T1D is an autoimmune disease caused by the T-cell-mediated destruction of insulin-producing pancreatic β-cells [1]. There are two subtypes of T1D [9]: 1A—autoimmune, including latent autoimmune diabetes of adults (LADA); and 1B—idiopathic. According to classical theory, the damaged β-cells expose self-antigens to antigen-presenting cells (APC) and thus initiate pathogenic processes through crosstalk with immune cells. But the exact mechanism of the autoimmune response in T1D is still unknown. This process is promoted by an incompletely understood interaction of various factors, such as age, genetic predisposition, and environmental triggers [10]. However, none of these factors is sufficient to develop early screening programs to identify the presymptomatic normoglycemic phase (prediabetes) [11] and to prevent disease progression to the symptomatic one. The typical manifestation of T1D with symptoms of hyperglycemia (polyuria, polydipsia, enuresis, weight loss, and blurred vision), sometimes even with diabetic ketoacidosis occurs with of 90% β-cells destruction [12]. As a result, the diagnosis is not confirmed until the late stages of absolute insulin deficiency when treatment options are limited to hormone replacement therapy and, in some cases, β-cell transplantation.
Table 1. Ongoing clinical trials of cell replacement therapies to treat patients with T1D.
| Clinical Trial NCT |
Company |
Product |
Cells |
Administration |
| NCT03163511 |
Viacyte |
VC-02 |
Pancreatic endoderm cells (PEC-01 cells) |
Subcutaneous in a protective device |
| NCT03513939 |
Sernova |
Cell Pouch |
Therapeutic cells including islets |
Abdominal musculature |
| NCT04786262 |
Vertex |
VX-880 |
Allogeneic fully differentiated insulin-producing islets |
Infused into the hepatic portal vein |
| NCT05565248 |
CRISPR Therapeutics AG in collaboration with Viacyte |
VCTX211 |
Allogeneic pancreatic endoderm cells (PEC211) genetically modified using CRISPR/Cas9 |
In a surgically implanted durable, removable, perforated device |
| NCT05791201 |
Vertex |
VX-264 |
Allogeneic fully differentiated insulin-producing islets |
In a surgically implanted channel array protective device |
Sernova Corp. (London, ON, Canada) has taken a different approach by introducing a new implantable device called Cell Pouch. The device is a scaffold made of non-degradable polymers, formed into small cylindrical chambers, which, when implanted into the abdominal muscle, grow vascularized tissue around the perimeter of removable plugs in as little as two weeks. After tissue engraftment, the plugs are removed, leaving fully formed tissue chambers with central cavities for the transplantation of therapeutic cells such as insulin-producing islets. The Cell Pouch forms a natural environment rich in microvessels that allows transplanted islets to engraft. According to data presented from a Phase I/II trial of the Cell Pouch system in patients with T1D, the first 5 patients who underwent islet transplantation achieved insulin independence for 6 to 38 months (ClinicalTrial NCT03513939).
An undoubted achievement of T1D cell therapy is the emergence of combination approaches using gene-edited allogeneic stem cells. A phase I/II trial of a combinatorial drug, VCTX211 (CRISPR Therapeutics AG (South Boston, MA, USA) in collaboration with Viacyte), consisting of two components, allogeneic pancreatic endoderm cells genetically modified with CRISPR/Cas9, and a removable perforated device designed to deliver and preserve these cells, is currently underway (ClinicalTrial NCT05565248). VCTX211 has two gene knockouts (B2M, TXNIP) and four insertions (PD-L1, HLA-E, TNFAIP3, and MANF) to improve functionality. Unlike the previous version of the drug, called VCTX210A, whose clinical trials have already been completed (ClinicalTrial NCT05210530), the current version of VCTX211, in addition to edits to reduce T- and NK-cell mediated immune rejection and protection against oxidative stress in the endoplasmic reticulum, was supplemented with the A20 (TNFAIP3) gene insert, which induces graft engraftment and protection against cytokine-induced apoptosis, and with the MANF gene insert, which enhances β-cell proliferation and protection against inflammatory stress.
