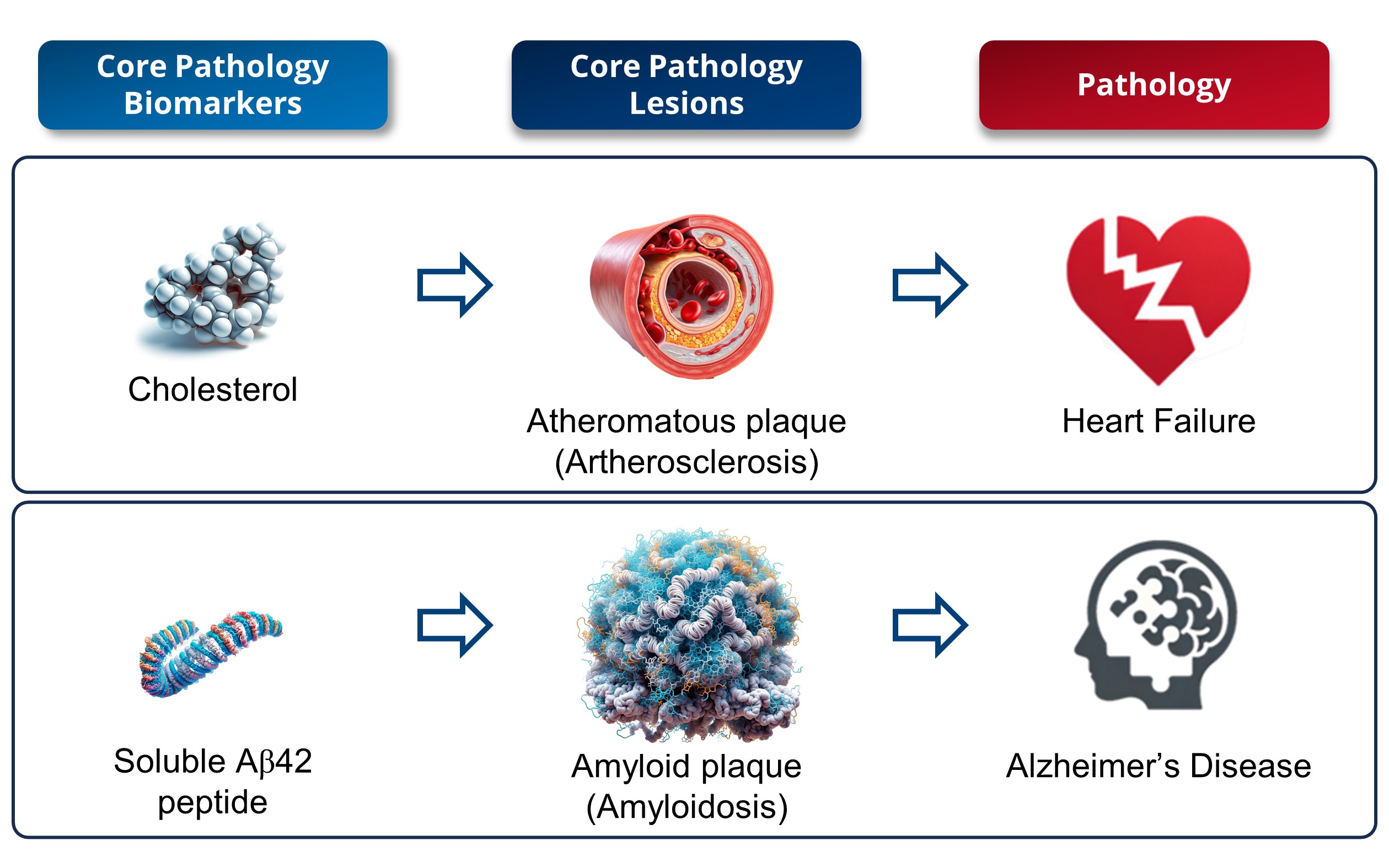
1. Interpretation of a Core Biomarker: Cholesterol against Amyloid Peptide (Figure 1)
In the field of medical research, significant parallels can be drawn between cholesterol in cardiovascular diseases and soluble amyloid peptide in AD, as their roles are similar in terms of risk factors and biomarkers. Serum cholesterol and low-density lipoprotein (LDL) are considered cardiovascular risk factors
[1], while amyloid levels are increasingly regarded as definitive biomarkers of AD.
