Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Benjamin Gellhaus | -- | 2080 | 2023-12-19 14:01:00 | | | |
| 2 | Rita Xu | Meta information modification | 2080 | 2023-12-20 02:47:43 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Gellhaus, B.; Böker, K.O.; Schilling, A.F.; Saul, D. IGF-1/PI3K/AKT/FOXO3 Axis in Sarcopenia. Encyclopedia. Available online: https://encyclopedia.pub/entry/52933 (accessed on 11 June 2026).
Gellhaus B, Böker KO, Schilling AF, Saul D. IGF-1/PI3K/AKT/FOXO3 Axis in Sarcopenia. Encyclopedia. Available at: https://encyclopedia.pub/entry/52933. Accessed June 11, 2026.
Gellhaus, Benjamin, Kai O. Böker, Arndt F. Schilling, Dominik Saul. "IGF-1/PI3K/AKT/FOXO3 Axis in Sarcopenia" Encyclopedia, https://encyclopedia.pub/entry/52933 (accessed June 11, 2026).
Gellhaus, B., Böker, K.O., Schilling, A.F., & Saul, D. (2023, December 19). IGF-1/PI3K/AKT/FOXO3 Axis in Sarcopenia. In Encyclopedia. https://encyclopedia.pub/entry/52933
Gellhaus, Benjamin, et al. "IGF-1/PI3K/AKT/FOXO3 Axis in Sarcopenia." Encyclopedia. Web. 19 December, 2023.
Copy Citation
The high prevalence of sarcopenia in an aging population has an underestimated impact on quality of life by increasing the risk of falls and subsequent hospitalization. Unfortunately, the application of the major established key therapeutic—physical activity—is challenging in the immobile and injured sarcopenic patient. Consequently, novel therapeutic directions are needed. The transcription factor Forkhead-Box-Protein O3 (FOXO3) may be an option, as it and its targets have been observed to be more highly expressed in sarcopenic muscle.
sarcopenia
FOXO3
skeletal muscle atrophy
aging
1. Introduction
According to the definition by Rosenberg, sarcopenia is characterized as a loss of muscle mass and function due to aging [1]. The European Working Group on Sarcopenia in Older People (EWGSOP2) published an updated definition (2018) for sarcopenia based on three main criteria: 1. Lower muscle strength is accompanied by 2. an impaired muscle quality and quantity and 3. lowered physical performance in the patient. A probable sarcopenia is identified by the first criterion and the diagnosis is confirmed by the second. If all three criteria are met by a patient, sarcopenia is diagnosed as severe. Moreover, sarcopenia is subdivided by its etiology into primary (aging) and secondary (disease/inflammation, inactivity, malnutrition) [2]. Remarkably, the lowered muscle strength became the major criterion. In 2010, the EWGSOP defined sarcopenia on lower muscle mass as the first, lower muscle strength as the second, and low physical performance as the third criterion. Here, depending on how many criteria were met, the stages presarcopenia (only criterion one), sarcopenia, and severe sarcopenia (all criteria) were defined [3].
Furthermore, it is of crucial importance to differentiate sarcopenia from muscle atrophy and cancer cachexia. Atrophy is defined as cell shrinkage leading to a decreased size of a tissue or an organ. Regarding muscle atrophy, the skeletal muscle fiber size is decreased [4]. In contrast to that, cancer cachexia is defined as a loss of muscle mass due to deregulated energy metabolism that is almost resistant to nutritional supplementation [5]. Basically, atrophy is part of both definitions (cancer cachexia and sarcopenia), but cancer cachexia is defined via metabolic imbalances, while sarcopenia focuses on the loss of function.