2. Sources for β-Cell Generation
Research on the production of insulin-producing cells in vitro has been underway since the opportunities offered by human pluripotent stem cells (PSCs) for regenerative medicine became clear. Sources of islet endocrine clusters for in vitro production of insulin-producing cells include embryonic stem cells (ESC), iPSCs, adult stem cells, and differentiated cells from mature tissues that can be transdifferentiated into insulin-producing cells.
Intensive research has been conducted to develop protocols for generating insulin-producing cells from stem cells, from the first seminal work describing the generation of definitive endoderm [
67] to the creation of stem cell-derived β-cells that secreted insulin in response to successive glucose challenges [
68] and the generation of monohormonal, insulin-expressing β-cells [
69,
70,
71,
72]. Present strategies for SC-islets are predominantly based on approaches that imitate normal pancreas development [
73]. A common principle for the various multi-stage protocols is that iPSCs cultured in 3D are differentiated by targeting key embryonic signaling pathways such as Nodal, WNT, RA, FGF, BMP, Notch, and Hippo (
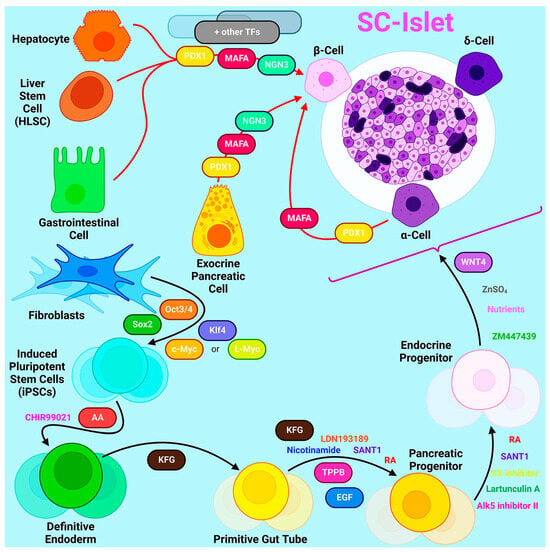
Figure 1). On average, this takes five to seven stages that last 20-30 days [
74], but functional maturation associated with transcriptional maturation can continue for months after cell transplantation [
75]. Recently, more than 600 genes have been identified whose expression increases 6 months after cell transplantation in vivo [
76].
Figure 1. The ways to produce β-cell from other cell types by differentiation or transdifferentiation.
The latest generation protocols describe the derivation of islet clusters from stem cells, in which other endocrine cell types such as α- and δ-cells are present along with the target β-cells. The role of α- and δ-cells in terms of β-cell function in the context of islet clusters, as well as their optimal ratio, currently remains unknown. Intestinal endocrine cells, termed enterochromaffin cells, are one of the most common off-target cell types in islet clusters. Other off-target cells can be hepatic, mesenchymal, and pancreatic exocrine cells [
69]. The presence of such off-target cell populations may negatively affect the functional activity of β-cells [
69]. In addition, although modern generation islet clusters are capable of secreting significant amounts of insulin in response to glucose stimulation in vitro, there are still fundamental transcriptional and epigenetic differences between artificially derived islet clusters and primary human islets. Since these differences primarily affect the activity of metabolic pathways and the functionality of islet clusters, studies are underway to better characterize cells derived from differentiation protocols. Multiomic studies show that β-cells derived by in vitro differentiation have the least similarity to their primary counterparts compared to other endocrine cell types [
76,
77]. Moreover, the chromatin of primary human islet cell types is more restricted than in islet-like clusters obtained in vitro, where the
INS gene remained open not only in β-cells but also in α-cells and δ-cells [
76]. However, the long-term culturing of SC-islets both in vitro and in vivo results in a transition of chromatin to a more closed state. By comparing scRNA-seq and snATAC-seq data, it was found that transplantation directs cells to their correct identity, and SC-islets become more similar to primary human islets compared to those formed in vitro [
77]. Thus, the detailed characterization of SC-islets using single-cell sequencing technologies sheds light on the unknown mechanisms controlling islet development and currently limiting islet maturation. The further optimization of differentiation protocols is needed to obtain fully mature islets with a minimum number of potentially unsafe off-target cell types in vitro and thereby increase the safety of transplantation of in vitro differentiated islets to patients.