Historical research has revealed that cholesterol is a key component of atherosclerotic plaques
[2], thus enhancing our understanding of heart diseases. Similarly to cholesterol’s role in atherosclerosis, studies have shown that amyloid peptides are primary constituents of amyloid plaques in AD
[3], thus enhancing our understanding of its pathology. However, while elevated levels of LDL-c or serum cholesterol are markers of atherosclerosis, serum cholesterol levels or the presence of atherosclerotic plaques are merely risk factors for the development of cardiovascular diseases
[1]. Similarly, reduced levels of soluble amyloid in biofluids (CSF and blood) reliably indicate the presence of brain amyloid plaques, while levels of soluble amyloid in biofluids or amyloid plaques are risk factors for the onset of AD dementia symptoms
[4][5][6][7][8][9]. Like cholesterol, the soluble peptide Aβ42 exerts physiological functions
[10]. In both cases, it is a supra-physiological excess that is responsible for the pathological effects. Yet, it is important to note that the toxicity associated with an excess of soluble amyloid Aβ42 peptides is comparatively greater than that resulting from elevated blood cholesterol levels.
There will be significant differences in treatment strategies for heart diseases and AD. Statins, which are effective at lowering cholesterol levels, differ from anti-amyloid antibodies in terms of administration (oral vs. intravenous injection), cost (a few dozen dollars vs. over USD 20,000 annually), and side effects (few vs. significant side effects). Consequently, prescribing statins to as many hypercholesterolemia patients as possible is viewed as fulfilling a medical need and posing minimal risk to both the patient and the healthcare system. However, this widespread approach, which is feasible with statins, is not viable with anti-amyloid antibodies. Treating all amyloid-positive individuals is not feasible due to the high rate of over-medication (over 20%) associated with secondary risks and significant treatment costs. This underscores the need for precision medicine in AD focused on developing specific biomarkers for personalized and cost-effective therapeutic approaches. The aim is to manage AD more effectively and balance benefits and risks while considering the financial implications for healthcare systems.
2. A potential Confirmation Bias in the Search for Alzheimer’s Biomarkers
Cochrane reviews unveiled a diagnostic test accuracy range for biomarkers related to the deregulation of amyloid metabolism to predict the patients who will develop symptoms of AD dementia
[4][5][6][7][8][9]. The median performance of these tests, predicated on an estimate of amyloid plaque presence, stands at
81.5% sensitivity (95% CI: 67–96%) and
66.5% specificity (95% CI: 50–72%). Consequently, given an AD prevalence of 60% among the population with cognitive impairment (MCI and dementia), a diagnosis exclusively anchored in amyloid metabolism deregulation computes to
21.5% false positive rate. While existing studies have uniformly determined that amyloid-based diagnostic tests for AD lack specificity, the research community remains disproportionately focused on amyloid and tau species for AD diagnosis. This trend overlooks the potential long-term implications of this issue. It is hypothesized that such a persistent focus might be influenced by confirmation bias in the field.
Confirmation bias is an important concept in psychology and cognitive sciences. It refers to the human tendency to seek, interpret, and favor information that confirms pre-existing beliefs or hypotheses while ignoring, minimizing, or rejecting information that contradicts them. In other words, when faced with new information or experiences, the human mind tends to prefer those that align with our preconceived ideas and accepts them more easily. This can occur consciously or unconsciously, and it can influence decision making, the evaluation of evidence, and our perception of the world around us.
