 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Svetlana V. Pylina | -- | 3822 | 2023-12-20 19:09:22 | | | |
| 2 | Wendy Huang | Meta information modification | 3822 | 2023-12-22 09:19:48 | | | | |
| 3 | Wendy Huang | Meta information modification | 3822 | 2023-12-26 03:39:31 | | |
Video Upload Options
Type 1 diabetes mellitus (T1D) is an autoimmune disease caused by the destruction of insulin-producing β-cells in the pancreas by cytotoxic T-cells. To date, there are no drugs that can prevent the development of T1D. Insulin replacement therapy is the standard care for patients with T1D. This treatment is life-saving, but is expensive, can lead to acute and long-term complications, and results in reduced overall life expectancy. This has stimulated the research and development of alternative treatments for T1D. In this research, potential therapies for T1D are considered using cellular regenerative medicine approaches with a focus on CRISPR/Cas-engineered cellular products. However, CRISPR/Cas as a genome editing tool has several drawbacks that should be considered for safe and efficient cell engineering. In addition, cellular engineering approaches themselves pose a hidden threat. The purpose of this research is to critically discuss novel strategies for the treatment of T1D using genome editing technology.
1. Introduction
T1D is an autoimmune disease caused by the T-cell-mediated destruction of insulin-producing pancreatic β-cells [1]. There are two subtypes of T1D [2]: 1A—autoimmune, including latent autoimmune diabetes of adults (LADA); and 1B—idiopathic. According to classical theory, the damaged β-cells expose self-antigens to antigen-presenting cells (APC) and thus initiate pathogenic processes through crosstalk with immune cells. But the exact mechanism of the autoimmune response in T1D is still unknown. This process is promoted by an incompletely understood interaction of various factors, such as age, genetic predisposition, and environmental triggers [3]. However, none of these factors is sufficient to develop early screening programs to identify the presymptomatic normoglycemic phase (prediabetes) [4] and to prevent disease progression to the symptomatic one. The typical manifestation of T1D with symptoms of hyperglycemia (polyuria, polydipsia, enuresis, weight loss, and blurred vision), sometimes even with diabetic ketoacidosis occurs with of 90% β-cells destruction [5]. As a result, the diagnosis is not confirmed until the late stages of absolute insulin deficiency when treatment options are limited to hormone replacement therapy and, in some cases, β-cell transplantation.
| Clinical Trial NCT | Company | Product | Cells | Administration |
|---|---|---|---|---|
| NCT03163511 | Viacyte | VC-02 | Pancreatic endoderm cells (PEC-01 cells) | Subcutaneous in a protective device |
| NCT03513939 | Sernova | Cell Pouch | Therapeutic cells including islets | Abdominal musculature |
| NCT04786262 | Vertex | VX-880 | Allogeneic fully differentiated insulin-producing islets | Infused into the hepatic portal vein |
| NCT05565248 | CRISPR Therapeutics AG in collaboration with Viacyte | VCTX211 | Allogeneic pancreatic endoderm cells (PEC211) genetically modified using CRISPR/Cas9 | In a surgically implanted durable, removable, perforated device |
| NCT05791201 | Vertex | VX-264 | Allogeneic fully differentiated insulin-producing islets | In a surgically implanted channel array protective device |
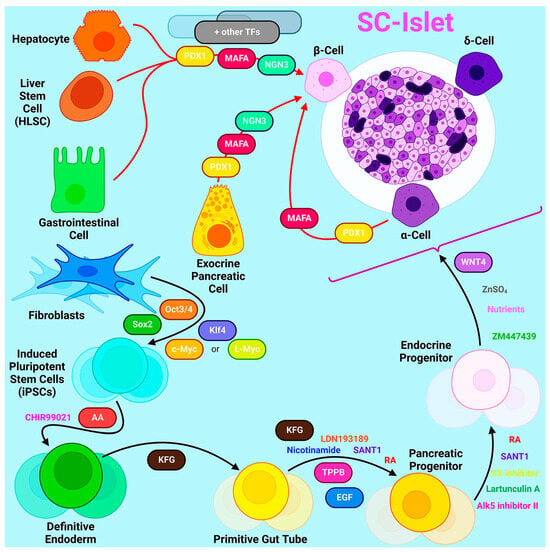
2. Sources for β-Cell Generation
3. HLA Haplotype Banks
4. CRISPR/Cas-Based Cell Therapy
