto, L.W.; Weller, B.; Steiner, G. Interaction between free fatty acids and insulin in the acute control of very low density lipoprotein production in humans. J. Clin. Investig. 1995, 95, 158–166.
- Puri, P.; Baillie, R.A.; Wiest, M.M.; Mirshahi, F.; Choudhury, J.; Cheung, O.; Sargeant, C.; Contos, M.J.; Sanyal, A.J. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 2007, 46, 1081–1090.
- Arteel, G.E. Beyond reasonable doubt: Who is the culprit in lipotoxicity in NAFLD/NASH? Hepatology 2012, 55, 2030–2032.
- Zou, Y.; Li, S.; Xu, B.; Guo, H.; Zhang, S.; Cai, Y. Inhibition of Proprotein Convertase Subtilisin/Kexin Type 9 Ameliorates Liver Fibrosis via Mitigation of Intestinal Endotoxemia. Inflammation 2020, 43, 251–263.
- Carroll, R.G.; Zaslona, Z.; Galvan-Pena, S.; Koppe, E.L.; Sevin, D.C.; Angiari, S.; Triantafilou, M.; Triantafilou, K.; Modis, L.K.; O’Neill, L.A. An unexpected link between fatty acid synthase and cholesterol synthesis in proinflammatory macrophage activation. J. Biol. Chem. 2018, 293, 5509–5521.
- Syed-Abdul, M.M.; Le, N.T.; Jacome-Sosa, M.; Hu, Q.; Oxler, B.M.; Bingham, K.; Arreola, R.; Juboori, A.M.A.; Gaballah, A.H.; Bartholow, B.D.; et al. Tailgate study: A pilot study measuring the impact of food and alcohol intake on whole-body and liver metabolism. FASEB J. 2018, 32, 760–766.

