 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Anita Kornicka | -- | 8131 | 2023-12-21 11:22:43 | | | |
| 2 | Lindsay Dong | -126 word(s) | 8005 | 2023-12-22 03:36:05 | | |
Video Upload Options
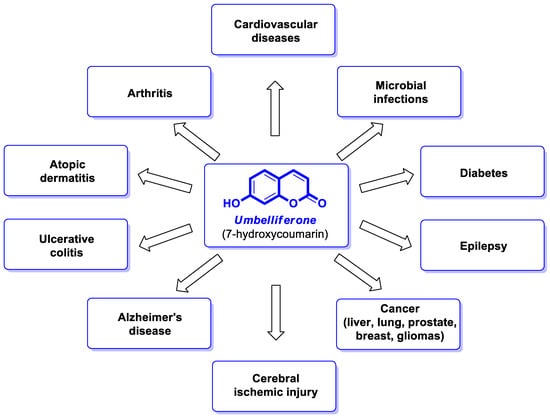
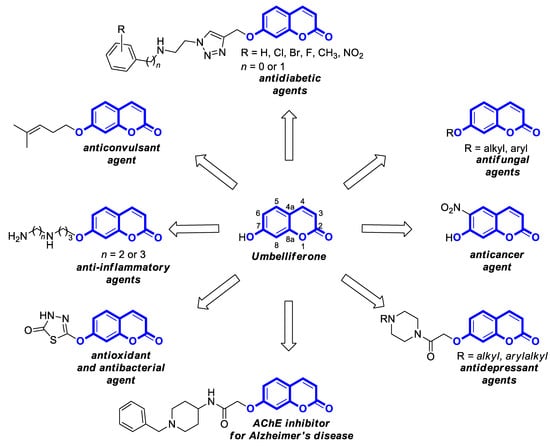
Umbelliferone (UMB), known as 7-hydroxycoumarin, hydrangine, or skimmetine, is a naturally occurring coumarin in the plant kingdom, mainly from the Umbelliferae family that possesses a wide variety of pharmacological properties. In addition, the use of nanoparticles containing umbelliferone may improve anti-inflammatory or anticancer therapy. Also, its derivatives are endowed with great potential for therapeutic applications due to their broad spectrum of biological activities such as anti-inflammatory, antioxidant, neuroprotective, antipsychotic, antiepileptic, antidiabetic, antimicrobial, antiviral, and antiproliferative effects.
1. Introduction
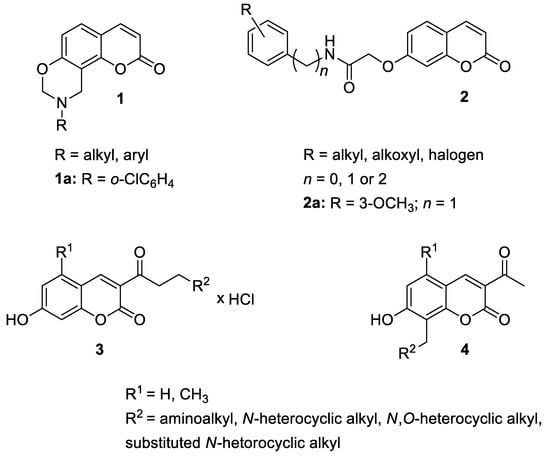
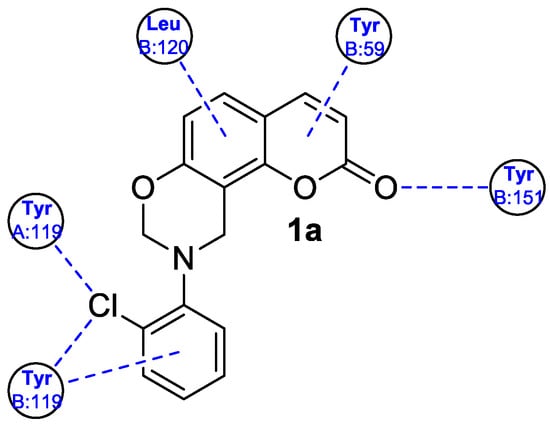
2. Anti-Inflammatory Activity
2.1. Anti-Inflammatory Properties of Umbelliferone
2.2. Synthetic 7-Hydroxycoumarin-Based Compounds as Anti-Inflammatory Agents
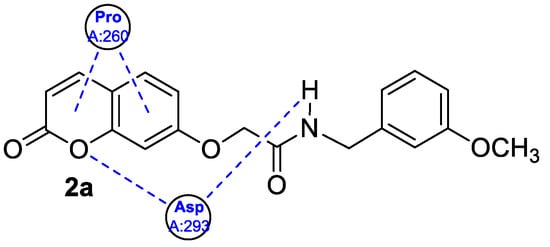
3. Antioxidant Activity
3.1. Antioxidant Properties of Umbelliferone
3.2. Synthetic 7-Hydroxycoumarin-Based Compounds as Antioxidant Agents
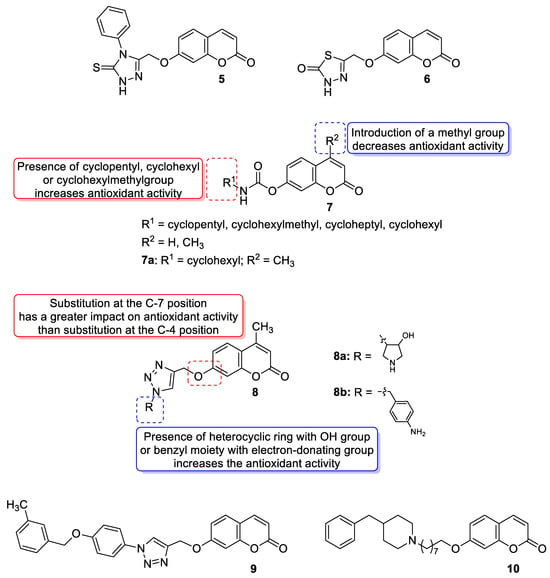
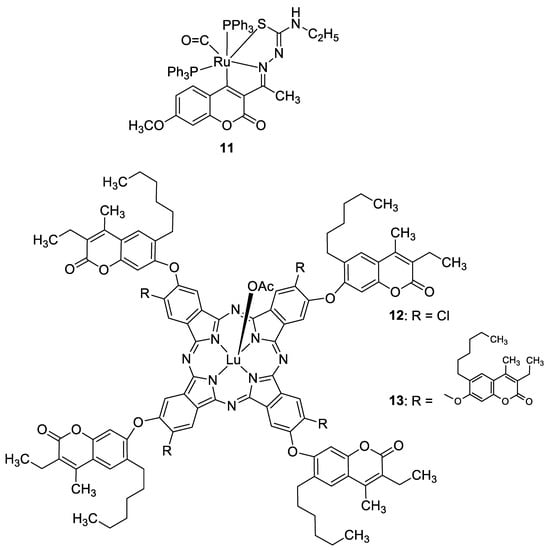
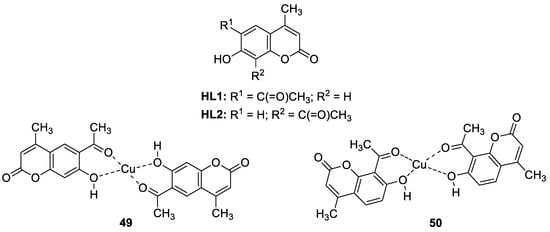
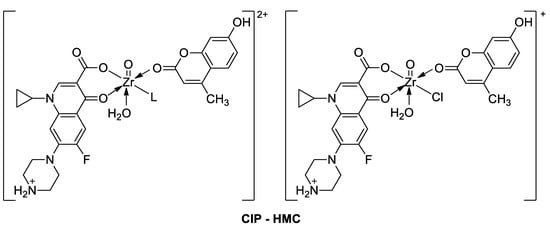
3.3. Metal Complexes with 7-Hydroxycoumarin-Based Compounds as Antioxidant Agents
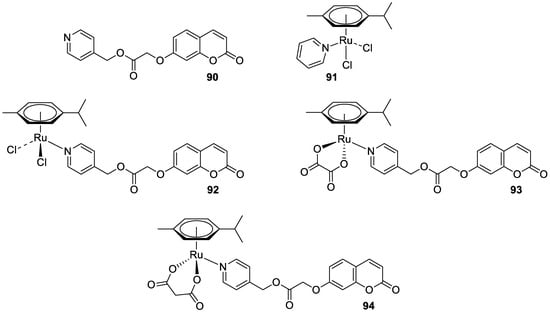
Recently, it was demonstrated that the radical scavenging ability of novel 3-acetyl-7-methoxy-4N-substituted thiosemicarbazones may be increased by ruthenium chelation [37]. The best radical scavenging properties have been shown by Ru(II) complex 11 (Figure 7), which displayed an antioxidant potency with about a fifteen-fold lower IC50 value than standard vitamin C in the DPPH model (IC50 = 5.28 µM vs. IC50 = 98.72 µM).
