 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Natalia V. Belosludtseva | -- | 7371 | 2023-12-18 16:49:44 | | | |
| 2 | Catherine Yang | Meta information modification | 7371 | 2023-12-19 01:40:11 | | |
Video Upload Options
Amyotrophic lateral sclerosis (ALS) is a fatal multisystem disease characterized by progressive death of motor neurons, loss of muscle mass, and impaired energy metabolism. The morphology and dynamics of mitochondrial network, quality control mechanisms, motility, and overall mitochondrial function are closely interrelated pathways that play a fundamental role in the dyshomeostasis of mitochondria in neurons, surrounding glial cells, myocytes, and many other cell types across peripheral tissues. Molecular mechanisms responsible for altered mitostasis in ALS-affected cells contribute to excitotoxicity, oxidative stress, energy deficiency and, ultimately, the death of motoneurons and other cells. Damaged mitochondria can release a number of proapoptotic factors and inflammatory response activators called damage-related molecular patterns, which creates a cycle of direct communication between mitochondrial disorders, inflammation, and cell degeneration.
1. Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease that leads to severe disability due to increasing muscle weakness, atrophy, spasticity and, ultimately, to paralysis of striated muscles and early death, usually within 3–5 years after the onset of symptoms [1][2][3]. ALS belongs to the group of motor neuron diseases and is characterized by steadily progressive degeneration and death of motor neurons of the cerebral cortex, brainstem and anterior horns of the spinal cord (upper and lower motor neurons, correspondingly). The death of the upper motor neurons leads to spasticity and increased excitability, while the loss of the lower motor neurons directly innervating skeletal muscles causes increasing muscular atrophy and subsequent progressive paralysis [4][5]. Impaired energy metabolism, loss of muscle and adipose tissue mass, and significant weight loss are also common and severe symptoms of ALS, caused by both an abnormally high metabolic rate in the organism and inadequate caloric intake [6][7].
Mitochondria play a crucial role in energy, ionic, and redox metabolisms, as well as the maintenance of cellular homeostasis, including the balance of protein metabolism (proteostasis) and RNA metabolism (ribostasis), which are essential for the structural integrity and functioning of all components of the neuromuscular system under pathophysiological conditions [8][9][10]. Recent studies point to the development of defects in “mitochondrial homeostasis” in ALS as a possible pathogenetic pathway that deserves a more detailed study [7][8][11]. Mitochondrial homeostasis or mitostasis refers to the maintenance of the adequate quantity and quality of the organelles and their internal environment, which is dynamically regulated by oxidative phosphorylation, mitochondrial dynamics (the fission/fusion processes), trafficking, and quality control systems (mitochondrial biogenesis and clearance) [12].
In recent years, it has become obvious that the steadily progressive death of motor neurons and the increase in myopathy in ALS are directly related to the dysregulation of mitochondrial homeostasis and bioenergetic stress. Numerous studies revealed that the accumulation of abnormal mitochondria is closely correlated with the onset of denervation in ALS patients and many in vitro and in vivo models of ALS [13][14][15]. Studies showed that several ALS-related genes, including the most well-known SOD1 and the recently identified SQSTM1, OPTN, and TBK1, play a direct role in the regulation of mitochondrial function [16]. For example, the mutant protein SOD1 can accumulate on the outer membrane of mitochondria [17] and cause defects in the functioning of respiratory chain complexes [18]. It is noteworthy that ALS-linked mutations in OPTN, SQSTM1, and TBK1 can interfere with selective degradation of mitochondria via autophagy (mitophagy) and lead to inefficient turnover of damaged mitochondria, which may contribute to the progression of the neurodegenerative disease [19][20]. Furthermore, the significance of maintaining a pool of functional mitochondria in ALS was confirmed by the data on neuroprotective effects of a number of mitochondria-targeted compounds (in particular, antioxidants MtoQ, Mito-CP, SS-31, coenzyme A, and others) [21][22][23][24][25][26][27]. Therefore, mitochondrial malfunction seems to be due to the mutation of several genes associated with ALS and plays a crucial role in the pathogenesis of the disease. However, the molecular details relating to the complex relationship between mitochondrial abnormalities and specific alterations in cells leading to neuromuscular degeneration are still poorly understood.