CRISPR/Cas-mediated genome editing and iPSC technology enable a new class of cell replacement therapies for the treatment of T1D. It is likely that their combination will accelerate the development of a β-cell replacement drug aimed at treating diabetes without the need for immunosuppression in a deviceless approach. However, the drawbacks of both cell technologies and CRISPR/Cas-editors must be carefully considered. β-cells obtained through differentiation or transdifferentiation protocols should be purified from immature β-like, non-targeted, or undifferentiated cells that may adversely affect β-cell activity. Allogeneic β-cells differentiated from genetically engineered low-immunogenic iPSCs are a cheap and rapid option for the treatment of T1D. However, their level of protection from the immune system is currently insufficient, resulting in significant levels of immune rejection in patients. The use of classical CRISPR/Cas system tools delivered in viral vectors can lead to the creation of off-target edits and insertional mutagenesis, which can lead to the cancerous transformation of low-immunogenic β-cells. This problem can be addressed by using high-fidelity Cas9 variants or advanced Cas9-based tools such as prime editors, delivering CRISPR/Cas systems as mRNA or RNA complexes, and carefully planning genome editing experiments to reduce the number of successive rounds of editing and cell clone selection to a minimum. To control the possible cancerous transformation of engineered β-cells, inducible non-immunogenic and cell state-independent suicide genes, such as inducible caspase 9, can be introduced. In addition, safe harbors for the introduction of protective genes should be carefully explored and selected so as not to disrupt the functions of surrounding genes. Perhaps the list of hidden problems and their solutions is not complete and further studies and clinical trials will uncover additional difficulties associated with hypoimmunogenic engineered β-cells. However, the current knowledge already allows us to foresee serious problems, the solution of which will greatly reduce the chance of turning engineered β-cells into a “Trojan horse” that will bring hidden problems to T1D patients after transplantation.
References
- Norris, J.M.; Johnson, R.K.; Stene, L.C. Type 1 diabetes-early life origins and changing epidemiology. Lancet Diabetes Endocrinol. 2020, 8, 226–238.
- American Diabetes Association Professional Practice Committee. 6. Glycemic targets: Standards of medical care in diabetes-2022. Diabetes Care 2022, 45, S83–S96.
- Li, Y.; Sun, F.; Yue, T.T.; Wang, F.X.; Yang, C.L.; Luo, J.H.; Rong, S.J.; Xiong, F.; Zhang, S.; Wang, C.Y. Revisiting the antigen-presenting function of beta cells in T1D pathogenesis. Front. Immunol. 2021, 12, 690783.
- Insel, R.A.; Dunne, J.L.; Atkinson, M.A.; Chiang, J.L.; Dabelea, D.; Gottlieb, P.A.; Greenbaum, C.J.; Herold, K.C.; Krischer, J.P.; Lernmark, A.; et al. Staging presymptomatic type 1 diabetes: A scientific statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care 2015, 38, 1964–1974.
- Atkinson, M.A. The pathogenesis and natural history of type 1 diabetes. Cold Spring Harb. Perspect. Med. 2012, 2, a007641.
- Shapiro, A.M.; Lakey, J.R.; Ryan, E.A.; Korbutt, G.S.; Toth, E.; Warnock, G.L.; Kneteman, N.M.; Rajotte, R.V. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N. Engl. J. Med. 2000, 343, 230–238.
- Shapiro, A.M.J.; Thompson, D.; Donner, T.W.; Bellin, M.D.; Hsueh, W.; Pettus, J.; Wilensky, J.; Daniels, M.; Wang, R.M.; Brandon, E.P.; et al. Insulin expression and C-peptide in type 1 diabetes subjects implanted with stem cell-derived pancreatic endoderm cells in an encapsulation device. Cell Rep. Med. 2021, 2, 100466.
- Ramzy, A.; Thompson, D.M.; Ward-Hartstonge, K.A.; Ivison, S.; Cook, L.; Garcia, R.V.; Loyal, J.; Kim, P.T.W.; Warnock, G.L.; Levings, M.K.; et al. Implanted pluripotent stem-cell-derived pancreatic endoderm cells secrete glucose-responsive C-peptide in patients with type 1 diabetes. Cell Stem Cell 2021, 28, 2047–2061.
- D’Amour, K.A.; Agulnick, A.D.; Eliazer, S.; Kelly, O.G.; Kroon, E.; Baetge, E.E. Efficient differentiation of human embryonic stem cells to definitive endoderm. Nat. Biotechnol. 2005, 23, 1534–1541.
- Pagliuca, F.W.; Millman, J.R.; Gurtler, M.; Segel, M.; Van Dervort, A.; Ryu, J.H.; Peterson, Q.P.; Greiner, D.; Melton, D.A. Generation of functional human pancreatic beta cells in vitro. Cell 2014, 159, 428–439.