In this instance, once the diagnostic principles based on AD’s biological markers (indicative of amyloid metabolism homeostasis disruption and suggesting amyloid plaque presence) were established, researchers quickly adopted these criteria. They began using them for AD diagnosis in research settings, including patient selection for clinical trials, even before thoroughly assessing the diagnostic value of these criteria. Thus, in patients for whom AD symptoms are not suspected, analysis of core biomarkers linked to amyloid deposits is not considered a priority
[11]. This makes it difficult for clinicians to estimate the specificity of core biomarkers in this cognitively impaired, non-AD population. For patients suspected of having AD because of their symptoms, biomarkers can be measured, but only as exclusion criteria. Amyloid-positive patients suspected of having AD will thus be diagnosed with AD, while amyloid-negative patients will be excluded for AD diagnosis. At the time of diagnosis, the clinician does not have the clinical elements—in particular, the cognitive decline towards more advanced stages that will follow the diagnosis—to establish the false-negative rate of amyloid biomarkers. Under these conditions, it remains complex for clinicians in their current practice to obtain an informed estimate of the use of biomarkers of deregulation of amyloid metabolism homeostasis.
The addition of these biomarkers to the diagnostic decision-making process has increased confidence in the veracity of the diagnosis for physicians, as it is easier to interpret. Thus, the evaluation criterion became the diagnosis of truth: an AD patient must be amyloid-positive, and other brain pathologies must be amyloid-negative. This circular reasoning led to confirmation of the hypothesis that, based on these criteria, all patients diagnosed as non-AD were all negative and AD patients were all amyloid-positive in clinical routine. Although scientific data on the diagnostic performance of amyloid biomarkers were available, clinicians did not take them into account, as they were convinced of the veracity of the diagnosis based exclusively on the biomarkers to which they had access. Subsequently, the search for newer, more specific biomarkers appeared to take a secondary role, as biomarkers for amyloid deposits were deemed potentially adequate. However, this confirmation bias is far from trivial, and it could have important practical consequences for the search for new diagnostic biomarkers of pre-dementia AD.
A confirmation bias lies in the ability to analyze experimental data through the prism of pre-established hypothesis. The post-mortem analysis of brains of centenarians (aged between 100 and 111 years) without dementia symptoms revealed similar levels of amyloid and tau protein buildup as those seen in AD patients. Thus, 55% of the centenarians studied had an NIA amyloid stage score greater than or equal to 2 and 83% had a Braak NFT stage score greater than or equal to III. A large proportion of centenarians spontaneously present a deregulation of amyloid and tau metabolism homeostasis, resulting in the appearance of brain lesions characteristic of AD in the absence of any cognitive symptoms
[12]. The presence of these lesions is thus not sufficient to make an AD diagnosis, which confirms the low specificity of these lesions to AD. In order not to invalidate the biological diagnosis, an argument put forward is that these people would have developed AD dementia if they had lived long enough. This argument rests on the principle that the lack of empirical evidence is not synonymous with empirical refutation. Therefore, the dependence on indemonstrable hypotheses tends to reinforce a pre-existing theory.
Demonstrating our hypothesis that confirmation bias may influence the perceived diagnostic utility of amyloid plaques in AD is difficult, yet it could provide some explanation for the prevailing tendency to attribute AD to its biological components despite known specificity constraints.
3. The Impact on the Search for AD Biomarkers and Treatments
3.1. Challenges in Precision Medicine: Treatment Complexity in Alzheimer’s Disease
The amyloid confirmation bias could have profound implications for the quest to identify diagnostic biomarkers for AD. The immediate consequence is a skewed focus on pinpointing biomarkers that align with amyloid status. As anti-amyloid treatments gain approval, AD management enters a precision medicine phase: administering the right drug to the right patient at the right time. Paradoxically, tests reliant on detecting amyloid deposits contradict the principles of precision medicine. They tend to mistakenly diagnose >20% of the patients without AD, thus leading to treatment for those who inherently will not respond to disease-modifying therapies (DMTs). While the impact of amyloid loads on memory issues in non-AD brain disorders is largely unexplored, scientific evidence challenges the notion that amyloid plaques are the primary cause of symptoms in these conditions. For instance, in disorders like Parkinson’s disease
[13], Lewy body dementia
[14], cortical basal syndrome
[15], post-stroke neurodegeneration
[16], schizophrenia
[17], alcohol-related cognitive disorders
[18], and late-life depression
[19], which are all linked to mild cognitive impairment (MCI), the presence of amyloid deposits does not seem causally tied to memory problems. Instead, these deposits might stem from typical brain aging due to soluble amyloid clearance defects. Consequently, reducing amyloid levels in these patients might not effectively slow cognitive decline. The exclusive use of biological diagnosis to prescribe treatments, particularly anti-amyloid treatments, will insidiously tend to apply the same treatment to all amyloid-positive individuals, whether or not they have AD, and whether or not they respond to treatment.