This research provides an update on recent progress in the study of the causes and progression of dyshomeostasis of mitochondrial processes in the context of ALS, with particular emphasis on new understanding of the role of mitochondrial quality control systems and remodeling of the mitochondrial network in the development of ALS. A comprehensive characterization of mitochondrial dysfunction in ALS-affected cells and its underlying molecular mechanisms may provide insight into novel therapeutic approaches, which may have a significant impact on current symptomatic therapies and personalized treatment programs for patients with ALS.
2. Pathophysiology of ALS
ALS is the third leading neurodegenerative disease worldwide with a frequency of 2–2.5 per 10 thousand people per year and a prevalence of about 5 cases per 100 thousand people. Most cases of ALS (90–95%) are sporadic (sALS), while about 10% of cases are inherited in an autosomal dominant manner and, more rarely, in an autosomal recessive or X-linked manner and belong to familial forms of ALS (fALS) [7]. Two forms of the disease—sALS and fALS—are clinically indistinguishable, since in both cases similar pathological signs develop. Respiratory muscle denervation and ventilatory failure are the cause of lethal paralysis in most cases of ALS [2][7][28].
ALS was initially referred to as multiple proteinopathies, suggesting that mutant proteins in some cases of ALS may exhibit prion-like activity and induce the formation of intracellular deposits. Indeed, excessive accumulation and aggregation of mutant proteins occur at the initial stages of the pathological process, and then normally functioning cellular proteins can be involved in this process [1][28]. This phenomenon was found to be exacerbated by changes in two major protein clearance pathways, the ubiquitin-proteasome system and autophagy [16][28]. Then, it was observed that some ALS-related proteins are regulators of the metabolism of mRNAs and noncoding RNAs (ncRNAs) [29][30], leading to a further interpretation of the disease as an RNA-mediated neuropathology, which better reflects its heterogeneity.
Currently, mutations in more than 40 genes involved in various cellular processes have been linked to ALS, including proteostasis, ribostasis, DNA repair, cytoskeletal dynamics, and trafficking [31][32][33]. Among them, mutations in four genes, namely C9ORF72 (chromosome 9 open reading frame 72), SOD1 (Cu/n superoxide dismutase 1), TARDBP (transactive response DNA-binding protein 43 kDa), and FUS (fused in sarcoma), account for up to 70% of fALS. Other ALS-associated mutations were found in genes encoding a variety of proteins: ALS2, SETX, SPG1 1, VAPB, ANG, FIG 4, SIG-MA1, HNRNPA1, SQSTM1, VCP, PFN1, OPTN [31][32], TLS [34], UBQLN2 [35], and others [31]. Furthermore, an increasing number of studies report the presence of two or more mutations in different genes in ALS patients, suggesting the oligogenic inheritance of the disease in some cases [31]. In about 20% of fALS cases, the disease occurs due to mutations in the gene encoding the antioxidant enzyme SOD1, which in a healthy cell catalyzes the dismutation of superoxide anions into molecular oxygen and hydrogen peroxide [36][37]. Mutations in this gene were the very first to be discovered and most studied in genetic models of ALS.
Patients who do not have affected relatives are classified as having sALS [38]. In the cases of sALS, in addition to genetic mutations, environmental factors are of great importance, including smoking, toxins, heavy metals, pesticides, traumatic brain injuries, viral infections, etc. [39]. This may explain the difficulties in finding individual risk factors in ALS patients and bring the pathology closer to oncological diseases, since both diseases are apparently due to the consistent impact of many factors [38][40][41].