2. PI3K/AKT Pathway
In general, an upregulation of the PI3K/AKT pathway results in inactivation of FOXO3 by phosphorylation. This is governed by IGF-1 as an activator of growth factor receptor protein tyrosine kinases in anabolic situations. In catabolic situations, the absence of IGF-1 results in active FOXO3 that enters the nucleus to bind to the promotor region of the ubiquitin ligases Atrogin-1 and Murf-1 [6][7][8][9] (Figure 1). Both (ATROGIN-1 and MURF-1) are E3 ubiquitin ligases that cause polyubiquitination of proteins and result in proteasomal degradation [10]. Autophosphorylation of an activated growth factor receptor protein tyrosine kinase, located at the cell membrane, results in recruiting phosphoinositide 3-kinase (PI3K). This kinase converts phosphatidylinositol-4,5-bisphosphate (PIP2), a membrane-bound molecule, to phosphatidylinositol-3,4,5-trisphosphate (PIP3). Because of its ability to bind signaling molecules containing a certain pleckstrin homology, PIP3 brings both the serine–threonine protein kinase (AKT) and the phosphoinositide-dependent kinase 1 (PDK1) together. If PDK1 is physically close to its downstream target AKT, AKT is activated via phosphorylation. Activated AKT phosphorylates and prevents FOXO3 from entering the nucleus [11] (Figure 1). More specifically, AKT phosphorylates FOXO3 at Ser253, S315, and T32 in vitro and in vivo. The phosphorylated FOXO3 is bound by the protein 14-3-3, which isolates FOXO3 as inactive in the cytoplasm [12]. Notably, there are other proteins that regulate Foxo3 both negatively and positively (see [13]). Two examples of positive regulation are demonstrated: At first, MAPK-activated protein kinase 5 can phosphorylate and activate FOXO3 in response to DNA damage [14]. Also, the AMP-activated protein kinase activates the transcriptional activity of FOXO3 through phosphorylation at low energy levels [15]. By comparing the contrary regulatory mechanisms of FOXO3, the targetability of Foxo3 itself is highlighted.
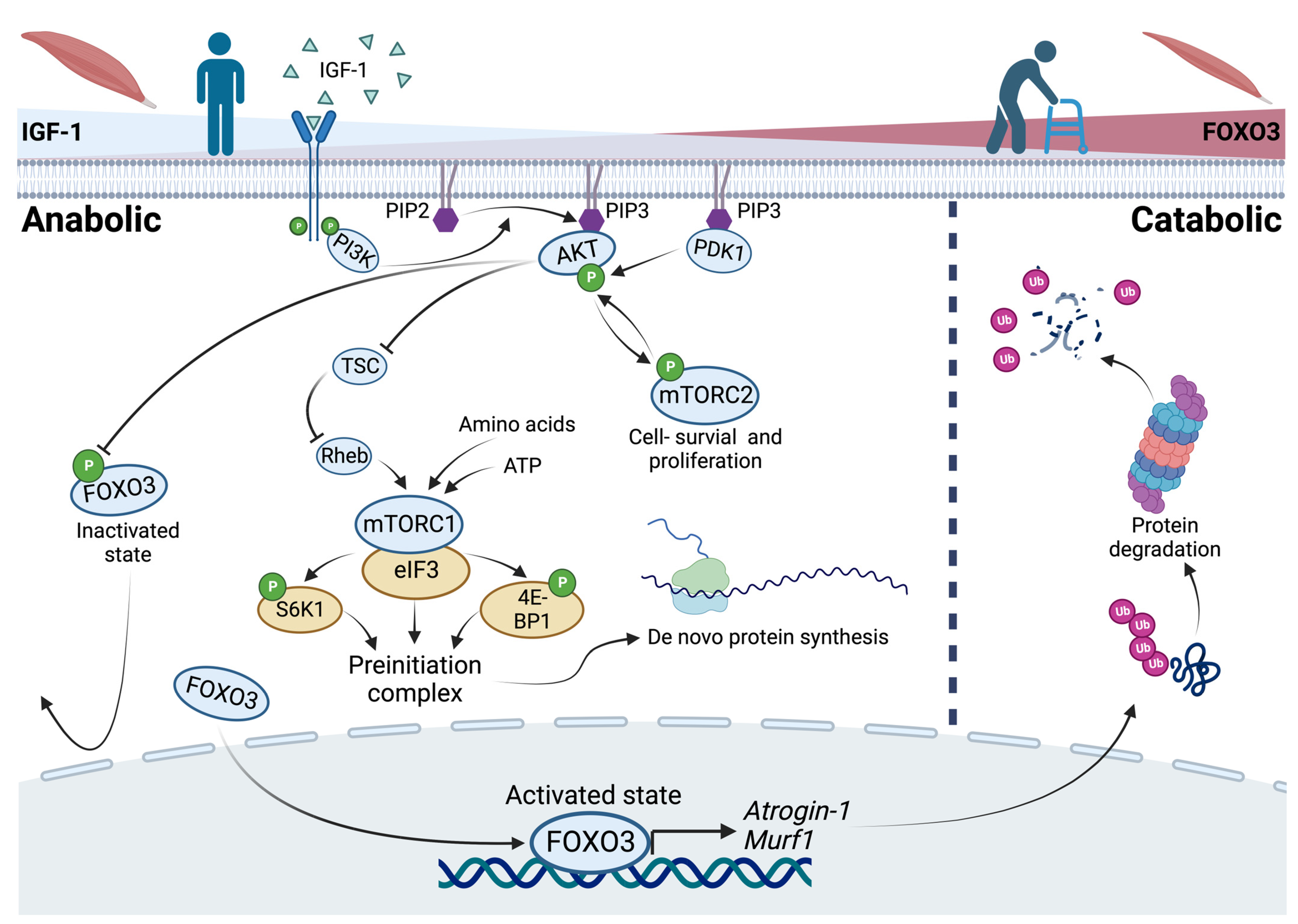
Figure 1. The PI3K/AKT pathway and the role of FOXO3 in anabolic and catabolic conditions as a reflection of changes in the catabolic state, like aging. In anabolic situations, recruitment of phosphoinositide-3-kinases (PI3K) to an activated growth factor receptor results in the conversion of phosphatidylinositol-4,5-bisphosphate (PIP2) to phosphatidylinositol-3,4,5-trisphosphate (PIP3) bringing the serine–threonine protein kinase (AKT) and phosphoinositide-dependent kinase 1 (PDK1) together. AKT inhibits the Forkhead-Box Protein O3 (FOXO3) by phosphorylation. Further, AKT promotes cell survival via the mammalian target of rapamycin complex 2 (mTORC2) and indirectly promotes protein synthesis via the mammalian target of rapamycin complex 1 (mTORC1). In catabolic situations, FOXO3 regulates the expression of its downstream targets Atrogin-1 and Murf-1 as ubiquitin ligases that cause proteasomal degradation of proteins.