3.2. Expanding Alzheimer’s Understanding beyond Amyloid: Challenges and Opportunities
Relying on a biological diagnosis rooted in amyloid plaque presence affects the efficacy demonstration of new treatments. Amyloid-based tests lead to the inclusion of non-responders in clinical trials due to low AD specificity, thus reducing statistical power in evaluating treatment effectiveness. To emulate cancer diagnosis successes, where highly specific early tests guide timely, tailored treatment
[20], researchers must develop diagnostics that uphold this standard.
Reducing AD diagnosis to a mere biological definition constrains the discovery of unique AD pathological mechanisms. Studies driven by the biological definition compare amyloid-negative, cognitively normal subjects to amyloid-negative dementia patients, thus neglecting AD’s complexity. About 41% of cognitively normal, elderly patients test positive for amyloid spontaneously
[21], and 15% of individuals clinically diagnosed with AD dementia are amyloid-negative
[22]. Consequently, these studies elucidate amyloid-status-induced mechanisms rather than true AD mechanisms, thus reinforcing confirmation bias.
This confirmation bias oversimplifies AD complexity to amyloid and tau brain lesions. Amyloid metabolism deregulation is not binary but rather a nuanced spectrum. Even slight deregulation in an individual could trigger AD dementia without crossing arbitrary positivity thresholds, which underscores the need to explore resistance/resilience mechanisms against amyloid metabolism disruption. Understanding protective mechanisms could yield alternatives to anti-amyloid antibodies. Additionally, the focus on cerebral lesions disregards peripheral pathological mechanisms. Exploring peripheral mechanisms might unveil novel, lower-side-effect, anti-Alzheimer’s approaches. Indeed, the side effects associated with anti-amyloid antibodies, such as enlarged cerebral ventricles, cerebral edema, and microhemorrhages
[23], may stem from their cerebral mode of action. These side effects could potentially arise from increased mechanical permeabilization of the blood–brain barrier due to heightened osmotic pressure
[24] caused by a higher peripheral concentration of anti-amyloid antibodies relative to their cerebral concentration (<2% of the injected amount)
[25]. Alternatively, they might result from an off-target effect on cerebral amyloid angiopathy (CAA), which could increase the Blood–Brain Barrier (BBB)’s permeability and cerebral edema
[26]. Utilizing drugs with a primarily peripheral mode of action could mitigate these side effects, as their effectiveness does not depend on their presence in the brain, thereby preserving the integrity of the BBB. Furthermore, such drugs may allow for lower dosages compared to anti-amyloid antibodies, thus potentially reducing off-target biological effects. Developing peripherally acting drugs could potentially offer low-side-effect therapeutic options.
Finally, reducing AD diagnosis to a biological basis could hinder active treatment development at the asymptomatic stage. Using cognitive impairment absence and positive amyloid tests as inclusion criteria means only around 30% of participants in a primary prevention trial will suffer AD-related cognitive decline. With such a low responder rate, demonstrating cognitive decline reduction becomes highly unlikely.