At the moment, there are several hypotheses about the site of origin of ALS in the body [3][42][43]. Among them, there are “dying forward” and “dying back” hypotheses of the development of ALS—depending on which structures in the chain “upper motor neuron—lower motor neuron—skeletal muscle” are affected first (Figure 1). According to the “dying-forward” hypothesis, the pathological process begins with the upper motor neurons and moves in an anterograde direction from the central nervous system via glutamate-mediated excitotoxic processes, resulting in anterior horn cell metabolic deficit [42]. According to the “dying-back” hypothesis, the pathological process begins in the skeletal muscle and/or neuromuscular junction [44][45][46], which leads to the launch of degeneration of the lower and then the upper motor neurons [47].
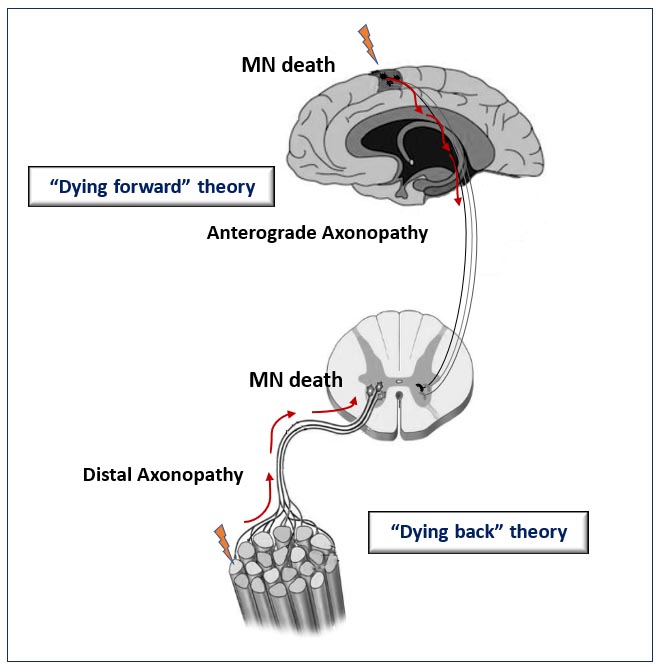
Figure 1. Schematic representation illustrating the theories for ALS onset. The “dying-forward” and “dying-back” hypotheses propose that pathological changes from the site of ALS origin move in the anterograde or retrograde direction, respectively, leading to the death of motor neuron (MN) cells.
Recent data suggest that the death of motor neurons is the result of pathogenetic processes occurring not only in motor neurons, but also in glial cells (astrocytes and microglia) and muscle fibers [4][48]. Moreover, studies demonstrated that ALS is not simply a disease of motor neurons, but rather a complex, multisystemic, and multifactorial disease that involves sensory neurons, interneurons, glial cells, and many other peripheral tissues [4][7][49].
The last few years of research in the field of genetics and the use of a number of in vitro and in vivo models of ALS have greatly advanced our understanding of the mechanisms of disease-associated changes in neuronal mitochondrial homeostasis and stress response. The most commonly studied ALS-linked mutations are SOD1 point mutations and variants, GGGGCC24+ hexanucleotide repeat expansion (HRE) in the C9ORF72 gene, TARDBP point mutations and variants, and mutations in the FUS and TBK1 genes. The main disease-causing mutations responsible for disturbances in mitochondrial homeostasis in neurons in genetic in vivo and in vitro models of the pathology are summarized in Table 1.