3. Post-Translational Modification of FOXO3: Acetylation and Deacetylation
Another regulatory mechanism of FOXO3 involves post-translational modification through acetylation and corresponding deacetylation. The CBP/p300 coactivator mediates acetylation, while SIRT1 and SIRT2 mediate deacetylation [16]. However, acetylation of FOXO3 has been reported to induce its cytosolic translocation and consequent proteasomal degradation in C57BL/6J mice in vivo [17].
On the other hand, it was found that SIRT1-mediated deacetylation of FOXO3 increases its activity in the nucleus accumbens of C57BL/6J mice in vivo [18]. Remarkably, SIRT1-mediated deacetylation of FOXO3 also leads to a decrease in its activity in vitro [19]. Another study confirmed that FOXO3 activity was reduced by SIRT1 deacetylation as well as by SIRT2 in vitro [20]
In conclusion, the post-translational modification of FOXO3 appeared to be intricate. Although its acetylation seemed to reduce its activity, deacetylation has been observed to both increase and decrease FOXO3 activity [16].
4. De Novo Protein Synthesis via mTOR Signaling
In vitro studies revealed that IGF-1 caused myotube hypertrophy in C2C12 myoblasts (an immortalized myoblast cell line used to study myogenesis) to be mediated via the PI3K/AKT/mTOR pathway, which was prevented by applying Rapamycin, an inhibitor of the mammalian target of rapamycin (mTOR) [21]. The protein kinase mTOR is contained in two complexes with different functions. The first one, mTORC1, mediates cell growth and is sensitive to rapamycin. The other one, mTORC2, mediates cell survival and proliferation but is insensitive to rapamycin [22]. Increased ATP or amino acid levels, predominantly in anabolic conditions, positively regulate the activity of mTORC1 independently [23][24]. Activated mTORC1 binds to eIF3 and phosphorylates S6K1 and 4E-BP1, leading to an assembly of the preinitiation complex and therefore de novo protein synthesis in vitro [25][26]. Another positive but indirect regulator of mTORC1 is AKT itself. AKT phosphorylates the tuberous sclerosis complex (TSC), leading to a release from the GTPase Rheb which in turn activates mTORC1 [27]. In conclusion, a positive effect of mTORC1 in terms of myotube hypertrophy and protein synthesis is described. In contrast, an in vivo mouse model revealed that mTORC1 inhibition by rapamycin had positive effects on age-related muscle loss, whereas TSC knockout mice (higher levels of active mTORC1) showed a sarcopenic muscle fiber pattern due to impaired stability of the neuromuscular junction [28]. This implies that in addition to atrophy, the etiology of denervation plays an important role in sarcopenia. However, mTORC2 mediates the IGF-1 signaling as a direct target of AKT. Moreover, PDK1 phosphorylates and activates AKT, yet activates mTORC2 by phosphorylation. Via a positive feedback loop, the activation of AKT is boosted by phosphorylation of mTORC2 [29][30].