Transdifferentiation into β-cells can occur from a variety of sources including hepatocytes, gastrointestinal cells, exocrine pancreatic cells, and other endocrine cell types. Since these cells share common precursors with β-cells and therefore similar developmental pathways and epigenetic profiles, transdifferentiation from these cell types is most appropriate [
78]. There are two different approaches to the transdifferentiation of cells: through genetic modifications in the cell or by exposing cells to certain molecules to activate certain signaling pathways. One of the most common ways to obtain insulin-producing cells is the exogenous overexpression of certain transcription factors.
In 2008, Zhou et al. first identified three key transcription factors (MafA, Pdx1, and Ngn3) that induce the reprogramming of fully differentiated exocrine cells into β-cells, demonstrating the possibility of targeted reprogramming of adult cells without reverting to a pluripotent state [
79]. Pdx1, also known as pancreatic and duodenal homeobox transcription factor 1, is required for β-cell maturation and the maintenance of their metabolism and proliferation. MafA binds to an enhancer that regulates insulin gene expression in pancreatic cells. Neurogenin-3 (Ngn3) is a transcription factor common to endocrine progenitors, so all endocrine cells originate from an Ngn3-positive progenitor.
Since α-cells are the second most abundant type of pancreatic endocrine cell that can be transformed into insulin-producing cells, the greatest amount of research has focused on studying transdifferentiation from α-cells. In murine models of diabetes, it has been shown that α-cells increase their size and proliferation rate during the development of the disease, while the level of apoptosis is significantly reduced [
80]. Moreover, the number of glucagon/Pdx1-positive cells increases in these mice during diabetes, indicating a possible transition of α-cells to β-cells in vivo. Given these facts and the common origin of α- and β-cells, the transdifferentiation of α-cells may seem to be a promising approach to the treatment of diabetes.
Additionally, there have been reports of transdifferentiation possibilities in vivo, in addition to attempts made to create insulin-producing cells in vitro. Recently, it was demonstrated that α-cells can be transdifferentiated into β-cells in mice in vivo by delivering Pdx1 and MafA with a promoter specific for human α-cells using adeno-associated virus (AAV). Transduction was performed on mice with chemically induced and autoimmune diabetes, and resulted in decreased hyperglycemia in both cases. An increase in proliferation rate was also proven to enhance the effect of viral transduction and help prevent further disease progression [
81]. Thus, the resulting insulin-producing cells were not really β-cells but β-like cells, because they expressed specific markers of the two cell types.
Except the attempts of creating insulin-producing cells in vitro, opportunities of in vivo transdifferentiation were also reported. Recently, it has been demonstrated that α-cells can be transdifferentiated into β-cells in mice in vivo via delivering Pdx1 and MafA with human α-cell specific promoter using AAV. The transduction was held in mice with chemically induced and autoimmune diabetes and resulted in a decreased hyperglycemia in both cases. An increased proliferation rate was also proven to enhance the effect of viral transduction and contributed to the prevention of the further development of the disease [
82]. Thus, the in vivo transdifferentiation approach showed higher efficiency, which may be a consequence of the presence of a microenvironment and certain endogenous signals.
Similar strategies of exogenous overexpression were also applied to other cell types. For example, the simultaneous overexpression of MafA, Pdx1, and Ngn3 allowed the transformation of Sox9-positive mouse liver cells into insulin-secreting cells [
83]. However, the transdifferentiation of hepatocytes has limitations due to their low proliferative capacity [
84]. To increase the efficiency of transdifferentiation, the researchers used human liver stem cells, which have shown the ability to form islet-like structures. Although the resulting islet-like structures were immature, they displayed an insulin response to glucose and controlled hyperglycemia in murine models of diabetes [
84].