Table 1. The main ALS-causing mutations responsible for disturbances in mitostasis in neuronal and other cells in genetic in vitro and in vivo models of the pathology.
|
Disease-causing mutations |
Encoding proteins and their function |
Commonly used in vivo or in vitro models |
Most common disturbances in mitochondrial homeostasis |
References |
|
In vivo models |
||||
|
SOD1 point mutations and variants |
Cu/Zn superoxide dismutase 1 (SOD1) protein catalyzes the dismutation of the superoxide radical into hydrogen peroxide and molecular oxygen; it plays a crucial role in the cellular homeostasis of ROS. |
Cu/Zn superoxide dismutase 1 (SOD1)-G93A mouse models; G93R-mSOD1 zebrafish model
|
Vacuolar degeneration of mitochondria; Disruption of the cristae and disintegration of the mitochondrial structure; Dysfunction of Oxidative Phosphorylation; Mitochondrial Ca2+ uptake disorder; Mitochondria distribution impairment; Reduced mitochondrial density |
|
|
A GGGGCC24+ hexanucleotide repeat expansion (HRE) in the C9ORF72 gene |
The function of C9ORF72 remains unknown, but it has been suggested that it plays a role in protein trafficking. |
FVB-C9orf72 mouse model; Rodent models of C9orf72; C9orf72 knockdown zebrafish model |
Defective mitochondrial bioenergetic function; Mitochondrial fragmentation; Reduced protein level of the ATP synthase complex
|
|
|
TARDBP point mutations and variants |
TAR DNA-binding protein 43 (TDP-43) protein participates in transcriptional regulation through RNA/DNA and protein–protein interactions, RNA processing and splicing regulation |
TDP43-Q331K mouse model TDP-43G298S transgenic mice (20 different mouse models of TDP-43) TDP43-A315T zebrafish model
|
Abnormal mitochondrial transport and morphology |
|
|
FUS and TBK1 variants |
Fused in sarcoma (FUS) protein is a DNA/RNA-binding protein that participates in in DNA damage, mRNA splicing, transport, transcription, and translation. TNF receptor–associated family member–associated NF-κB activator (TANK)-binding kinase 1 (TBK1) protein plays the cellular role in autophagy, mitophagy and glial immune responses. |
Drosophila models of FUS-related ALS hFUSWT transgenic mice; FUS(1-359) transgenic mice Tbk1fl/flNestin-Cre mice |
Mitochondrial fragmentation; Damaged mitochondrial cristae; Hyperfused and elongated mitochondria
|
|
|
In vitro models |
||||
|
Mutations in SOD1, FUS, TARDBP, C9orf72, SFPQ, and others |
Human induced pluripotent stem cells (hiPSCs) iPSC- motoneurons, astrocytes and microglia derived from patients with SOD1, FUS, TARDBP, C9orf72 ad other mutations |
Disorders in mitochondrial morphology and movement; Impairment of mitochondrial calcium buffering; Disruption of endoplasmic reticulum (ER)-mitochondria tethering and signaling
|
||
|
Mutations in SOD1, FUS, KIF5A, PFN1, and others
|
Cultured murine primary neurons co-transfected with mutant fALS-related proteins; human cells expressing mutant derivatives
|
Altered mitochondrial shape and size Reduced mitochondrial movement Matrix swelling and other structural defects in mitochondria Decreased mitochondrial respiration Decrease in the number of mitochondria
|
||
3. Molecular Mechanisms Responsible for Altered Mitostasis in ALS
3.1. Alterations in Morphology of Individual Mitochondria and Dynamics of the Mitochondrial Network in ALS
3.2. Functional Signs of Mitochondrial Damage in ALS
3.3. Modifications in the Mitochondrial Genome in ALS
3.4. Disorders in Mitochondrial Clearance and Replacement in ALS
3.5. Defects in Mitochondrial Trafficking in ALS
4. Strategies of Targeted Mitochondrial Therapy for ALS
Currently available drugs for the treatment of ALS have insufficient efficacy and are predominantly compensatory, symptomatic. In clinical practice, only two drugs are approved by the Food and Drug Administration for the treatment of ALS: riluzole (Rilutek, Teglutik), which suppresses excessive motor neuron firing by blocking the release of glutamate, and edaravone (Radicava, Radicut), which acts by neutralizing free radicals. Studies showed that riluzole slightly (by 2–3 months) prolongs the average life expectancy of patients with ALS without affecting its quality [128], while edaravone slows the rate of functional decline at the early stages of disease progression [129].