4. Breaking out of Confirmation Bias
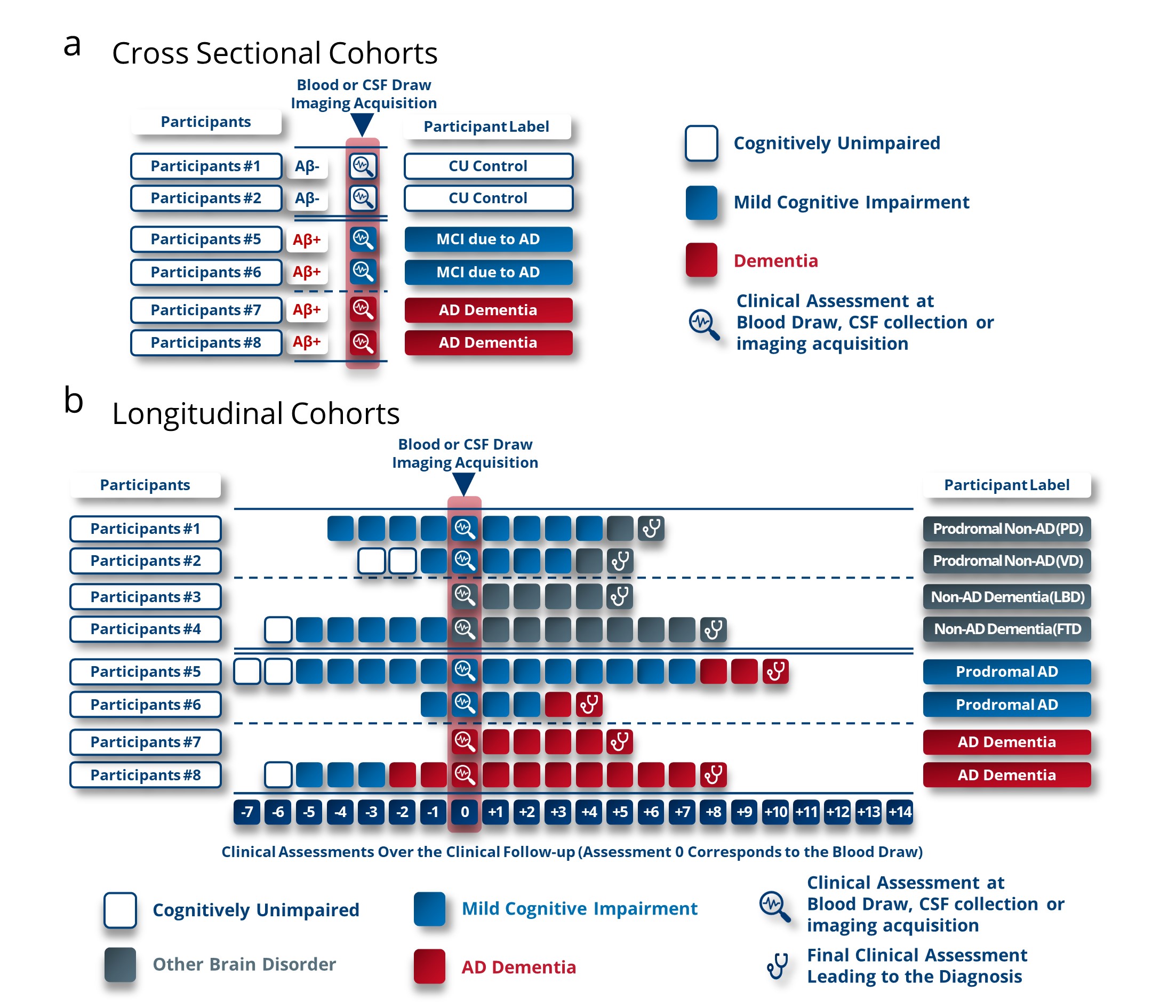
To counteract a potential confirmation bias and its detrimental impact on future research endeavors, we present a set of straightforward recommendations. At the outset, we emphasize the critical need to reevaluate participant inclusion criteria and categorization protocols in the initial phases of biomarker discovery phases. The practice of using cross-sectional clinical groups consisting of cognitively unimpaired (CU) individuals testing negative for amyloid, alongside individuals labeled as having dementia due to AD based on a cognitive impairment and amyloid positivity, poses a significant obstacle to unveiling novel biomarkers or panels specific for AD and not for amyloid status (Figure 2a). The hindrance arises from the fact that approximately 21% of participants are misdiagnosed with AD based solely on amyloid testing, thus impeding the identification of distinct biomarkers indicative of AD.
