Metabolic-associated steatotic liver disease (MASLD) is a cluster of pathological conditions primarily developed due to the accumulation of ectopic fat in the hepatocytes. During the severe form of the disease, i.e., metabolic-associated steatohepatitis (MASH), accumulated lipids promote lipotoxicity, resulting in cellular inflammation, oxidative stress, and hepatocellular ballooning. If left untreated, the advanced form of the disease progresses to fibrosis of the tissue, resulting in irreversible hepatic cirrhosis or the development of hepatocellular carcinoma. Although numerous mechanisms have been identified as significant contributors to the development and advancement of MASLD, altered lipid metabolism continues to stand out as a major factor contributing to the disease.
- MASLD
- MASH
- cholesterol
- DNL
- fatty acid oxidation
1. Metabolic-Associated Steatotic Liver Disease
2. Pathophysiological Changes at the Molecular Level in MASLD
Serological studies and numerous genomic studies performed on hepatocytes sourced from patients diagnosed with MASLD and individuals undergoing bariatric surgery consistently reveal a noticeable increase in several biochemical parameters [15] and the upregulation of key enzymes integral to the de novo lipogenesis (DNL) pathway [16][17][18][19][20][21][16,17,18,19,20,21]. As a master regulator of the DNL pathway, sterol regulatory element-binding protein 1c (SREBP1c), primarily activated by insulin, exhibited a significant increase in MASLD patients compared to those without MASLD [17][20][22][23][24][25][17,20,22,23,24,25], underscoring its central role in governing this metabolic process. Contrary to these findings, single-cell RNA sequencing (scRNA-seq) combined with computational network analyses to explore lipid signatures in mice with MASLD showed that, despite its traditional role as a driver of lipid synthesis, high SREBP1 expression is not predictive of hepatic lipid accumulation in non-alcoholic fatty liver disease (NAFLD); instead, the study identifies the constitutive androstane receptor (CAR) as a key player in regulating functional modules associated with cholesterol homeostasis, bile acid metabolism, fatty acid metabolism, and estrogen response, demonstrating its correlation with steatohepatitis in human livers [26]. Notably, among the subsequent enzymes regulated by SREBP1c, the isoforms of acetyl-CoA carboxylase (ACC) exhibited a remarkable increase, i.e., a more than eight-fold increase in expression in MASLD patients compared to those with normal liver profiles [16][17][19][16,17,19]. In addition to an increase in DNL enzyme expression, patients with MASLD also exhibited altered expression of FA binding protein (FABP), FA transport protein (FATP) [16][17][21][27][28][16,17,21,27,28], and CD36 [16][21][28][16,21,28]—genes responsible for FA uptake. Moreover, genes associated with triacylglycerols (TAGs) synthesis, including diacylglycerol o-acyltransferase 2 (DGAT2) [19] and microsomal triglyceride transfer protein (MTTP) [16][17][29][16,17,29], along with genes impacting very-low-density lipoprotein (VLDL) kinetics, exemplified by apolipoprotein B100 (apoB100) [16][17][29][16,17,29], exhibited increased expression