- Veres, A.; Faust, A.L.; Bushnell, H.L.; Engquist, E.N.; Kenty, J.H.; Harb, G.; Poh, Y.C.; Sintov, E.; Gurtler, M.; Pagliuca, F.W.; et al. Charting cellular identity during human in vitro beta-cell differentiation. Nature 2019, 569, 368–373.
- Velazco-Cruz, L.; Song, J.; Maxwell, K.G.; Goedegebuure, M.M.; Augsornworawat, P.; Hogrebe, N.J.; Millman, J.R. Acquisition of Dynamic Function in Human Stem Cell-Derived beta Cells. Stem Cell Rep. 2019, 12, 351–365.
- Sharon, N.; Vanderhooft, J.; Straubhaar, J.; Mueller, J.; Chawla, R.; Zhou, Q.; Engquist, E.N.; Trapnell, C.; Gifford, D.K.; Melton, D.A. Wnt Signaling Separates the Progenitor and Endocrine Compartments during Pancreas Development. Cell Rep. 2019, 27, 2281–2291.
- Rosado-Olivieri, E.A.; Anderson, K.; Kenty, J.H.; Melton, D.A. YAP inhibition enhances the differentiation of functional stem cell-derived insulin-producing beta cells. Nat. Commun. 2019, 10, 1464.
- Chen, S.; Du, K.; Zou, C. Current progress in stem cell therapy for type 1 diabetes mellitus. Stem Cell Res. Ther. 2020, 11, 275.
- Helman, A.; Melton, D.A. A Stem Cell Approach to Cure Type 1 Diabetes. Cold Spring Harb. Perspect. Biol. 2021, 13, a035741.
- Balboa, D.; Barsby, T.; Lithovius, V.; Saarimaki-Vire, J.; Omar-Hmeadi, M.; Dyachok, O.; Montaser, H.; Lund, P.E.; Yang, M.; Ibrahim, H.; et al. Functional, metabolic and transcriptional maturation of human pancreatic islets derived from stem cells. Nat. Biotechnol. 2022, 40, 1042–1055.
- Augsornworawat, P.; Hogrebe, N.J.; Ishahak, M.; Schmidt, M.D.; Marquez, E.; Maestas, M.M.; Veronese-Paniagua, D.A.; Gale, S.E.; Miller, J.R.; Velazco-Cruz, L.; et al. Single-nucleus multi-omics of human stem cell-derived islets identifies deficiencies in lineage specification. Nat. Cell Biol. 2023, 25, 904–916.
- Zhu, H.; Wang, G.; Nguyen-Ngoc, K.V.; Kim, D.; Miller, M.; Goss, G.; Kovsky, J.; Harrington, A.R.; Saunders, D.C.; Hopkirk, A.L.; et al. Understanding cell fate acquisition in stem-cell-derived pancreatic islets using single-cell multiome-inferred regulomes. Dev. Cell 2023, 58, 727–743.e711.
- Wang, W.; Zhang, C. Targeting beta-cell dedifferentiation and transdifferentiation: Opportunities and challenges. Endocr. Connect. 2021, 10, R213–R228.
- Zhou, Q.; Brown, J.; Kanarek, A.; Rajagopal, J.; Melton, D.A. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature 2008, 455, 627–632.
- Bru-Tari, E.; Cobo-Vuilleumier, N.; Alonso-Magdalena, P.; Dos Santos, R.S.; Marroqui, L.; Nadal, A.; Gauthier, B.R.; Quesada, I. Pancreatic alpha-cell mass in the early-onset and advanced stage of a mouse model of experimental autoimmune diabetes. Sci. Rep. 2019, 9, 9515.
- Furuyama, K.; Chera, S.; van Gurp, L.; Oropeza, D.; Ghila, L.; Damond, N.; Vethe, H.; Paulo, J.A.; Joosten, A.M.; Berney, T.; et al. Diabetes relief in mice by glucose-sensing insulin-secreting human alpha-cells. Nature 2019, 567, 43–48.
- Guo, P.; Zhang, T.; Lu, A.; Shiota, C.; Huard, M.; Whitney, K.E.; Huard, J. Specific reprogramming of alpha cells to insulin-producing cells by short glucagon promoter-driven Pdx1 and MafA. Mol. Ther. Methods Clin. Dev. 2023, 28, 355–365.
- Banga, A.; Akinci, E.; Greder, L.V.; Dutton, J.R.; Slack, J.M. In Vivo reprogramming of Sox9+ cells in the liver to insulin-secreting ducts. Proc. Natl. Acad. Sci. USA 2012, 109, 15336–15341.
- Navarro-Tableros, V.; Gai, C.; Gomez, Y.; Giunti, S.; Pasquino, C.; Deregibus, M.C.; Tapparo, M.; Pitino, A.; Tetta, C.; Brizzi, M.F.; et al. Islet-Like Structures Generated In Vitro from Adult Human Liver Stem Cells Revert Hyperglycemia in Diabetic SCID Mice. Stem Cell Rev. Rep. 2019, 15, 93–111.