4. Umbelliferone and 7-Hydroxycoumarin-Based Compounds Acting in the Central Nervous System (CNS)
4.1. Neurodegenerative Disorders
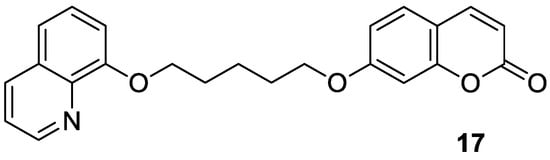
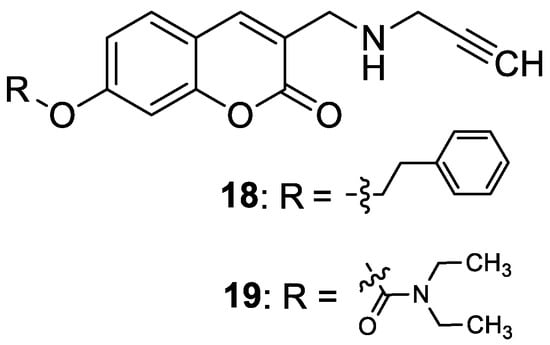
4.2. Neuropsychiatric Diseases
4.2.1. Synthetic 7-Hydroxycoumarin-Based Compounds Targeting Monoamine Oxidase (MAO) and D-Amino Acid Oxidase (DAAO)
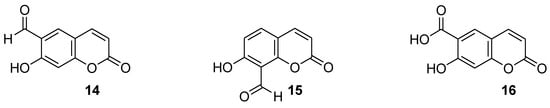
Recently, Seong et al. reported 6-formylumbelliferone derivative 14 and its isomeric analogue 15, presented in Figure 8, as highly selective hMAO-A inhibitors [47]. The higher selectivity and inhibitory activity towards hMAO-A exhibited 7-hydroxy-2-oxo-2H-chromene-6-carbaldehyde (14) with an IC50 value of 3.23 μM for hMAO-A and an IC50 value of 15.31 μM for hMAO-B. Enzyme kinetic studies revealed that both 6-formylumbelliferone 14 and 8-formylumbelliferone 15 are competitive hMAO inhibitors. These investigations were supported by molecular docking studies. Data revealed that compounds 14 and 15 dock well into the active sites of recombinant human monoamine oxidase A and B. The formyl group of 14 interacts strongly with substrate binding site (SBS) residues Tyr444 and Tyr197 of hMAO-A via water-mediated hydrogen bonds, whereas Phe352 and Tyr407 residues are involved in hydrophobic noncovalent π-π T-shaped (perpendicular T-shaped) and π-π stacking interactions. Hydroxycoumarin derivatives 14 and 15 demonstrated a neuroprotective effect due to their antilipid peroxidation and anti-Aβ25–35 (amyloid β self-assembly) aggregation activity in rat brain tissue.
4.2.2. Synthetic 7-Hydroxycoumarin-Based Compounds Targeting Serotonin Receptors
4.3. Antiepileptic Agents
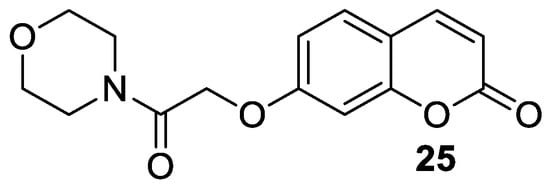
5. Umbelliferone and 7-Hydroxycoumarin-Based Compounds as Antidiabetic Agents
Extracts of widely cultivated plants, such as Musa species (banana flower ethanolic extracts) containing umbelliferone, were identified as potential antidiabetic herbal remedies in the management of diabetes and associated complications. Isolated umbelliferone increased the activity of crucial enzymes involved in glucose utilization and the glycolytic activity of the liver in alloxan-induced diabetic rats [52].
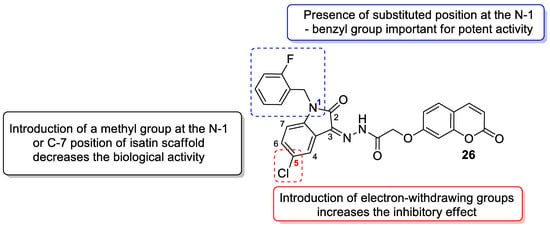
6. Chemotherapeutic Activity
6.1. Antimicrobial Properties of Umbelliferone and 7-Hydroxycoumarin-Based Compounds
The search for 7-hydroxycoumarin-based compounds as antimicrobial agents has developed due to the rapid growth of the drug resistance of microbes. The antimicrobial activity of parent 7-hydroxycoumarin—umbelliferone of various origins—was reported several times in in vitro studies [14]. Pure 7-hydroxycoumarin showed activity against Bacillus cereus with a MIC and MBC value of 62.5 µg/mL. However, this coumarin exhibited rather moderate effectiveness against other enteropathogenic bacterial species of Gram-negative Escherichia coli, Shigella sonnei, and Salmonella typhimurium, as well as Gram-positive Enterococcus faecalis and Staphylococcus aureus. In addition, high concentrations were often required to inhibit the growth of most species tested (MIC = 500–1000 µg/mL) [14].
Recently, the 7-hydroxycoumarin moiety was explored for its ability to inhibit biofilm formation by pathogenic bacterial strains. Firstly, the biofilm inhibitory properties of umbelliferone were shown at a concentration of 50 µg/mL against uropathogenic E. coli [56].