Difficulties in developing effective genetic therapy are associated with the multifactorial nature of the disease [31]. The identification of novel mechanisms of ALS pathogenesis in recent years has resulted in growing numbers of proposed therapeutic approaches that are used to plan clinical trials. According to ClinicalTrials.gov, over 770 ALS-related clinical trials have been registered to date, but strategies for targeted mitochondrial therapy in ALS are limited. Clinical trials of the combination of dextromethorphan and quinidine (Nuedexta), which affect the demethylation of P450-containing systems in the inner membrane of mitochondria and in the endoplasmic reticulum, have revealed an improvement in the bulbar function (speech, swallowing, and salivation) in patients with ALS (ClinicalTrials.gov identifier NCT01806857). Pridopidine (PL-101), a selective and potent Sigma-1 receptor agonist that affects MAM contacts, is being investigated in the HEALEY ALS Platform Trial (NCT04615923).
Table 2. Potential pharmacological strategies for mitochondrial therapy in ALS.
|
Pharmacological agent |
Molecular Targets |
Therapeutic Effect |
References |
|
Pridopidine (PL-101) |
Sigma-1 receptor agonist, MAM contacts, mitochondrial calcium homeostasis |
Enhancement of bulbar and speech function in ALS patients |
HEALEY ALS Platform Trial identifier NCT04615923
|
|
Nuedexta (Dextromethorphan and Quinidine) |
Effectors of P450-containing systems in the inner membrane of mitochondria and in the endoplasmic reticulum |
Improvement in bulbar function (speech, swallowing, and salivation) |
ClinicalTrials.gov identifier NCT01806857 |
|
VAR10303 |
Iron chelator, free radical catcher |
Stimulation of mitochondrial biogenesis and an increase in animal life expectancy |
[130] |
|
Sodium butyrate |
Broad-spectrum antioxidant |
Stimulation of mitochondrial biogenesis and an increase in animal life expectancy |
[131] |
|
Compound R13 |
Precursor of 7,8-dihydroxyflavone, selective activator of the TrkB signaling pathway |
Activation of mitochondrial biogenesis, suppression of the development of mitochondrial dysfunction and an increase in animal life expectancy |
[132] |
|
Caprylic triglyceride |
Substance metabolized into energy-rich ketone bodies |
Restoration of energy metabolism, mitochondrial biogenesis and improvement of motor function in SOD1 mice |
[133] |
|
MitoQ |
Mitochondria-targeted antioxidant |
Improvement in mitochondrial function and neuroprotective effects in animal and cellular models of ALS |
[134] |
|
Cholest-4-en-3-one, oxime (TRO19622) |
Inhibitor of the components of the mPTP opening: the voltage-dependent anion channel and the translocator protein |
Delay in the onset of disease symptoms and increase in animal survival |
[135] |
|
Mito-CP |
Mitochondria-targeted antioxidant |
Improvement in mitochondrial function and neuroprotective effects in animal and cellular models of ALS |
[136] |
|
Elamipretide (SS-31) |
Mitochondrion-targeted antioxidant |
Improvement in mitochondrial dysfunction, synaptic and memory impairment |
[137] |
|
Coenzyme Q10 |
Electron carrier of electron transport chain and free-radical-scavenging antioxidant |
Slowdown in the decline of mitochondrial function in animal and cellular models of ALS |
[131] |
|
Mitochonic Acid 5 |
Inductor of ATP synthase dimer formation |
Increase in local ATP production and cell survival in various mitochondrial diseases |
[132] |
4.1. Mitochondria-Specific Form of Autophagy as a Potential Target for ALS Treatment
4.2. PGC-1α as a Drug Target for ALS Treatment
4.3. Mitochondria-Targeted Antioxidants
4.4. Mitochondrial Biogenetics and Energy Metabolism as Targets of Therapeutic High-Fat Diet in ALS
5. Conclusions
References
- Rowland, L.P.; Shneider, N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700.