levels. Furthermore, genes related to the oxidation of FAs, including peroxisome proliferator-activated receptor gamma (PPAR-γ) [27] and carnitine palmitoyltransferase 1 (CPT1) [16][27][16,27], were upregulated, while PPAR-γ coactivator 1α (PGC-1α) was downregulated [30]. It is worth noting that Moore et al. reported decreased expression of FA oxidation genes in MASLD patients [31]. MASH patients, in comparison with MASLD patients, exhibited lower expression of peroxisome proliferator-activated receptor α (PPAR-α), MTTP, and apoB100, but no changes were observed in SREBP1c, FASN, DGAT1 and 2, FABP and FATP, and CD36 [32][33][34][32,33,34].3. Lipid Synthesis in MASLD
3.1. Fatty Acids
As shown in Figure 1, FA synthesis is a complex biochemical process responsible for the synthesis of FAs, utilizing glycerol and carbon molecules. When FAs are generated from non-lipid sources, notably carbohydrates, this metabolic pathway is termed DNL [35][36][37,38]. Serving as the master regulator, SREBP1c controls the activation of enzymes of the DNL pathway, collectively governing the complex process of FA synthesis at the molecular level through their specific functions. The activation of SREBP-1c has been demonstrated to be regulated by insulin concentrations, with higher insulin levels leading to increased SREBP-1c activation [22]. At the molecular level, crucial enzymes involved in FA synthesis include ACC, which catalyzes acetyl-CoA carboxylation, a pivotal step in FA synthesis. FASN plays an orchestrating role in the intricate assembly of FAs, ensuring the formation of these essential molecules. SCD-1 is responsible for facilitating desaturation reactions, crucial for modifying FA chains to confer specific properties.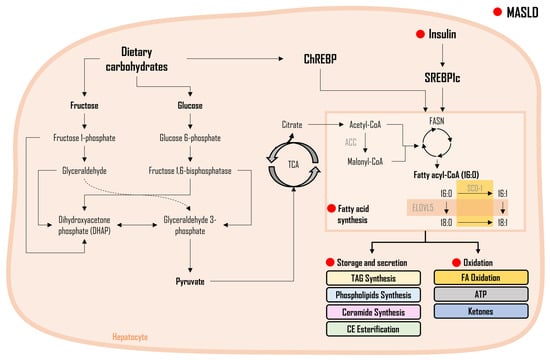
Saturated vs. Unsaturated FAs in MASLD
3.2. Triacylglycerol and Diacylglycerol
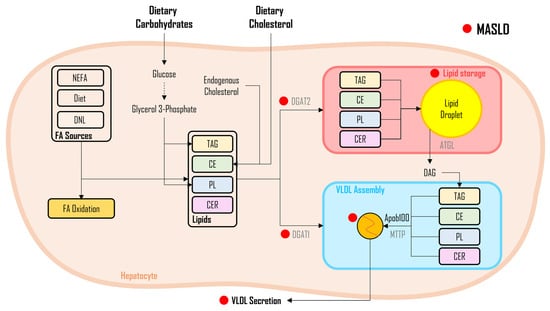
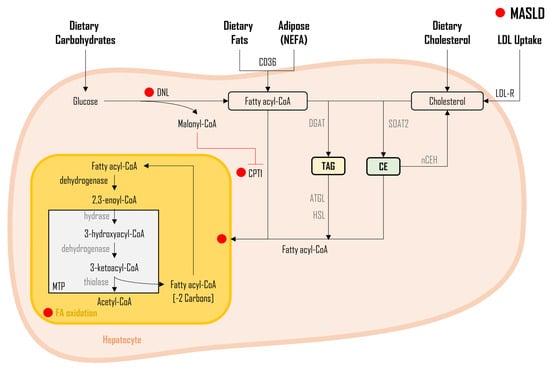
As illustrated in Figure 2, three distinct sources of FAs DNL from dietary carbohydrates, non-esterified FAs (NEFA) released from adipose tissue, and FAs obtained from the diet, undergo a sequence of enzymatic conversions, resulting in the formation of fatty acyl-CoA molecules. Subsequently, fatty acyl-CoA undergoes a series of enzymatic reactions, with glycerol-3-phosphate acyltransferase (GPAT) facilitating their conjugation to glycerol-3-phosphate, leading to the synthesis of lysophosphatidate. Next, another fatty acyl-CoA is incorporated into the process through the enzymatic activity of 1-acylglycerol-3-phosphate-O-acyltransferase (AGPAT), resulting in the formation of phosphatidate. The generated phosphatidate molecules, in turn, undergo enzymatic modification via phosphohydrolase-1 (PPH-1), resulting in the production of diglycerides (DAG). It is noteworthy that DAGs can also be derived from monoglycerides (MAG), with monoacylglycerol acyltransferase (MGAT) playing a pivotal role, although this specific pathway is not depicted in Figure 2.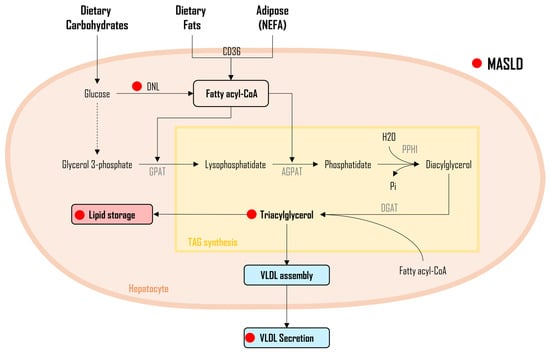
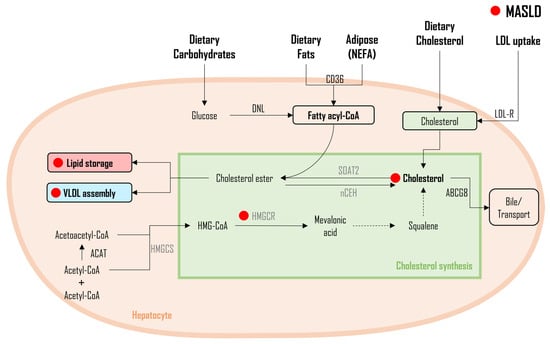
3.3. Cholesterol
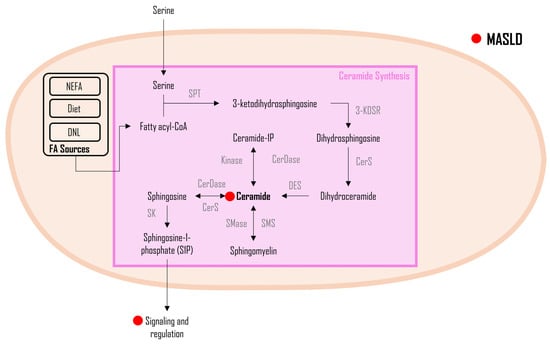
3.4. Ceramides
4. Lipid oxidation in MASLD
FA Oxidation
5. Lipid Flux and Storage in MASLD