6.1.1. Synthetic 7-Hydroxycoumarin-Based Compounds as Antibacterial and Antifungal Agents
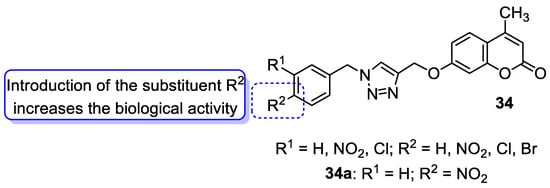
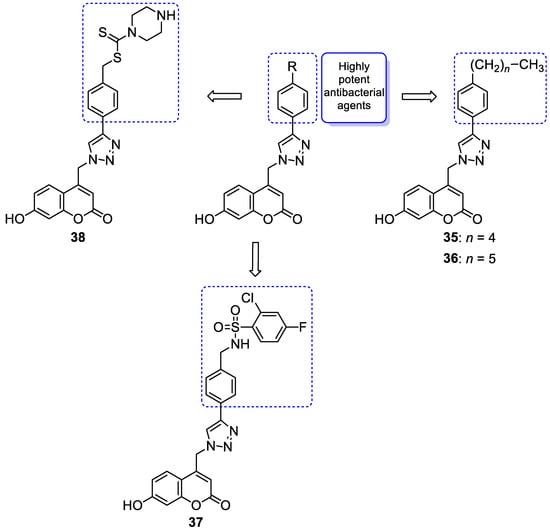
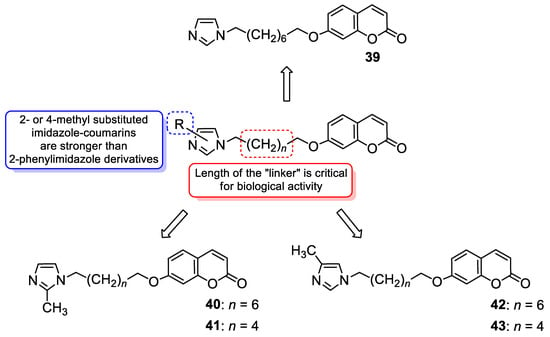
6.1.2. Metal Complexes of 7-Hydroxycoumarin-Based Compounds as Antibacterial and Antifungal Agents
6.2. Synthetic 7-Hydroxycoumarin-Based Compounds as Antituberculosis Agents
6.3. Synthetic 7-Hydroxycoumarin-Based Compounds as Antimalarial Agents
6.4. Umbelliferone and 7-Hydroxycoumarin-Based Compounds as Antiviral Agents
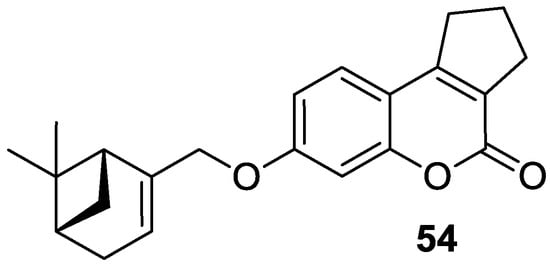
6.5. Umbelliferone and 7-Hydroxycoumarin-Based Compounds as Anticancer Agents
6.5.1. Synthetic 7-Hydroxycoumarin-Based Compounds as Histone Deacetylase (HDAC) Inhibitors
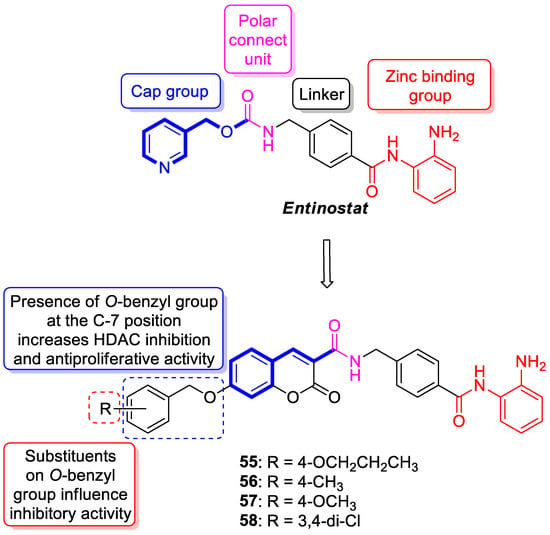
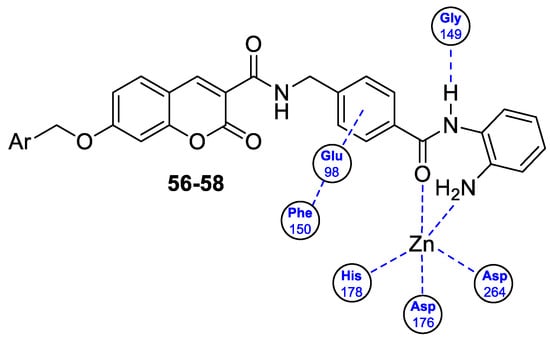
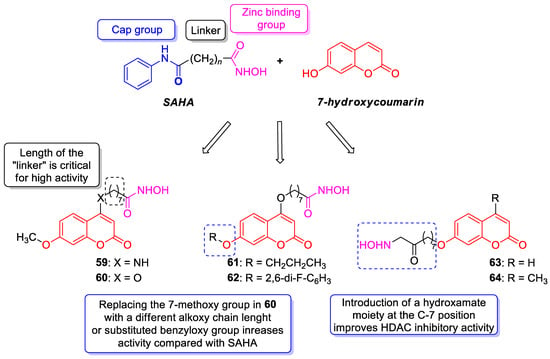
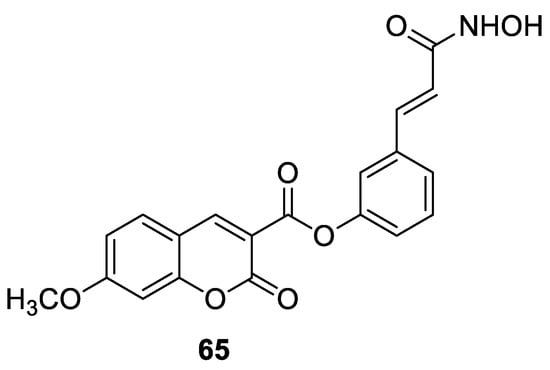
6.5.2. Synthetic 7-Hydroxycoumarin-Based Compounds as Androgen Receptor (AR) Antagonists
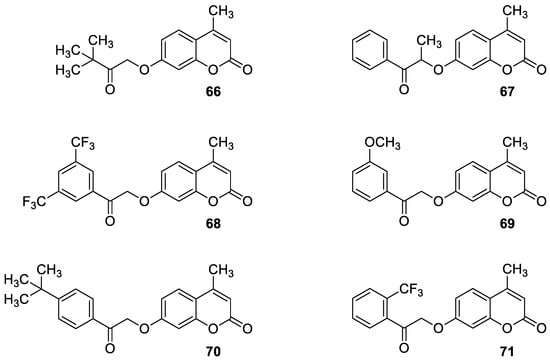
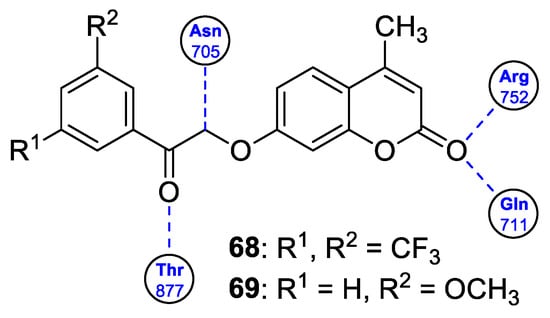
6.5.3. Synthetic 7-Hydroxycoumarin-Based Compounds as Inhibitors of the PIK3/Akt Signaling Pathway
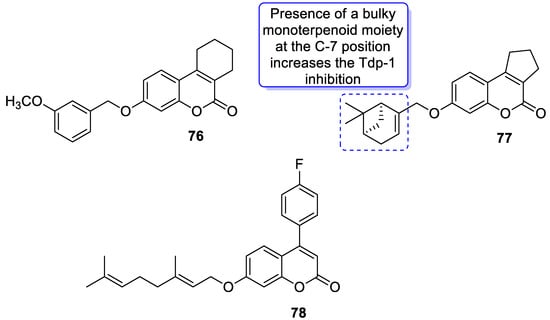
6.5.4. Monoterpene-Coumarin Hybrids as Tyrosyl-DNA Phosphodiesterase 1 (Tdp1) Inhibitors
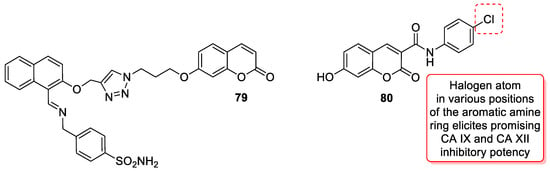
6.5.5. Synthetic 7-Hydroxycoumarin-Based Compounds as Carbonic Anhydrase (CA) Inhibitors
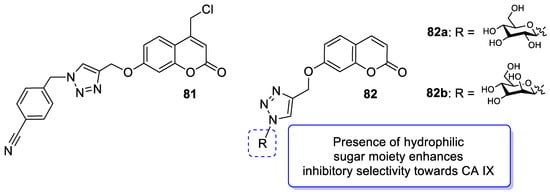
6.5.6. Synthetic 7-Hydroxycoumarin-Based Compounds as Cyclooxygenase-2 (COX-2) and 5-Lipoxygenas (5-LOX) Inhibitors
The development of novel antitumor drugs based on the inhibition of cyclooxygenase-2 (COX-2) has been an important part of antitumor drug development, because COX has proven to be a promising target in the design of antitumor agents. There is a growing understanding that several inflammatory mediators, such as cytokines, chemokines, and growth factors, may promote cancer formation and progression by controlling the tumor microenvironment.