Figure 2. Labelling method proposed for participants in the discovery phases of new Alzheimer’s disease biomarkers to reduce the confirmation bias observed. (a) Current criteria for inclusion of participants in the discovery or validation phases of Alzheimer’s biomarkers. Confirmation bias is reflected here in these criteria, which guide the results and thus the biomarkers to be correlated with amyloid status and not with pathological status (AD or non-AD). (b) Proposed criteria for labelling participants in Alzheimer’s biomarker discovery or validation studies. These criteria are based on cohorts with longitudinal follow-up to enable labelling based on cognitive characterization of participants at an advanced stage of brain pathology. We also suggest not including cognitively unaltered participants as a reference group and instead including patients with brain pathologies other than AD to determine performance as a differential diagnosis. These were the criteria used in studies referenced in Cochrane reviews. PD, Parkinson’s Disease; VD, vascular dementia; LBD, Lewy body Dementia; FTD, frontotemporal dementia.
To surmount this challenge, a viable solution involves categorizing patients according to their clinical diagnoses in the advanced stages of diverse brain disorders. Leveraging longitudinal cohorts becomes a pivotal strategy in this context (Figure 2b). One of the advantages of labeling patients based on a longitudinal cognitive decline lies in the alignment between the AD decline-related biomarkers and the need for clinical trials and the prescription of current or future treatments. Therefore, the discovery of new biomarkers based on retrospective cognitive labeling will highlight biomarkers that can identify patients who will progress towards AD dementia symptoms. This will improve the assessment of anti-Alzheimer’s therapies’ performance by including pre-demented patients who will progress towards AD dementia symptoms. These patients will also be the ones who should be prioritized for treatment compared to patients who will not experience cognitive decline (stable MCI) or who will develop symptoms other than AD dementia symptoms. Furthermore, the consideration of participant types is crucial. For robust identification of differential biomarkers of AD pre-dementia, it is essential to perform comparative analyses between CSF, blood or imaging biomarkers of individuals at the MCI stage, who have been followed longitudinally until they clinically progress to AD dementia or another neurodegenerative disorder (Figure 2b). This spectrum of cognitive disorders should encompass a wide range to encapsulate the utmost diversity within these alternative pathologies. In the discovery phase of new biomarkers, the array of disorders that could serve as a benchmark group includes, but is not limited to, frontotemporal dementia, Lewy body dementia, Parkinson’s disease, corticobasal degeneration, epilepsy, isolated amyloidosis, primary progressive aphasia, psychological or psychiatric disorders, traumatic brain injury, stroke, and vascular dementia. To ensure the discovery of broadly applicable biomarkers, it is advisable to undertake these investigative stages across a minimum of two independent cohorts, thereby mitigating the risk of identifying biomarkers with limited generalizability.
The identification of new biomarkers, made possible by unbiased labeling criteria, will facilitate a more accurate recognition of AD patients in the pre-dementia stages. These novel biomarkers or panel of biomarkers can be used in combination with amyloid status to prioritize patients for treatment with anti-amyloid antibodies for whom the risk of misdiagnosis is really low. They will also enable the development of new therapeutic strategies that are independent of an anti-amyloid mode of action. Additionally, they will lead to a more precise identification of AD patients even in the asymptomatic stage and could support the validation of primary prevention therapeutic approaches.