- Hardiman, O. Management of respiratory symptoms in ALS. J. Neurol. 2011, 258, 359–365.
- Mukhamedyarov, M.A.; Khabibrakhmanov, A.N.; Zefirov, A.L. Early dysfunctions in amyotrophic lateral sclerosis: Pathogenetic mechanisms and role in the initiation of the disease. Biochem. Suppl. Ser. A Membr. Cell Biol. 2020, 14, 261–266.
- Boillee, S.; Velde, C.V.; Cleveland, D.W. ALS: A disease of motor neurons and their nonneuronal neighbors. Neuron 2006, 52, 39–59.
- Bruijn, L.; Miller, T.M.; Cleveland, D.W. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu. Rev. Neurosci. 2004, 27, 723–749.
- Maksimovic, K.; Youssef, M.; You, J.; Sung, H.-K.; Park, J. Evidence of metabolic dysfunction in amyotrophic lateral sclerosis (ALS) patients and animal models. Biomolecules 2023, 13, 863.
- Sirozh, O.; Saez-Mas, A.; Lafarga, V.; Fernandez-Capetillo, O. Basic concepts and emergent disease mechanisms of amyotrophic lateral sclerosis. Encycl. Cell Biol. 2023, 6, 644–665.
- Jishi, A.; Qi, X. Altered mitochondrial protein homeostasis and proteinopathies. Front. Mol. Neurosci. 2022, 15, 867935.
- Casanova, A.; Wevers, A.; Navarro-Ledesma, S.; Pruimboom, L. Mitochondria: It is all about energy. Front. Physiol. 2023, 14, 1114231.
- Lim, S.M.; Nahm, M.; Kim, S.H. Proteostasis and ribostasis impairment as common cell death mechanisms in neurodegenerative diseases. J. Clin. Neurol. 2023, 19, 101–114.
- Gao, F.; Zhang, J. Mitochondrial quality control and neurodegenerative diseases. Neuronal Signal. 2018, 2, NS20180062.
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, mitochondrial homeostasis, and cell fate. Front. Cell Dev. Biol. 2020, 8, 467.
- Borthwick, G.M.; Johnson, M.A.; Ince, P.G.; Shaw, P.J.; Turnbull, D.M. Mitochondrial enzyme activity in amyotrophic lateral sclerosis: Implications for the role of mitochondria in neuronal cell death. Ann. Neurol. 1999, 46, 787–790.
- Granatiero, V.; Manfredi, G. Mitochondrial transport and turnover in the pathogenesis of amyotrophic lateral sclerosis. Biology 2019, 8, 36.
- Günther, R.; Pal, A.; Williams, C.; Zimyanin, V.L.; Liehr, M.; von Neubeck, C.; Krause, M.; Parab, M.G.; Petri, S.; Kalmbach, N.; et al. Alteration of mitochondrial integrity as upstream event in the pathophysiology of SOD1-ALS. Cells 2022, 11, 1246.
- Evans, C.S.; Holzbaur, E.L.F. Autophagy and mitophagy in ALS. Neurobiol. Dis. 2019, 122, 35–40.
- Vande Velde, C.; Miller, T.M.; Cashman, N.R.; Cleveland, D.W. Selective association of misfolded ALS-linked mutant SOD1 with the cytoplasmic face of mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 4022–4027.
- Mattiazzi, M.; D’Aurelio, M.; Gajewski, C.D.; Martushova, K.; Kiaei, M.; Beal, M.F.; Manfredi, G. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J. Biol. Chem. 2002, 277, 29626–29633.
- Wong, Y.C.; Holzbaur, E.L. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448.
- Moore, A.S.; Holzbaur, E.L. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc. Natl. Acad. Sci. USA 2016, 113, E3349–E3358.