Similar to COX-2, lipoxygenases (LOXs) are pro-inflammatory enzymes associated with arachidonic acid (AA) cascade. In this pathway, AA is transformed into hydroxyeicosatetraenoic acids derivatives (HETEs) and leukotrienes (LTs), which play a major key role in the development and progression of human cancers as a result of LOX activation. In particular, the overexpression of 5-LOX has been shown to have significant effects on the cell cycle, preventing apoptosis and stimulating angiogenesis.
In this line, Shen et al. designed a novel COX-2 and 5-LOX dual inhibitor composed of the 1-(4-sulfamoylphenyl)-5-(3,4,5-trimethoxyphenyl)-1H-pyrazole and 7-hydroxycoumarin moieties [94]. A high selectivity level has been observed for compound 83 towards enzyme subtypes based on its IC50 values of 0.23 µM for COX-2 and 0.87 µM for 5-LOX, making the tested compound superior to celecoxib as a positive control for COX-2 (IC50 = 0.41 µM) and zileuton for 5-LOX (IC50 = 1.35 µM).
6.5.7. Metal Complexes of 7-Hydroxycoumarin-Based Compounds as Anticancer Agents
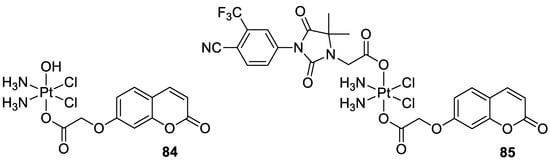
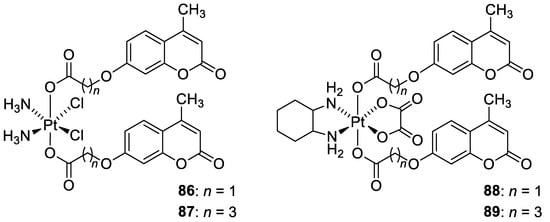
In 2018, Hua et al. described cou-platin (84, Figure 36) composed of 7-hydroxycoumarin and a platinum(IV) moiety derived from cisplatin as more potent towards a variety of cancer cells than cisplatin (IC50 = 0.08–2.46 µM vs. IC50 = 1.86–9.34 µM) [95]. The mechanistic studies with the use of human colon carcinoma HCT116 cells revealed that new Pt-binding molecule 84 is able to inhibit cancer cell growth via activation of cell apoptosis and inhibition of the ERK/MAPK signaling pathway.
7. Umbelliferone and 7-Hydroxycoumarin-Based Compounds as Probes and Sensors
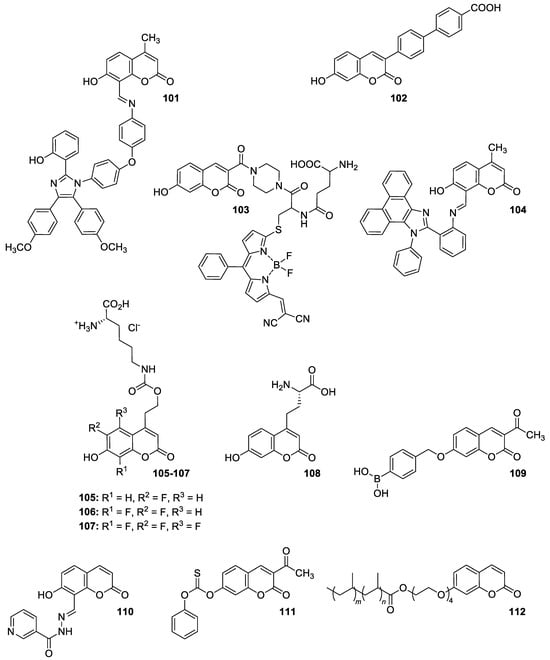
Levin et al. [100] synthesized novel dyad molecule 101 shown in Figure 40 composed of two different fluorophores: 1,2,4,5-tetraarylimidazole and 8-arylazomethinocoumarin. Because of the presence of both proton (hydrogen) donating/accepting groups in the structure, the designed molecule exhibits multiple fluorescence with maxima at 450 nm and 535 nm as a result of excited-state intramolecular proton transfer (ESIPT).
8. Conclusions
References
- Sharifi-Rad, J.; Cruz-Martins, N.; López-Jornet, P.; Pons-Fuster Lopez, E.; Harun, N.; Yeskaliyeva, B.; Beyatli, A.; Sytar, O.; Shaheen, S.; Sharopov, F.; et al. Natural coumarins: Exploring the pharmacological complexity and underlying molecular mechanisms. Oxid. Med. Cell. Longev. 2021, 2021, 6492346.
- Dawidowicz, A.L.; Bernacik, K.; Typek, R. Umbelliferone instability during an analysis involving its extraction process. Monatsh. Chem. 2018, 149, 1327–1340.
- Mazimba, O. Umbelliferone: Sources, chemistry and bioactivities review. Bull. Fac. Pharm. Cairo Univ. 2017, 55, 223–232.
- Radha, G.V.; Sadhana, B.; Trideva Sastri, K.; Ganapaty, S. Biooactive umbelliferone and its derivatives: An update. J. Pharmacogn. Phytochem. 2019, 8, 59–66.
- Lin, Z.; Zheng, H. Umbelliferon: A review of its pharmacology, toxicity and pharmacokinetics. Inflammopharmacology 2023, 31, 1731–1750.
- Fylaktakidou, K.C.; Hadjipavlou-Litina, D.J.; Litinas, K.E.; Nicolaides, D.N. Natural and synthetic coumarin derivatives with anti-inflammatory/antioxidant activities. Curr. Pharm. Des. 2004, 10, 3813–3833.
- Emami, S.; Dadashpour, S. Current developments of coumarin-based anti-cancer agents in medical chemistry. Eur. J. Med. Chem. 2015, 102, 611–630.
- Pan, Y.; Liu, T.; Wang, X.; Sun, J. Research progress of coumarins and their derivatives in the treatment of diabetes. J. Enzyme Inhib. Med. Chem. 2022, 37, 616–628.
- Genovese, S.; Epifano, F.; Curini, M.; Dudra-Jastrzebska, M.; Luszczki, J.J. Prenyloxyphenylpropanoids as a novel class of anticonvulsive agents. Bioorg. Med. Chem. Lett. 2009, 19, 5419–5422.
- Alipour, M.; Khoobi, M.; Moradi, A.; Nadri, H.; Moghadam, F.H.; Emami, S.; Hasanpour, Z.; Foroumadi, A.; Shafiee, A. Synthesis and anticholinesterase activity of new 7-hydroxycoumarin derivatives. Eur. J. Med. Chem. 2014, 82, 536–544.
- Wang, X.; Zhou, H.; Wang, X.; Lei, K.; Wang, S. Design, synthesis, and in vivo and in silico evaluation of coumarin derivatives with potential antidepressant effects. Molecules 2021, 26, 5556.