Fiber Type Composition during Aging
In the aging of skeletal muscle, different types of fibers follow different paths, which is important to understand for the development of a specific molecular therapy. A comparative study analyzing human M. vastus lateralis biopsies of younger (23–31 years) and older (68–70 years) men identified myosin heavy chain (MHC) type I fibers as constant in size upon aging, but MHC type IIa and IIx fibers decreasing in size in older subjects [31]. The phenomenon of a type II fiber size decrease in aging was identified to be reversed when resistance training (RT) was applied. As a result of 6 months of training, type II fiber size was increased by RT, but type I fibers remained constant in size in younger (23 years) compared to older (71 years) men [32]. The different muscle types were also rigorously studied in a mouse model. Fast-twitch gastrocnemius muscle (type II) was identified to undergo the highest impairment by aging-dependent atrophy, while slow-twitch (type I) soleus muscle remained unaffected. This finding mirrors the observed human fiber type II atrophy due to aging, as described above [33]. As another response to exercise, the peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) is more highly expressed in human and rat muscle in in vivo models after endurance training [34][35][36].
Interestingly, PGC-1α transgenic mice are protected from denervation and fasting-induced muscle atrophy and show a reduced expression of genes required for glycolysis and oxidative phosphorylation [37]. This suggests that PGC-1α creates a milieu typically preferred by type I fibers. Consequently, if the PGC-1α gene was placed downstream of the muscle creatine kinase and subsequently selectively expressed in skeletal and cardiac muscle, type II muscle fibers switched and became type I fibers, indicated by the expression of typical genes for type I fibers (such as troponin I, myoglobin, and cytochrome C) and showed a higher fatigue resistance, induced by electrical stimulation [38][39]. Interestingly, PGC-1α is able to block FOXO3 binding to its responsive element on the Atrogin-1 promotor [37]. Both ubiquitin ligases (Atrogin-1 and Murf-1) have been identified to be expressed more highly and selectively in cardiac and skeletal muscle during muscle atrophy in an in vivo mouse model [40]. Further, skeletal muscle atrophy was observed to be more severe in glycolytic (IId/x and IIb) compared to oxidative (I and IIa) fibers in vivo [41]. These findings suggest a link between FOXO3 and its target genes (Atrogin-1 and Murf-1) and therefore the PI3K/AKT pathway to sarcopenia, highlighting FOXO3 as a possible target.
5. The Influence of Physical Activity and Aging upon the Expressional Profile of FOXO3 in Humans
Foxo3 content and its target genes have also been studied in the context of physical activity. Here, in mice with unilaterally immobilized hindlimbs, immobilization caused an increase in FOXO3 acetylation and a reduction in gastrocnemius muscle weight in vivo. However, this increase was reversible. Within a few days of unrestricted movement, FOXO3 acetylation levels decreased again. The authors conclude that acetylation of FOXO3 promotes muscle atrophy, while deacetylation of FOXO3 increases muscle regeneration potential, highlighting the importance of physical activity [42].