- Klein, D.; Alvarez-Cubela, S.; Lanzoni, G.; Vargas, N.; Prabakar, K.R.; Boulina, M.; Ricordi, C.; Inverardi, L.; Pastori, R.L.; Dominguez-Bendala, J. BMP-7 Induces Adult Human Pancreatic Exocrine-to-Endocrine Conversion. Diabetes 2015, 64, 4123–4134.
- Nakatsuji, N.; Nakajima, F.; Tokunaga, K. HLA-haplotype banking and iPS cells. Nat. Biotechnol. 2008, 26, 739–740.
- Okita, K.; Matsumura, Y.; Sato, Y.; Okada, A.; Morizane, A.; Okamoto, S.; Hong, H.; Nakagawa, M.; Tanabe, K.; Tezuka, K.; et al. A more efficient method to generate integration-free human iPS cells. Nat. Methods 2011, 8, 409–412.
- Gourraud, P.A.; Gilson, L.; Girard, M.; Peschanski, M. The role of human leukocyte antigen matching in the development of multiethnic “haplobank” of induced pluripotent stem cell lines. Stem Cells 2012, 30, 180–186.
- Taylor, C.J.; Peacock, S.; Chaudhry, A.N.; Bradley, J.A.; Bolton, E.M. Generating an iPSC bank for HLA-matched tissue transplantation based on known donor and recipient HLA types. Cell Stem Cell 2012, 11, 147–152.
- Abberton, K.M.; McDonald, T.L.; Diviney, M.; Holdsworth, R.; Leslie, S.; Delatycki, M.B.; Liu, L.; Klamer, G.; Johnson, P.; Elwood, N.J. Identification and Re-consent of Existing Cord Blood Donors for Creation of Induced Pluripotent Stem Cell Lines for Potential Clinical Applications. Stem Cells Transl. Med. 2022, 11, 1052–1060.
- Lee, S.; Huh, J.Y.; Turner, D.M.; Lee, S.; Robinson, J.; Stein, J.E.; Shim, S.H.; Hong, C.P.; Kang, M.S.; Nakagawa, M.; et al. Repurposing the Cord Blood Bank for Haplobanking of HLA-Homozygous iPSCs and Their Usefulness to Multiple Populations. Stem Cells 2018, 36, 1552–1566.
- Ichise, H.; Nagano, S.; Maeda, T.; Miyazaki, M.; Miyazaki, Y.; Kojima, H.; Yawata, N.; Yawata, M.; Tanaka, H.; Saji, H.; et al. NK Cell Alloreactivity against KIR-Ligand-Mismatched HLA-Haploidentical Tissue Derived from HLA Haplotype-Homozygous iPSCs. Stem Cell Rep. 2017, 9, 853–867.
- Nowak, I.; Majorczyk, E.; Wisniewski, A.; Pawlik, A.; Magott-Procelewska, M.; Passowicz-Muszynska, E.; Malejczyk, J.; Ploski, R.; Giebel, S.; Barcz, E.; et al. Does the KIR2DS5 gene protect from some human diseases? PLoS ONE 2010, 5, e12381.
- Wang, R.; Sun, Y.; Kuang, B.H.; Yan, X.; Lei, J.; Lin, Y.X.; Tian, J.; Li, Y.; Xie, X.; Chen, T.; et al. HLA-Bw4 in association with KIR3DL1 favors natural killer cell-mediated protection against severe COVID-19. Emerg. Microbes Infect. 2023, 12, 2185467.
- Total Number of Donors and Cord Blood Units. Available online: https://statistics.wmda.info (accessed on 27 October 2023).
- Rim, Y.A.; Park, N.; Nam, Y.; Ham, D.S.; Kim, J.W.; Ha, H.Y.; Jung, J.W.; Jung, S.M.; Baek, I.C.; Kim, S.Y.; et al. Recent progress of national banking project on homozygous HLA-typed induced pluripotent stem cells in South Korea. J. Tissue Eng. Regen. Med. 2018, 12, e1531–e1536.
- Umekage, M.; Sato, Y.; Takasu, N. Overview: An iPS cell stock at CiRA. Inflamm. Regen. 2019, 39, 17.
- Yoshida, S.; Kato, T.M.; Sato, Y.; Umekage, M.; Ichisaka, T.; Tsukahara, M.; Takasu, N.; Yamanaka, S. A clinical-grade HLA haplobank of human induced pluripotent stem cells matching approximately 40% of the Japanese population. Med 2023, 4, 51–66.