5. Alternative Biomarkers and More Possibilities
The development of novel imaging, CSF, or blood biomarkers that do not predict amyloid status but rather the cognitive progression towards symptoms of AD dementia would enable the implementation of the precision medicine needed to manage AD. Biomarkers not directly related to amyloid status are being evaluated, including, in particular, neuronal damage biomarkers (Neurofilament light chain protein (NfL), S100b and neuron-specific enolase (NSE)), biomarkers of neuro-inflammation (Glial fibrillary acidic protein (GFAP), Triggering receptor expressed on myeloid cells 2 (TREM2), chitinase 3-L1 (YKL-40), and Cytokines-chemokines) and other reactional biomarkers (Neurogranin) and markers of metabolic response (apolipoproteins, neurotrophic factors, intestinal and obesity markers, and diabetes and glycemic markers)
[27]. However, most of these biomarkers are not specific to AD but are deregulated in the context of other neurodegenerative diseases
[27]. This significantly reduces their potential as differential biomarkers.
An alternative is the discovery of multiomic biomarker panels (genomic, proteomic, lipidomic, and metabolic biomarkers). These biomarkers could then be used to train machine learning algorithms
[28] capable of predicting which patients will develop symptoms of AD dementia from MCI or even an asymptomatic stage. The composition of these panels may vary and may potentially include or exclude amyloid peptides, p-tau proteins, or APOE genotyping. However, the development of such multiomic panels is still in nascent stages, with limited studies conducted to date.
One notable study identified a 10-metabolite panel (comprising PC diacyl aa C36:6, PC aa C38:0, PC aa C38:6, PC aa C40:1, PC aa C40:2, PC aa C40:6, PC acyl-alkyl ae C40:6, lysoPC a C18:2, Propionyl AC (C3), and C16:1-OH). Initially, plasma samples were taken from cognitively healthy individuals. These individuals were then clinically followed over several years to monitor cognitive changes. The study employed this metabolite panel to train an algorithm that successfully differentiated between those who remained cognitively unimpaired and those who converted to MCI or AD dementia with over 90% accuracy (AUC = 0.92, sensitivity/specificity of 90%/90%)
[29]. However, this discovery was based on a single cohort without external validation. Subsequent validation in two independent cohorts, the Baltimore Longitudinal Study of Aging (BLSA) and the Age, Gene/Environment Susceptibility–Reykjavik Study (AGES-RS), did not replicate these findings. The analysis in these cohorts yielded significantly lower accuracy (BLSA, AUC = 0.642, sensitivity/specificity of 51.6%/65.7%; AGES-RS, AUC = 0.395, sensitivity/specificity of 47.0%/36.0%)
[30]. Further machine learning analysis of 187 metabolite concentrations in the BLSA cohort indicated only moderate predictive value, which did not translate effectively to the AGES-RS samples
[30]. These results underscore the importance of conducting the discovery phase of biomarker research using multiple independent cohorts to ensure the generalizability of findings and to minimize the risk of developing non-generalizable biomarker panels.
The potential of multiomic biomarker panels analyzed through artificial intelligence algorithms remains an underexplored avenue in AD research. This approach holds promise for identifying individuals with high specificity who are likely to develop cognitive symptoms of AD. Early identification would render these individuals eligible for targeted anti-AD treatments. Importantly, such a stratified approach could optimize the patient selection process by focusing on those who stand to benefit the most, thereby ensuring a favorable benefit-to-risk balance in therapeutic interventions. The clear advantage of this multiomic signature approach is that it could include biomarkers involved in various biological pathways, such as amyloid metabolism, tau metabolism, oxidative stress, inflammation, bioenergetics, blood coagulation, lipid metabolism, or immune response. This would not only enable the prediction of patients who are likely to develop AD dementia but also offer a tailored profile of each patient. This profile could identify the biological pathways specifically deregulated in each patient, thereby facilitating a more personalized therapeutic approach (likely a combination of treatments) best suited to their condition. However, a significant limitation in identifying such multiomic signatures is the challenge of accessing sufficient clinically followed patient samples until the onset of dementia during discovery phases. One potential solution to this obstacle of limited patient samples could be the pre-identification of biomarker panels in animal models, followed by their study in a smaller set of patient samples
[31].
 +1 credit
+1 credit