Two in vivo human studies of younger (20 years) and older (70 years) healthy male and female subjects independently showed that ATROGIN1 and MURF1 expression did not change during aging [43][44]. Further investigations showed that ubiquitin expression did not change in human rectus abdominis muscles either [45]. However, the overall ubiquitin protein content showed an age-dependent increase. Ubiquitin protein levels were increased in older (70–79 years) human quadriceps muscle biopsies compared to younger (20–29 years) subjects’ biopsies [46]. In contrast, another study identified increased levels of FOXO3 and MURF1 (but not ATROGIN1) in older healthy females (85 years) compared to younger females (23 years) in M. vastus lateralis biopsies. After a single session of RT, the FOXO3 expression remained unchanged, whereas ATROGIN1 expression was markedly increased in older subjects, and MURF1 accumulated in both groups [47]. A follow-up study in older women (70 years) performing long-term training (12 weeks on a cycle ergometer) revealed decreased FOXO3 expression levels with no significant effect on the protein levels and no change in expression of ATROGIN1 and MURF1 [48] (Table 1). An additional comparative study investigated 12 weeks of RT with a focus on FOXO3 protein content in younger (24 years) and older (85 years) females. In the untrained state, older subjects showed lower levels of cytosolic phosphorylated FOXO3 (P-FOXO3) and therefore less inactivated FOXO3. After the training period, increased levels of total nuclear FOXO3 were observed in older subjects. On the other side, younger subjects showed higher levels of P-FOXO3 in response to RT. These results indicate an increase in total nuclear FOXO3 due to aging and impaired nuclear phosphorylation and thus inactivation in response to resistance training, which may attenuate the beneficial effect of physical activity [49] (Table 1).
Table 1. FOXO3 levels upon different (training) conditions.
| FOXO3 Levels | Condition | Model | Reference |
|---|---|---|---|
| FOXO3 ↓ after 12 weeks on a cycle ergometer in older women |
Long-term training | Human | [48] |
| FOXO3 phosphorylation ↓ before and total nuclear FOXO3 ↑ after 12 weeks of RT in older females |
RT | Human | [49] |
| FOXO3 ↑ in older healthy females with FOXO3 expression ↔ after a single session of RT |
Aging + RT | Human | [47] |
| FOXO3 acetylation↑ due to hindlimb immobilization |
Immobilization | Mice | [42] |
| No age-dependent downregulation of the PI3K-AKT pathway | Aging | Mice | [50] |
In summary, these results suggest that the aging muscle is atrophic through increased protein content of Ubiquitin and nuclear FOXO3. Moreover, it suggests an imbalanced anabolic/catabolic interaction, linking FOXO3 as a potential molecular target for the clinical treatment of sarcopenia.
Therapeutical Targets
To evaluate the targetability of the PI3K/AKT pathway, the current prospects can be subdivided into an AKT-dependent and an AKT-independent treatment strategy.
References
- Rosenberg, I.H. Sarcopenia: Origins and Clinical Relevance. J. Nutr. 1997, 127, 990S–991S.
- Cruz-Jentoft, A.J.; Bahat, G.; Bauer, J.; Boirie, Y.; Bruyère, O.; Cederholm, T.; Cooper, C.; Landi, F.; Rolland, Y.; Sayer, A.A.; et al. Sarcopenia: Revised European Consensus on Definition and Diagnosis. Age Ageing 2019, 48, 16–31.
- Cruz-Jentoft, A.J.; Baeyens, J.P.; Bauer, J.M.; Boirie, Y.; Cederholm, T.; Landi, F.; Martin, F.C.; Michel, J.-P.; Rolland, Y.; Schneider, S.M.; et al. Sarcopenia: European Consensus on Definition and Diagnosis. Age Ageing 2010, 39, 412–423.
- Bonaldo, P.; Sandri, M. Cellular and Molecular Mechanisms of Muscle Atrophy. Dis. Models Mech. 2013, 6, 25–39.
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and Classification of Cancer Cachexia: An International Consensus. Lancet Oncol. 2011, 12, 489–495.
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo Transcription Factors Induce the Atrophy-Related Ubiquitin Ligase Atrogin-1 and Cause Skeletal Muscle Atrophy. Cell 2004, 117, 399–412.
- Sacheck, J.M.; Ohtsuka, A.; McLary, S.C.; Goldberg, A.L. IGF-I Stimulates Muscle Growth by Suppressing Protein Breakdown and Expression of Atrophy-Related Ubiquitin Ligases, Atrogin-1 and MuRF1. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E591–E601.