- Vasconcelos, J.F.; Teixeira, M.M.; Barbosa-Filho, J.M.; Agra, M.F.; Nunes, X.P.; Giulietti, A.M.; Ribeiro-dos-Santos, R.; Soares, M.B.P. Effects of umbelliferone in a murine model of allergic airway inflammation. Eur. J. Pharmacol. 2009, 609, 126–131.
- Zinovieva, M.L.; Zhminko, P.G. Single and repeat dose toxicity study of 7-hydroxycoumarin, ethanol, and their mixture in rats. J. Pharm. Pharmacol. 2017, 5, 237–244.
- Cruz, L.F.; de Figueiredo, G.F.; Pedro, L.P.; Amorin, Y.M.; Andrade, J.T.; Passos, T.F.; Rodrigues, F.F.; Souza, I.L.A.R.; Gonçalves, T.P.R.; Dos Santos Lima, L.A.R.; et al. Umbelliferone (7-hydroxycoumarin): A non-toxic antidiarrheal and antiulcerogenic coumarin. Biomed. Pharmacother. 2020, 129, 110432.
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218.
- Kishore, N.; Kumar, P.; Shanker, K.; Kumar Verma, A. Human disorders associated with inflammation and the evolving role of natural products to overcome. Eur. J. Med. Chem. 2019, 179, 272–309.
- Grover, J.; Jachak, S.M. Coumarins as privileged scaffold for antiinflammatory drug development. RSC Adv. 2015, 5, 38892–38905.
- Rostom, B.; Karaky, R.; Kassab, I.; Veitía, M.S.-I. Coumarins derivatives and inflammation: Review of their effects on the inflammatory signaling pathways. Eur. J. Pharmacol. 2022, 922, 174867.
- Di Stasi, L.C. Natural coumarin derivatives activating Nrf2 signaling pathway as lead compounds for the design and synthesis of intestinal anti-inflammatory drugs. Pharmaceuticals 2023, 16, 511.
- Lee, J.H.; Cho, S.H. Korean red ginseng extract ameliorates skin lesions in NC/ Nga mice: An atopic dermatitis model. J. Ethnopharmacol. 2011, 133, 810–817.
- Akdis, C.A.; Akdis, M. Mechanisms and treatment of allergic disease in the big picture of regulatory T cells. J. Allergy Clin. Immunol. 2009, 123, 735–746.
- Huang, Y.; Li, W.; Su, Z.; Kong, A.T. The complexity of the Nrf2 pathway: Beyond the antioxidant response. J. Nutr. Biochem. 2015, 26, 1401–1413.
- Younas; Khan, A.; Shehzad, O.; Seo, E.K.; Onder, A.; Khan, S. Anti-allergic activities of umbelliferone against histamine- and picryl chloride- induced ear edema by targeting Nrf2/iNOS signaling in mice. BMC Complement. Med. 2021, 21, 215.
- Bansal, Y.; Sethi, P.; Bansal, G. Coumarin: A potential nucleus for anti-inflammatory molecules. Med. Chem. Res. 2013, 22, 3049–3060.
- Zhang, H.-J.; Li, Y.-F.; Cao, Q.; Tian, Y.-S.; Quan, Z.-S. Pharmacological evaluation of 9,10-dihydrochromenooxazin-2(8H)-one derivatives as potent anti-inflammatory agent. Pharmacol. Rep. 2017, 69, 419–425.
- Lawrence, T.; Willoughby, D.A.; Gilroy, D.W. Anti-inflammatory lipid mediators and insights into the resolution of inflammation. Nat. Rev. Immunol. 2002, 2, 787–793.
- Mu, C.; Wu, M.; Li, Z. Anti-inflammatory effect of novel 7-substituted coumarin derivatives through inhibition of NF-κB signaling pathway. Chem. Biodivers. 2019, 16, e1800559.
- Gao, F.; Tao, D.; Ju, C.; Yang, B.-B.; Bao, X.-Q.; Zhang, D.; Zhang, T.-T.; Li, L. Regioselectivity of aminomethylation in 3-acetyl-7-hydroxycoumarins: Mannich bases and Betti bases. New J. Chem. 2021, 45, 9864–9871.
- Sharifi-Rad, M.; Kumar, N.V.A.; Zucca, P.; Varoni, E.M.; Dini, L.; Panzarini, E.; Rajkovic, J.; Fokou, P.V.T.; Azzini, E.; Peluso, I.; et al. Lifestyle, oxidative stress and antioxidants: Back and forth in the pathophysiology of chronic diseases. Front. Physiol. 2020, 11, 694.
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations od antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689.
- Al-Majedy, Y.K.; Al-Amiery, A.; Kadhum, A.A.H.; Mohamad, A.B. Antioxidant activities of 4-methylumbelliferone derivatives. PLoS ONE 2016, 11, e0156625.
- Al-Majedy, Y.K.; Al-Duhaidahawi, D.; Al-Azawi, K.; Al-Amiery, A.A.; Kadhum, A.A.H.; Mohamad, A.B. Coumarins as potential antioxidant agents complemented with suggested mechanisms and approved by molecular modeling studies. Molecules 2016, 21, 135.
- Kurt, B.Z.; Gazioglu, I.; Kandas, N.O.; Sonmez, F. Synthesis, anticholinesterase, antioxidant, and anti-aflatoxigenic activity of novel coumarin carbamates. ChemistrySelect 2018, 3, 3978–3983.
- Joy, M.N.; Bodke, Y.D.; Telkar, S.; Bakulev, V.A. Synthesis of coumarins linked with 1,2,3-triazoles under microwave irradiation and evaluation of their antimicrobial and antioxidant activity. J. Mex. Chem. Soc. 2020, 64, 53–73.
- Kaushik, C.P.; Chahal, M. Synthesis, antimalarial and antioxidant activity of coumarin appended 1,4-disubstituted 1,2,3-triazoles. Mon. Chem. Chem. Mon. 2021, 152, 1001–1012.
- Kecel-Gunduz, S.; Budama-Kilinic, Y.; Bicak, B.; Gok, B.; Belmen, B.; Aydogan, F.; Yolacan, C. New coumarin derivative with potential antioxidant activity: Synthesis, DNA binding and in silico studies (Docking, MD, ADMET). Arab. J. Chem. 2023, 16, 104440.
- Kalaiarasi, G.; Rajkumar, S.R.J.; Dharani, S.; Małecki, J.G.; Prabhakaran, R. An investigation on 3-acetyl-7-methoxy-coumarin Shiff bases and their Ru(II) metallates with potent antiproliferative activity and enhanced LDH and NO release. RSC Adv. 2018, 8, 1539–1561.
- Özdemir, M.; Köksoy, B.; Yalçin, B.; Taşkin, T.; Selçuki, N.A.; Salan, Ü.; Durmuş, M.; Bulut, M. Novel lutetium(III) phthalocyanine-coumarin dyades; synthesis, characterization, photochemical, theoretical and antioxidant activity. Inorg. Chem. Acta 2020, 517, 120145.
- Ali, M.Y.; Jannat, S.; Jung, H.A.; Choi, R.J.; Roy, A.; Choi, J.S. Anti-Alzheimer’s disease potential of coumarins from Angelica decursiva and Artemisia capillaris and structure-activity analysis. Asian Pac. J. Trop. Med. 2016, 9, 103–111.
- Ali, M.Y.; Seong, S.H.; Reddy, M.R.; Seo, S.Y.; Choi, J.S.; Jung, H.A. Kinetics and molecular docking studies of 6-formyl umbelliferone isolated from Angelica decursiva as an inhibitor of cholinesterase and BACE1. Molecules 2017, 22, 1604.