- Miquel, E.; Cassina, A.; Martínez-Palma, L.; Souza, J.M.; Bolatto, C.; Rodríguez-Bottero, S.; Logan, A.; Smith, R.A.; Murphy, M.P.; Barbeito, L.; et al. Neuroprotective effects of the mitochondria-targeted antioxidant MitoQ in a model of inherited amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2014, 70, 204–213.
- Bordet, T.; Buisson, B.; Michaud, M.; Drouot, C.; Galéa, P.; Delaage, P.; Akentieva, N.P.; Evers, A.S.; Covey, D.F.; Ostuni, M.A.; et al. Identification and characterization of cholest-4-en-3-one, oxime (TRO19622), a novel drug candidate for amyotrophic lateral sclerosis. J. Pharmacol. Exp. Ther. 2007, 322, 709–720.
- Cassina, P.; Cassina, A.; Pehar, M.; Castellanos, R.; Gandelman, M.; de León, A.; Robinson, K.M.; Mason, R.P.; Beckman, J.S.; Barbeito, L.; et al. Mitochondrial dysfunction in SOD1G93A-bearing astrocytes promotes motor neuron degeneration: Prevention by mitochondrial-targeted antioxidants. J. Neurosci. 2008, 28, 4115–4122.
- Zhao, W.; Xu, Z.; Cao, J.; Fu, Q.; Wu, Y.; Zhang, X.; Long, Y.; Zhang, X.; Yang, Y.; Li, Y.; et al. Elamipretide (SS-31) improves mitochondrial dysfunction, synaptic and memory impairment induced by lipopolysaccharide in mice. J. Neuroinflamm. 2019, 16, 230.
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933.
- Mehta, A.R.; Walters, R.; Waldron, F.M.; Pal, S.; Selvaraj, B.T.; Macleod, M.R.; Hardingham, G.E.; Chandran, S.; Gregory, J.M. Targeting mitochondrial dysfunction in amyotrophic lateral sclerosis: A systematic review and meta-analysis. Brain Commun. 2019, 1, fcz009.
- Zhao, J.; Wang, X.; Huo, Z.; Chen, Y.; Liu, J.; Zhao, Z.; Meng, F.; Su, Q.; Bao, W.; Zhang, L.; et al. The impact of mitochondrial dysfunction in amyotrophic lateral sclerosis. Cells 2022, 11, 2049.
- Brown, R.H.; Al-Chalabi, A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2017, 377, 1602.
- Ferraiuolo, L.; Kirby, J.; Grierson, A.J.; Sendtner, M.; Shaw, P.J. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011, 7, 616–630.
- Laneve, P.; Tollis, P.; Caffarelli, E. RNA deregulation in amyotrophic lateral sclerosis: The noncoding perspective. Int. J. Mol. Sci. 2021, 22, 20285.
- Suzuki, N.; Nishiyama, A.; Warita, H.; Aoki, M. Genetics of amyotrophic lateral sclerosis: Seeking therapeutic targets in the era of gene therapy. J. Hum. Genet. 2023, 68, 131–152.
- Ticozzi, N.; Tiloca, C.; Morelli, C.; Colombrita, C.; Poletti, B.; Doretti, A.; Maderna, L.; Messina, S.; Ratti, A.; Silani, V. Genetics of familial amyotrophic lateral sclerosis. Arch. Ital. Biol. 2011, 149, 65–82.
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in the C9ORF72 is the cause of the chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268.
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208.
- Deng, H.X.; Chen, W.; Hong, S.T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ASL and ALS/dementia. Nature 2011, 477, 211–215.
- Barber, S.C.; Mead, R.J.; Shaw, P.J. Oxidative stress in ALS: A mechanism of neurodegeneration and a therapeutic target. Biochim. Biophys. Acta 2006, 1762, 1051–1067.