- Latres, E.; Amini, A.R.; Amini, A.A.; Griffiths, J.; Martin, F.J.; Wei, Y.; Lin, H.C.; Yancopoulos, G.D.; Glass, D.J. Insulin-like Growth Factor-1 (IGF-1) Inversely Regulates Atrophy-Induced Genes via the Phosphatidylinositol 3-Kinase/Akt/Mammalian Target of Rapamycin (PI3K/Akt/mTOR) Pathway. J. Biol. Chem. 2005, 280, 2737–2744.
- Bollinger, L.M.; Witczak, C.A.; Houmard, J.A.; Brault, J.J. SMAD3 Augments FoxO3-Induced MuRF-1 Promoter Activity in a DNA-Binding-Dependent Manner. Am. J. Physiol. Cell Physiol. 2014, 307, C278–C287.
- Bodine, S.C.; Baehr, L.M. Skeletal Muscle Atrophy and the E3 Ubiquitin Ligases MuRF1 and MAFbx/Atrogin-1. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E469–E484.
- Cantley, L.C. The Phosphoinositide 3-Kinase Pathway. Science 2002, 296, 1655–1657.
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt Promotes Cell Survival by Phosphorylating and Inhibiting a Forkhead Transcription Factor. Cell 1999, 96, 857–868.
- Eijkelenboom, A.; Burgering, B.M.T. FOXOs: Signalling Integrators for Homeostasis Maintenance. Nat. Rev. Mol. Cell Biol. 2013, 14, 83–97.
- Kress, T.R.; Cannell, I.G.; Brenkman, A.B.; Samans, B.; Gaestel, M.; Roepman, P.; Burgering, B.M.; Bushell, M.; Rosenwald, A.; Eilers, M. The MK5/PRAK Kinase and Myc Form a Negative Feedback Loop That Is Disrupted during Colorectal Tumorigenesis. Mol. Cell 2011, 41, 445–457.
- Greer, E.L.; Oskoui, P.R.; Banko, M.R.; Maniar, J.M.; Gygi, M.P.; Gygi, S.P.; Brunet, A. The Energy Sensor AMP-Activated Protein Kinase Directly Regulates the Mammalian FOXO3 Transcription Factor. J. Biol. Chem. 2007, 282, 30107–30119.
- Wang, X.; Hu, S.; Liu, L. Phosphorylation and Acetylation Modifications of FOXO3a: Independently or Synergistically? (Review). Oncol. Lett. 2017, 13, 2867–2872.
- Bertaggia, E.; Coletto, L.; Sandri, M. Posttranslational Modifications Control FoxO3 Activity during Denervation. Am. J. Physiol. Cell Physiol. 2012, 302, C587–C596.
- Ferguson, D.; Shao, N.; Heller, E.; Feng, J.; Neve, R.; Kim, H.-D.; Call, T.; Magazu, S.; Shen, L.; Nestler, E.J. SIRT1-FOXO3a Regulate Cocaine Actions in the Nucleus Accumbens. J. Neurosci. 2015, 35, 3100–3111.
- Motta, M.C.; Divecha, N.; Lemieux, M.; Kamel, C.; Chen, D.; Gu, W.; Bultsma, Y.; McBurney, M.; Guarente, L. Mammalian SIRT1 Represses Forkhead Transcription Factors. Cell 2004, 116, 551–563.
- Wang, F.; Chan, C.-H.; Chen, K.; Guan, X.; Lin, H.-K.; Tong, Q. Deacetylation of FOXO3 by SIRT1 or SIRT2 Leads to Skp2-Mediated FOXO3 Ubiquitination and Degradation. Oncogene 2012, 31, 1546–1557.
- Rommel, C.; Bodine, S.C.; Clarke, B.A.; Rossman, R.; Nunez, L.; Stitt, T.N.; Yancopoulos, G.D.; Glass, D.J. Mediation of IGF-1-Induced Skeletal Myotube Hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 Pathways. Nat. Cell Biol. 2001, 3, 1009–1013.
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976.
- Hara, K.; Yonezawa, K.; Weng, Q.-P.; Kozlowski, M.T.; Belham, C.; Avruch, J. Amino Acid Sufficiency and mTOR Regulate P70 S6 Kinase and eIF-4E BP1 through a Common Effector Mechanism. J. Biol. Chem. 1998, 273, 14484–14494.