- Karakaya, S.; Koca, M.; Sytar, O.; Duman, H. The natural phenolic compounds and their antioxidant and anticholinesterase potential of herb Leiotulus dasyanthus (K. Koch) Pimenov & Ostr. Nat. Prod. Res. 2020, 1303–1305.
- Decker, M. Hybrid Molecules for Drug Development; Elsevier Ltd.: New York, NY, USA, 2017; ISBN 9780081011188.
- Decker, M. Hybrid molecules incorporating natural products: Applications in cancer therapy, neurodegenerative disorders and beyond. Curr. Med. Chem. 2011, 18, 1464–1475.
- Spilovska, K.; Korabecny, J.; Sepsova, V.; Jun, D.; Hrabinova, M.; Jost, P.; Muckova, L.; Soukup, O.; Janockova, J.; Kucera, T.; et al. Novel tacrine-scutellarin hybrids as multipotent anti-Alzheimer’s agents: Design, synthesis and biological evaluation. Molecules 2017, 22, 1006.
- Hirbod, K.; Jalili-Baleh, L.; Nadri, H.; Ebrahimi, S.E.S.; Moradi, A.; Pakseresht, B.; Foroumadi, A.; Shafiee, A.; Khoob, M. Coumarin derivatives bearing benzoheterocycle moiety: Synthesis, cholinesterase inhibitory, and docking simulation study. Iran. J. Basic. Med. Sci. 2017, 20, 631–638.
- Mzezewa, S.C.; Omoruyib, S.I.; Zondagha, L.S.; Malana, S.F.; Ekpoband, O.E.; Joubert, J.J. Design, synthesis, and evaluation of 3,7-substituted coumarin derivatives asmultifunctional Alzheimer’s disease agents. Enzyme Inhib. Med. Chem. 2021, 36, 1606–1620.
- Seong, S.H.; Ali, M.Y.; Jung, H.A.; Cho, J.S. Umbelliferone derivatives exert neuroprotective effects by inhibiting monoamine oxidase A, self-amyloidβ aggregation, and lipid peroxidation. Bioorg. Chem. 2019, 92, 103293.
- Dhiman, P.; Malik, N.; Khatkar, A. Exploration of umbelliferone based derivatives as potent MAO inhibitors: Dry vs. wet lab evaluation. Curr. Top. Med. Chem. 2018, 18, 1857–1871.
- Bester, E.; Petzer, A.; Petzer, J.P. Coumarin derivatives as inhibitors of D-amino acid oxidase and monoamine oxidase. Bioorg. Chem. 2022, 123, 105791.
- Ostrowska, K.; Leśniak, A.; Czarnocka, Z.; Chmiel, J.; Bujalska-Zadrożny, M.; Trzaskowski, B. Design, synthesis, and biological evaluation of a series of 5- and 7-hydroxycoumarin derivatives as 5-HT1A serotonin receptor antagonists. Pharmaceuticals 2021, 14, 179.
- Yakovleva, E.E.; Myznikov, L.V.; Shabanov, P.D. Comparison of the anticonvulsant activities of substituted hydroxycoumarins and 4-butanoic acid. Pharm. Chem. J. 2020, 54, 904–908.
- Ramu, R.; Shirahatti, P.S.; Swamy, S.N.; Zameer, F.; Dhananjaya, B.L.; Prasad, M.N.N. Assessment of in vivo antidiabetic properties of umbelliferone and lupeol constituents of banana (Musa sp. var. Nanjangud Rasa Bale) flower in hyperglycaemic rodent model. PLoS ONE 2016, 11, e0151135.
- Khadrawy, S.M.; El Sayed, R.A. Umbelliferone attenuates diabetic cardiomyopathy by suppression of JAK/STAT signaling pathway through amelioration of oxidative stress and inflammation in rats. J. Biochem. Mol. Toxicol. 2023, 37, e23296.
- Ali, M.Y.; Zamponi, G.W.; Seong, S.H.; Jung, H.A.; Choi, J.S. 6-Formyl umbelliferone, a furanocoumarin from Angelica decursiva L., inhibits key diabetes-related enzymes and advanced glycation end-product formation. Molecules 2022, 27, 5720.
- Wang, G.; Wang, J.; He, D.; Li, X.; Li, J.; Peng, Z. Synthesis, in vitro evaluation and molecular docking studies of novel coumarin-isatin derivatives as α-glucosidase inhibitors. Chem. Biol. Drug Des. 2017, 89, 456–463.
- Lee, J.H.; Kim, Y.G.; Cho, H.S.; Ryu, S.Y.; Cho, M.H.; Lee, J. Coumarins reduce biofilm formation and the virulence of Escherichia coli O157:H7. Phytomedicine 2014, 21, 1037–1042.
- Darla, M.M.; Krishna, B.S.; Umamaheswara Rao, K.; Reddy, N.B.; Srivash, M.K.; Adeppa, K.; Sundar, C.S.; Reddy, C.S.; Misra, K. Synthesis and bio-evaluation of novel 7-hydroxy coumarin derivatives via Knoevenagel reaction. Res. Chem. Intermed. 2015, 41, 1115–1133.
- Sokol, I.; Toma, M.; Krnić, M.; Macan, A.M.; Drenjančević, D.; Liekens, S.; Raić-Malić, S.; Gazivoda Kraljević, T. Transition metal-catalyzed synthesis of new 3-substituted coumarin derivatives as antibacterial and cytostatic agents. Future Med. Chem. 2021, 13, 1865–1884.
- Farshori, N.N.; Banday, M.R.; Ahmad, A.; Khan, A.U.; Rauf, A. 7-Hydroxy-coumarin derivatives: Synthesis, characterization and preliminary antimicrobial activities. Med. Chem. Res. 2010, 20, 535–541.
- Soares, V.; Marini, M.B.; de Paula, L.A.; Gabry, P.S.; Amaral, A.C.F.; Malafaia, C.A.; Leal, I.C.R. Umbelliferone esters with antibacterial activity produced by lipase-mediated biocatalytic pathway. Biotechnol. Lett. 2020, 43, 469–477.
- Shaikh, M.H.; Subhedar, D.D.; Shingate, B.B.; Kalam Khan, F.A.; Sangshetti, J.N.; Khedkar, V.M.; Nawale, L.; Sarkar, D.; Navale, G.R.; Shinde, S.S. Synthesis, biological evaluation and molecular docking of novel coumarin incorporated triazoles as antitubercular, antioxidant and antimicrobial agents. Med. Chem. Res. 2016, 25, 790–804.
- Gazivoda Kraljević, T.; Harej, A.; Sedić, M.; Kraljević Pavelić, S.; Stepanić, V.; Drenjančević, D.; Talapko, J.; Raić-Malić, S. Synthesis, in vitro anticancer and antibacterial activities and in silico studies of new 4-substituted 1,2,3-triazole-coumarin hybrids. Eur. J. Med. Chem. 2016, 124, 794–808.
- Hu, Y.; Shen, Y.; Wu, X.; Tu, X.; Wang, G.-X. Synthesis and biological evaluation of coumarin derivatives containing imidazole skeleton as potential antibacterial agents. Eur. J. Med. Chem. 2018, 143, 958–969.
- El-Sherief, H.A.; Abuo-Rahma, G.E.-D.A.; Shoman, M.E.; Beshr, E.A.; Abdel-baky, R.M. Design and synthesis of new coumarin–chalcone/NO hybrids of potential biological activity. Med. Chem. Res. 2017, 26, 3077–3090.
- Şahin Gül, D.; Ogutcu, H.; Hayvalı, Z. Investigation of photophysical behaviours and antimicrobial activity of novel benzo-15-crown-5 substituted coumarin and chromone derivatives. J. Mol. Struct. 2020, 1204, 127569.