Along with transdifferentiation protocols based on epigenetic modifications, there are approaches to induce the transition into insulin-producing cells by exposing them to specific inhibitors and growth factors. Non-endocrine pancreatic cells can differentiate into β-like cells through the simultaneous inhibition of TGFb and activation of the BMP pathway through exposure to BMP7. However, the resulting cells lacked the necessary features and were unable to maintain glucose homeostasis after in vivo transplantation [
85]. Thus, the method of exposing cells to specific molecules was not quite effective.
Currently, there are no effective transdifferentiation protocols for obtaining mature β-cells, responding to glucose by secreting the required amount of insulin. However, inducing the overexpression of specific transcription factors has been shown to be more effective than exposing cells to low molecular weight inhibitors and inducers. In addition to incomplete β-cell differentiation, another challenge for T1D therapy may be allogeneic cell-derived graft rejection. In this respect, patient-specific cell products derived from autologous PSCs are an ideal option for transplantation. However, allografts are preferable to autografts, both in terms of cost and the speed of obtaining them. After all, the use of cell products in medicine involves the collection of thorough preclinical data for each individual cell line, which is associated with the significant expenditure of material resources and time. Thus, it seems reasonable to use allogeneic cell products, but then the problem of the rejection of transplanted cells becomes relevant. Currently, there are two approaches to its solution: the creation of biobanks of iPSC lines allowing the needs of the majority of patients to be covered, and the creation of immunoprivileged iPSC lines.
3. HLA Haplotype Banks
HLA-haplotype banks are now widely used throughout the world. Bone marrow and cord blood samples with different HLA-haplotypes are used in hematopoietic stem cell transplantation for patients suffering from leukemia and other blood diseases to minimize the impact of immune rejection. Therefore, one approach to reduce the cost of iPSC-based therapy is to use allogeneic donors homozygous for common HLA types. This approach does not require genome editing and allows the generation of iPSC lines and the creation of a clinical-grade iPSC bank covering a large percentage of the population.
In the case of the Japanese population, 30 iPSC lines homozygous for common HLA types selected from 15,000 donors were assumed to provide a three-locus (loci affecting graft rejection—HLA-A, HLA-B, and HLA-DR) match for 82.2% of recipients, whereas 50 iPSC lines selected from 24,000 donors provided coverage for 90.7% of the same population [
4]. Okita et al. reported that the screening of 160,000 donors resulted in the selection of 140 HLA-homozygous donors with iPSCs, providing a match for 90% of the Japanese population [
5]. Analytical data based on prospective HLA typing of large numbers of individuals in other ethnic populations are available. It has been estimated that 26,000 donors for European Americans and 110,000 donors for African Americans would need to be screened to achieve a match of 50% and 22% of the recipient population, respectively [
86].
In contrast to previous approaches based on randomly finding HLA types, Taylor et al. proposed an alternative approach based on the concept that iPSCs derived from adult donors with the desired homozygous HLA type could be identified from existing registries of voluntary stem cell donors. By calculating all theoretically possible homozygous HLA-A, -B, and -DR combinations, 405 combinations were found that together could provide an HLA match for all potential UK recipients. It was found that the most useful homozygous HLA-types present among the more than 17 million HLA-typed potential stem cell donors registered on the Bone Marrow Donors Worldwide website could provide a complete HLA match for 93.16% of the UK population [
87]. HLA–antigen matching rates are similar in countries with similar demographic majorities. The top four homozygous haplotypes found in the Australian population are identical to the top four haplotypes found in the United Kingdom [
88]. Similar modeling in Korea also revealed common homozygous HLA haplotypes with most Asian countries including Japan, China, Hong Kong, Taiwan, Vietnam, the Philippines, etc. [
89].