- Dennis, P.B.; Jaeschke, A.; Saitoh, M.; Fowler, B.; Kozma, S.C.; Thomas, G. Mammalian TOR: A Homeostatic ATP Sensor. Science 2001, 294, 102–1105.
- Holz, M.K.; Ballif, B.A.; Gygi, S.P.; Blenis, J. mTOR and S6K1 Mediate Assembly of the Translation Preinitiation Complex through Dynamic Protein Interchange and Ordered Phosphorylation Events. Cell 2005, 123, 569–580.
- Thoreen, C.C.; Chantranupong, L.; Keys, H.R.; Wang, T.; Gray, N.S.; Sabatini, D.M. A Unifying Model for mTORC1-Mediated Regulation of mRNA Translation. Nature 2012, 485, 109–113.
- Menon, S.; Dibble, C.C.; Talbott, G.; Hoxhaj, G.; Valvezan, A.J.; Takahashi, H.; Cantley, L.C.; Manning, B.D. Spatial Control of the TSC Complex Integrates Insulin and Nutrient Regulation of mTORC1 at the Lysosome. Cell 2014, 156, 771–785.
- Ham, D.J.; Börsch, A.; Lin, S.; Thürkauf, M.; Weihrauch, M.; Reinhard, J.R.; Delezie, J.; Battilana, F.; Wang, X.; Kaiser, M.S.; et al. The Neuromuscular Junction Is a Focal Point of mTORC1 Signaling in Sarcopenia. Nat. Commun. 2020, 11, 4510.
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and Regulation of Akt/PKB by the Rictor-mTOR Complex. Science 2005, 307, 1098–1101.
- Yang, G.; Murashige, D.S.; Humphrey, S.J.; James, D.E. A Positive Feedback Loop between Akt and mTORC2 via SIN1 Phosphorylation. Cell Rep. 2015, 12, 937–943.
- Klitgaard, H.; Zhou, M.; Schiaffino, S.; Betto, R.; Salviati, G.; Saltin, B. Ageing Alters the Myosin Heavy Chain Composition of Single Fibres from Human Skeletal Muscle. Acta Physiol. Scand. 1990, 140, 55–62.
- Nilwik, R.; Snijders, T.; Leenders, M.; Groen, B.B.L.; van Kranenburg, J.; Verdijk, L.B.; van Loon, L.J.C. The Decline in Skeletal Muscle Mass with Aging Is Mainly Attributed to a Reduction in Type II Muscle Fiber Size. Exp. Gerontol. 2013, 48, 492–498.
- Picard, M.; Ritchie, D.; Thomas, M.M.; Wright, K.J.; Hepple, R.T. Alterations in Intrinsic Mitochondrial Function with Aging Are Fiber Type-Specific and Do Not Explain Differential Atrophy between Muscles. Aging Cell 2011, 10, 1047–1055.
- Russell, A.P.; Feilchenfeldt, J.; Schreiber, S.; Praz, M.; Crettenand, A.; Gobelet, C.; Meier, C.A.; Bell, D.R.; Kralli, A.; Giacobino, J.-P.; et al. Endurance Training in Humans Leads to Fiber Type-Specific Increases in Levels of Peroxisome Proliferator-Activated Receptor-Gamma Coactivator-1 and Peroxisome Proliferator-Activated Receptor-Alpha in Skeletal Muscle. Diabetes 2003, 52, 2874–2881.
- Baar, K.; Wende, A.R.; Jones, T.E.; Marison, M.; Nolte, L.A.; Chen, M.; Kelly, D.P.; Holloszy, J.O. Adaptations of Skeletal Muscle to Exercise: Rapid Increase in the Transcriptional Coactivator PGC-1. FASEB J. 2002, 16, 1879–1886.
- Taylor, E.B.; Lamb, J.D.; Hurst, R.W.; Chesser, D.G.; Ellingson, W.J.; Greenwood, L.J.; Porter, B.B.; Herway, S.T.; Winder, W.W. Endurance Training Increases Skeletal Muscle LKB1 and PGC-1α Protein Abundance: Effects of Time and Intensity. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E960–E968.