- Nath, M.; Jairath, R.; Eng, G.; Song, X.; Kumar, A. Triorganotin(IV) derivatives of umbelliferone (7-hydroxycoumarin) and their adducts with 1,10-phenanthroline: Synthesis, structural and biological studies. J. Organomet. Chem. 2005, 690, 134–144.
- Yernule, N.G.; Bennikallu Hire Mathada, M. Preparation of octahedral Cu(II), Co(II), Ni(II) and Zn(II) complexes derived from 8-formyl-7-hydroxy-4-methylcoumarin: Synthesis, characterization and biological study. J. Mol. Struct. 2020, 1220, 128659.
- Klepka, M.T.; Drzewiecka-Antonik, A.; Wolska, A.; Rejmak, P.; Ostrowska, K.; Hejchman, E.; Kruszewska, H.; Czajkowska, A.; Młynarczuk-Biały, I.; Ferenc, W. Synthesis, structural studies and biological activity of new Cu(II) complexes with acetyl derivatives of 7-hydroxy-4-methylcoumarin. J. Inorg. Biochem. 2015, 145, 94–100.
- El-Attar, M.S.; Sadeek, S.A.; Abd El-Hamid, S.M.; Elshafie, H.S. Spectroscopic analyses and antimicrobial activity of novel ciprofloxacin and 7-hydroxy-4-methylcoumarin, the plant-based natural benzopyrone derivative. Int. J. Mol. Sci. 2022, 23, 8019.
- Chiang, C.-C.; Cheng, M.-J.; Peng, C.-F.; Huang, H.-Y.; Chen, I.-S. A novel dimeric coumarin analog and antimycobacterial constituents from Fatoua Pilosa. Chem. Biodivers. 2010, 7, 1728–1736.
- Batra, N.; Rajendran, V.; Wadi, I.; Ghosh, P.C.; Nath, M. Synthesis and antimalarial activity of sulfonamide-attached coumarin--triazoles. Indian J. Chem. 2020, 59B, 1545–1555.
- Khomenko, T.M.; Zarubaev, V.V.; Orshanskaya, I.R.; Kadyrova, R.A.; Sannikova, V.A.; Korchagina, D.V.; Volcho, K.P.; Salakhutdinov, N.F. Anti-influenza activity of monoterpene-containing substituted coumarins. Bioorg. Med. Chem. Lett. 2017, 27, 2920–2925.
- Hu, Y.; Chen, W.; Shen, Y.; Zhu, B.; Wang, G.-X. Synthesis and antiviral activity of coumarin derivatives against infectious hematopoietic necrosis virus. Bioorg. Med. Chem. Lett. 2019, 29, 1749–1755.
- Verdone, L.; Agricola, E.; Caserta, M.; Di Mauro, E. Histone acetylation in gene regulation. Brief. Funct. Genom. Proteom. 2006, 5, 209–221.
- Pramanik, S.D.; Kumar Halder, A.; Mukherjee, U.; Kumar, D.; Dey, Y.N. Potential of histone deacetylase inhibitors in the control and regulation of prostate, breast and ovarian cancer. Front. Chem. 2022, 10, 847.
- Abdizadeh, T.; Kalani, M.R.; Abnous, K.; Tayarani-Najaran, Z.; Khashyarmanesh, B.Z.; Abdizadeh, R.; Hadizadeh, F. Design, synthesis and biological evaluation of novel coumarin-based benzamides as potent histone deacetylase inhibitors and anticancer agents. Eur. J. Med. Chem. 2017, 132, 42–62.
- Yang, F.; Zhao, N.; Song, J.; Zhu, K.; Jiang, C.; Shan, P.; Zhang, H. Design, synthesis and biological evaluation of novel coumarin-based hydroxamate derivatives as histone deacetylase (Hdac) inhibitors with antitumor activities. Molecules 2019, 24, 2569.
- Zhao, N.; Yang, F.; Han, L.; Yuhua, Q.; Ge, D.; Zhang, H. Development of coumarin-based hydroxamates as histone deacetylase inhibitors with antitumor activities. Molecules 2020, 25, 717.
- Ding, J.; Liu, J.; Zhang, Z.; Guo, J.; Cheng, M.; Wan, Y.; Wang, R.; Fang, Y.; Guan, Z.; Jin, Y.; et al. Design, synthesis and biological evaluation of coumarin-based N-hydroxycinnamide derivatives as novel histone deacetylase inhibitors with anticancer activities. Bioorg. Chem. 2020, 101, 104023.
- Chang, C.; Lee, S.O.; Yeh, S.; Chang, T.M. Androgen receptor (AR) differential roles in hormone-related tumors including prostate, bladder, kidney, lung, breast and liver. Oncogene 2014, 33, 3225–3234.
- Voet, A.; Helsen, C.; Zhang, K.Y.J.; Claessens, F. The discovery of novel human androgen receptor antagonist chemotypes using a combined pharmacophore screening procedure. ChemMedChem 2013, 8, 644–651.
- Kandil, S.; Westwell, A.D.; McGuigan, C. 7-Substituted umbelliferone derivatives as androgen receptor antagonists for the potential treatment of prostate and breast cancer. Bioorg. Med. Chem. Lett. 2016, 26, 2000–2004.
- Ma, C.-C.; Liu, Z.-P. Design and synthesis of coumarin derivatives as novel PI3K inhibitors. Anti-Cancer Agents Med. Chem. 2017, 17, 395–403.
- Abdelnaby, R.M.; Rateb, H.S.; Ali, O.; Saad, A.S.; Nadeem, R.I.; Abou-Seri, S.M.; Amin, K.M.; Younis, N.S.; Abdelhady, R. Dual PI3K/Akt inhibitors bearing coumarin-thiazolidine pharmacophores as potential apoptosis inducers in MCF-7 cells. Pharmaceuticals 2022, 15, 428.
- Khomenko, T.; Zakharenko, A.; Odarchenko, T.; Arabshahi, H.J.; Sannikova, V.; Zakharova, O.; Korchagina, D.; Reynisson, J.; Volcho, K.; Salakhutdinov, N.; et al. New inhibitors of tyrosyl-DNA phosphodiesterase I (Tdp 1) combining 7-hydroxycoumarin and monoterpenoid moieties. Bioorg. Med. Chem. 2016, 24, 5573–5581.
- Khomenko, T.M.; Zakharenko, A.L.; Chepanova, A.A.; Ilina, E.S.; Zakharova, O.D.; Kaledin, V.I.; Nikolin, V.P.; Popova, N.A.; Korchagina, D.V.; Reynisson, J.; et al. Promising new inhibitors of tyrosyl-DNA phosphodiesterase I (Tdp 1) combining 4-arylcoumarin and monoterpenoid moieties as components of complex antitumor therapy. Int. J. Mol. Sci. 2020, 21, 126.
- Kurt, B.Z.; Sonmez, F.; Ozturk, D.; Akdemir, A.; Angeli, A.; Supuran, C.T. Synthesis of coumarin-sulfonamide derivatives and determination of their cytotoxicity, carbonic anhydrase inhibitory and molecular docking studies. Eur. J. Med. Chem. 2019, 183, 111702.
- Thacker, P.S.; Alvala, M.; Arifuddin, M.; Angeli, A.; Supuran, C.T. Design, synthesis and biological evaluation of coumarin-3-carboxamides as selective carbonic anhydrase IX and XII inhibitors. Bioorg. Chem. 2019, 86, 386–392.
- Thacker, P.S.; Goud, N.S.; Argulwar, O.S.; Soman, J.; Angeli, A.; Alvala, M.; Arifuddin, M.; Supuran, C.T. Synthesis and biological evaluation of some coumarin hybrids as selective carbonic anhydrase IX and XII inhibitors. Bioorg. Chem. 2020, 104, 104272.
- Wilkinson, B.L.; Bornaghi, L.F.; Houston, T.A.; Innocenti, A.; Supuran, C.T.; Poulsen, S.A. A novel class of carbonic anhydrase inhibitors: Glycoconjuate benzene sulfonamides prepared by “click-tailing”. J. Med. Chem. 2006, 49, 6539–6548.