At the same time, comparative HLA analysis shows that iPSC lines relevant in one country have limited use in ethnically diverse populations in other countries, although in some cases there is significant overlap in certain HLA haplotypes. The HLA-haplotypes found by Okita et al. (HLA-A*24:02; HLA-B*52:01; HLA-DRB1*15:02 and HLA-A*11:01; HLA-B*15:01; and HLA-DRB1*04:06) are present in 8.5% and 1.3% of the Japanese population, respectively, and iPSCs derived from these two donors match approximately 20% of Japanese individuals [
5]. However, theoretically, these HLA haplotypes may not be of much use in, for example, the UK population, as none of the haplotypes mentioned are part of the optimal combination of the most useful homozygous HLA types reported by Taylor et al. [
87].
In addition, HLA matching with homozygous donors does not guarantee protection against NK-cell attack. There are two HLA-C groups, namely HLA-C1 and HLA-C2, which bind to different inhibitory receptors, KIR2DL3 and KIR2DL1, respectively, on the surface of NK-cells [
90]. When using donor HLA homozygous iPSC lines, either C1/C1 or C2/C2, the phenomenon of the lack of self-recognition due to lack of inhibitory signaling will be observed for recipients with C1/C2. In the Japanese population, the frequency of C1 and C2 allotypes is 92.7:7.3. Therefore, the above homozygous iPSC lines would be applicable and mismatches would be rare. However, in other populations where the frequency of C2 is higher, this issue will be more significant. For example, in the Polish population, the ratio of C1 to C2 is approximately 6:4 [
91], as well as in Russia [
92].
The heterogeneity of ethnic populations indicates that there is no single universal set of the most useful homozygous HLA-types to maximize the coverage of the entire world. Each country selects optimal sets based on the prevalence of certain HLA haplotypes in its population. The World Marrow Donor Association currently estimates that 804,522 cord blood units are stored in cord blood banks worldwide [
93]. Collaboration with established biobanks allows the efficient identification of HLA-homozygous donors to prepare a stock of iPSC lines with the best immune compatibility in a given population [
88,
94,
95,
96]. Therefore, it should be understood that the establishment of a global network of iPSC banks is necessary to lay a solid foundation for future worldwide clinical cell therapy. At the same time, even with the selection of HLA-adequate donor material, immunosuppressive therapy after transplantation cannot always be completely avoided. However, this approach allows the dose and duration of immunosuppression to be reduced, which in itself is a significant advantage for patients.
4. CRISPR/Cas-Based Cell Therapy
CRISPR/Cas-mediated genome editing and iPSC technology enable a new class of cell replacement therapies for the treatment of T1D. It is likely that their combination will accelerate the development of a β-cell replacement drug aimed at treating diabetes without the need for immunosuppression in a deviceless approach. However, the drawbacks of both cell technologies and CRISPR/Cas-editors must be carefully considered. β-cells obtained through differentiation or transdifferentiation protocols should be purified from immature β-like, non-targeted, or undifferentiated cells that may adversely affect β-cell activity. Allogeneic β-cells differentiated from genetically engineered low-immunogenic iPSCs are a cheap and rapid option for the treatment of T1D. However, their level of protection from the immune system is currently insufficient, resulting in significant levels of immune rejection in patients. The use of classical CRISPR/Cas system tools delivered in viral vectors can lead to the creation of off-target edits and insertional mutagenesis, which can lead to the cancerous transformation of low-immunogenic β-cells. This problem can be addressed by using high-fidelity Cas9 variants or advanced Cas9-based tools such as prime editors, delivering CRISPR/Cas systems as mRNA or RNA complexes, and carefully planning genome editing experiments to reduce the number of successive rounds of editing and cell clone selection to a minimum. To control the possible cancerous transformation of engineered β-cells, inducible non-immunogenic and cell state-independent suicide genes, such as inducible caspase 9, can be introduced. In addition, safe harbors for the introduction of protective genes should be carefully explored and selected so as not to disrupt the functions of surrounding genes. Perhaps our list of hidden problems and their solutions is not complete and further studies and clinical trials will uncover additional difficulties associated with hypoimmunogenic engineered β-cells. However, our current knowledge already allows us to foresee serious problems, the solution of which will greatly reduce the chance of turning engineered β-cells into a “Trojan horse” that will bring hidden problems to T1D patients after transplantation.