- Sandri, M.; Lin, J.; Handschin, C.; Yang, W.; Arany, Z.P.; Lecker, S.H.; Goldberg, A.L.; Spiegelman, B.M. PGC-1α Protects Skeletal Muscle from Atrophy by Suppressing FoxO3 Action and Atrophy-Specific Gene Transcription. Proc. Natl. Acad. Sci. USA 2006, 103, 16260–16265.
- Lin, J.; Wu, H.; Tarr, P.T.; Zhang, C.-Y.; Wu, Z.; Boss, O.; Michael, L.F.; Puigserver, P.; Isotani, E.; Olson, E.N.; et al. Transcriptional Co-Activator PGC-1α Drives the Formation of Slow-Twitch Muscle Fibres. Nature 2002, 418, 797–801.
- Johnson, J.E.; Wold, B.J.; Hauschka, S.D. Muscle Creatine Kinase Sequence Elements Regulating Skeletal and Cardiac Muscle Expression in Transgenic Mice. Mol. Cell. Biol. 1989, 9, 3393–3399.
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of Ubiquitin Ligases Required for Skeletal Muscle Atrophy. Science 2001, 294, 1704–1708.
- Li, P.; Waters, R.E.; Redfern, S.I.; Zhang, M.; Mao, L.; Annex, B.H.; Yan, Z. Oxidative Phenotype Protects Myofibers from Pathological Insults Induced by Chronic Heart Failure in Mice. Am. J. Pathol. 2007, 170, 599–608.
- Chacon-Cabrera, A.; Gea, J.; Barreiro, E. Short- and Long-Term Hindlimb Immobilization and Reloading: Profile of Epigenetic Events in Gastrocnemius. J. Cell. Physiol. 2017, 232, 1415–1427.
- Léger, B.; Derave, W.; De Bock, K.; Hespel, P.; Russell, A.P. Human Sarcopenia Reveals an Increase in SOCS-3 and Myostatin and a Reduced Efficiency of Akt Phosphorylation. Rejuvenation Res. 2008, 11, 163–175B.
- Whitman, S.A.; Wacker, M.J.; Richmond, S.R.; Godard, M.P. Contributions of the Ubiquitin–Proteasome Pathway and Apoptosis to Human Skeletal Muscle Wasting with Age. Pflug. Arch. Eur. J. Physiol. 2005, 450, 437–446.
- Bossola, M.; Pacelli, F.; Costelli, P.; Tortorelli, A.; Rosa, F.; Doglietto, G.B. Proteasome Activities in the Rectus Abdominis Muscle of Young and Older Individuals. Biogerontology 2008, 9, 261.
- Cai, D.; Lee, K.K.H.; Li, M.; Tang, M.K.; Chan, K.M. Ubiquitin Expression Is Up-Regulated in Human and Rat Skeletal Muscles during Aging. Arch. Biochem. Biophys. 2004, 425, 42–50.
- Raue, U.; Slivka, D.; Jemiolo, B.; Hollon, C.; Trappe, S. Proteolytic Gene Expression Differs At Rest and After Resistance Exercise Between Young and Old Women. J. Gerontol. Ser. A 2007, 62, 1407–1412.
- Konopka, A.R.; Douglass, M.D.; Kaminsky, L.A.; Jemiolo, B.; Trappe, T.A.; Trappe, S.; Harber, M.P. Molecular Adaptations to Aerobic Exercise Training in Skeletal Muscle of Older Women. J. Gerontol. Ser. A 2010, 65A, 1201–1207.
- Williamson, D.L.; Raue, U.; Slivka, D.R.; Trappe, S. Resistance Exercise, Skeletal Muscle FOXO3A, and 85-Year-Old Women. J. Gerontol. A Biol. Sci. Med. Sci. 2010, 65A, 335–343.
- Sandri, M.; Barberi, L.; Bijlsma, A.Y.; Blaauw, B.; Dyar, K.A.; Milan, G.; Mammucari, C.; Meskers, C.G.M.; Pallafacchina, G.; Paoli, A.; et al. Signalling Pathways Regulating Muscle Mass in Ageing Skeletal Muscle. The Role of the IGF1-Akt-mTOR-FoxO Pathway. Biogerontology 2013, 14, 303–323.
More
Information
Subjects:
Nutrition & Dietetics; Orthopedics
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
619
Revisions:
2 times
(View History)
Update Date:
20 Dec 2023
Table of Contents




Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No