- Wilkinson, B.L.; Bornaghi, L.F.; Houston, T.A.; Innocenti, A.; Vullo, D.; Supuran, C.T.; Poulsen, S.A. Inhibition of membrane-associated carbonic anhydrase isozymes IX, XII and XIV with a library of glycoconjugate benzenesulfonamides. Bioorg. Med. Chem. Lett. 2007, 17, 987–992.
- Wilkinson, B.L.; Bornaghi, L.F.; Houston, T.A.; Innocenti, A.; Vullo, D.; Supuran, C.T.; Poulsen, S.A. Carbonic anhydrase inhibitors: Inhibition of isozymes I, II, and IX with triazole-linked O-glycosides of benzene sulfonamides. J. Med. Chem. 2007, 50, 1651–1657.
- Chu, N.; Wang, Y.; Jia, H.; Han, J.; Wang, X.; Hou, Z. Design, synthesis and biological evaluation of new carbohydrate-based coumarin derivatives as selective carbonic anhydrase IX inhibitors via “click” reaction. Molecules 2022, 27, 5464.
- Shen, F.-Q.; Wang, Z.-C.; Wu, S.-Y.; Ren, S.-Z.; Man, R.-J.; Wang, B.-Z.; Zhu, H.-L. Synthesis of novel hybrids of pyrazole and coumarin as dual inhibitors of COX-2 and 5-LOX. Bioorg. Med. Chem. Lett. 2017, 27, 3653–3660.
- Hua, W.; Zhao, J.; Hu, W.; Gou, S. Combination of 7-hydroxycoumarin in a platinum(IV) complex derived from cisplatin enhanced cytotoxicity with multiple mechanisms of action. J. Inorg. Biochem. 2018, 186, 17–23.
- Qin, X.; Fang, L.; Zhao, J.; Gou, S. Theranostic Pt(IV) conjugate with target selectivity for androgen receptor. Inorg. Chem. 2018, 57, 5019–5029.
- Wang, Q.; Chen, Y.; Li, G.; Liu, Z.; Ma, J.; Liu, M.; Li, D.; Han, J.; Wang, B. Synthesis and evaluation of bi-functional 7-hydroxycoumarin platinum(IV) complexes as antitumor agents. Bioorg. Med. Chem. 2019, 27, 2112–2121.
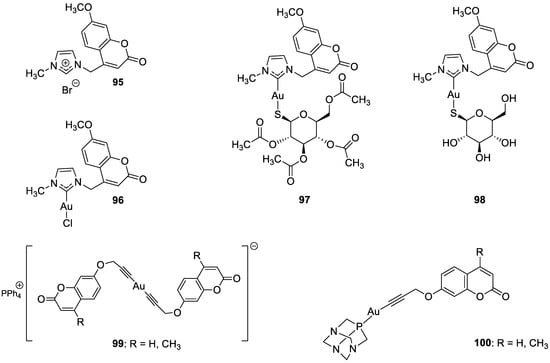
- Bertrand, B.; de Almeida, A.; van der Burgt, E.P.M.; Picquet, M.; Citta, A.; Folda, A.; Rigobello, M.P.; Le Gendre, P.; Bodio, E.; Casini, A. New gold(I) organometallic compounds with biological activity in cancer cells. Eur. J. Inorg. Chem. 2014, 27, 4532–4536.
- Arcau, J.; Andermark, V.; Aguiló, E.; Gandioso, A.; Moro, A.; Cetina, M.; Lima, J.C.; Rissanen, K.; Ott, I.; Rodríguez, L. Luminescent alkynyl-gold(I) coumarin derivatives and their biological activity. Dalton Trans. 2014, 43, 4426–4436.
- Levin, P.P.; Liubimov, A.V.; Shashkov, A.S.; Mardaleishvili, I.R.; Venidiktova, O.V.; Shienok, A.L.; Koltsova, L.S.; Astafiev, A.A.; Barachevsky, V.A.; Zaichenko, N.L. Multiple fluorescence of tetraarylimidazole and azomethinocoumarin dyad with dual excited-state intramolecular proton transfer. Dyes Pigm. 2020, 183, 108716.
- Xiao, Z.; Chen, D.; Song, S.; Vlag, R.; Wouden, P.; Merkerk, R.; Cool, R.H.; Hirsch, A.K.H.; Melgert, B.N.; Quax, W.J.; et al. 7-Hydroxycoumarins are affinity-based fluorescent probes for competitive binding studies of macrophage migration inhibitory factor. J. Med. Chem. 2020, 63, 11920–11933.
- Shi, B.; Zhang, Z.; Jin, Q.; Wang, Z.; Tang, J.; Xu, G.; Zhu, T.; Gong, X.; Tang, X.; Zhao, C. Selective tracking of ovarian-cancer-specific γ-glutamyltranspeptidase using a ratiometric two-photon fluorescent probe. J. Mater. Chem. B 2018, 6, 7439.
- Li, S.; Kan, W.; Zhao, B.; Liu, T.; Fang, Y.; Bai, L.; Wang, L. A fluorescent pH probe for an aqueous solution composed of 7-hydroxycoumarin, Schiff base and phenanthroimidazole moieties (PICO). Heterocycl. Commun. 2018, 24, 93–97.
- Shukla, L.; Moodie, L.W.K.; Kindahl, T.; Hedberg, C. Synthesis and spectroscopic properties of fluorinated coumarin lysine derivatives. J. Org. Chem. 2018, 83, 4792–4799.
- Gleason, P.R.; Kelly, P.I.; Grisingher, D.W.; Mills, J.H. An intrinsic FRET sensor of protein-ligand interactions. Org. Biomol. Chem. 2020, 18, 4079–4084.
- Gleason, P.R.; Kolbaba-Kartchner, B.; Henderson, J.N.; Stahl, E.P.; Simmons, C.R.; Mills, J.H. Structural origins of altered spectroscopic properties upon ligand binding in proteins containing a fluorescent noncanonical amino acid. Biochemistry 2021, 60, 2577–2585.
- Wang, K.; Yao, T.; Xue, J.; Guo, Y.; Xu, X. A novel fluorescent probe for the detection of hydrogen peroxide. Biosensors 2023, 13, 658.
- Zhu, G.; Huang, Y.; Wang, C.; Lu, L.; Sun, T.; Wang, M.; Tang, Y.; Shan, D.; Wen, S.; Zhu, J. A novel coumarin-based fluorescence chemosensor for Al3+ and its application in cell imaging. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2019, 210, 105–110.
- Li, X.; Duan, Q.; Yu, Y.; Wang, K.; Zhu, H.; Zhang, X.; Liu, C.; Jia, P.; Li, Z.; Sheng, W.; et al. A coumarin-based fluorescent probe for Hg2+ and its application in living cells and zebrafish. Luminescence 2020, 35, 941–946.
- Rojas-Montoyaa, S.M.; Vonlanthena, M.; Huerta-Roldána, J.M.; Aguilar-Ortíza, E.; Burillob, G.; Morales-Espinoza, E.G.; Rivera, E. Incorporation of photoluminescent 7-hydroxycoumarin units onto a polyethylene matrix by means of gamma radiation. Radiat. Phys. Chem. 2019, 163, 52–57.
- Stefanachi, A.; Leonetti, F.; Pisani, L.; Catto, M.; Carotti, A. Coumarin: A natural, privileged and versatile scaffold for bioactive compounds. Molecules 2018, 23, 250.
- Flores-Morales, V.; Villasana-Ruiz, A.P.; Garza-Veloz, I.; González-Delgado, S. Therapeutic effects of coumarins with different substitution patterns. Molecules 2023, 28, 2413.