- Barber, S.C.; Shaw, P.J. Oxidative stress in ALS: Key role in motor neuron injury and therapeutic target. Free Rad. Biol. Med. 2010, 48, 629–641.
- Zufiría, M.; Gil-Bea, F.J.; Fernández-Torrón, R.; Poza, J.J.; Muñoz-Blanco, J.L.; Rojas-García, R.; Riancho, J.; López de Munain, A. ALS: A bucket of genes, environment, metabolism and unknown ingredients. Progr. Neurobiol. 2016, 142, 104–112.
- Newell, M.E.; Adhikari, S.; Halden, R.U. Systematic and state-of the science review of the role of environmental factors in Amyotrophic Lateral Sclerosis (ALS) or Lou Gehrig’s Disease. Sci. Total Environ. 2022, 817, 152504.
- Bradley, W.G.; Borenstein, A.R.; Nelson, L.M.; Codd, G.A.; Rosen, B.H.; Stommel, E.W.; Cox, P.A. Is exposure to cyanobacteria an environmental risk factor for amyotrophic lateral sclerosis and other neurodegenerative diseases? Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 325–333.
- Alfahad, T.; Nath, A. Retroviruses and amyotrophic lateral sclerosis. Antivir. Res. 2013, 99, 180–187.
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955.
- Vidovic, M.; Müschen, L.H.; Brakemeier, S.; Machetanz, G.; Naumann, M.; Castro-Gomez, S. Current state and future directions in the diagnosis of amyotrophic lateral sclerosis. Cells 2023, 12, 736.
- Dupuis, L.; Gonzalez de Aguilar, J.L.; Echaniz-Laguna, A.; Eschbach, J.; Rene, F.; Oudart, H.; Halter, B.; Huze, C.; Schaeffer, L.; Bouillaud, F.; et al. Muscle mitochondrial uncoupling dismantles neuromuscular junction and triggers distal degeneration of motor neurons. PLoS ONE 2009, 4, e5390.
- Dupuis, L.; Loeffler, J.P. Neuromuscular junction destruction during amyotrophic lateral sclerosis: Insights from transgenic models. Curr. Opin. Pharmacol. 2009, 9, 341–346.
- Eisen, A. The dying forward hypothesis of ALS: Tracing its history. Brain Sci. 2021, 11, 300.
- Dadon-Nachum, M.; Melamed, E.; Offen, D. The “dyingback” phenomenon of motor neurons in ALS. J. Mol. Neurosci. 2011, 43, 470–477.
- Pikatza-Menoio, O.; Elicegui, A.; Bengoetxea, X.; Naldaiz-Gastesi, N.; López de Munain, A.; Gerenu, G.; Gil-Bea, F.J.; Alonso-Martín, S. The skeletal muscle emerges as a new disease target in amyotrophic lateral sclerosis. J. Pers. Med. 2021, 11, 671.
- Rubio, M.A.; Herrando-Grabulosa, M.; Navarro, X. Sensory involvement in amyotrophic lateral sclerosis. Int. J. Mol. Sci. 2022, 23, 15521.
- Higgins, C.M.; Jung, C.; Xu, Z. ALS-associated mutant SOD1G93A causes mitochondrial vacuolation by expansion of the intermembrane space and by involvement of SOD1 aggregation and peroxisomes. BMC Neurosci. 2003, 4, 16.
- Vande Velde, C.; McDonald, K.K.; Boukhedimi, Y.; McAlonis-Downes, M.; Lobsiger, C.S.; Hadj, S.B.; Zandona, A.; Julien, J.P.; Shah, S.B.; Cleveland, D.W. Misfolded SOD1 associated with motor neuron mitochondria alters mitochondrial shape and distribution prior to clinical onset. PLoS ONE 2011, 6, e22031.
- Moller, A.; Bauer, C.S.; Cohen, R.N.; Webster, C.P.; De Vos, K.J. Amyotrophic lateral sclerosis-associated mutant SOD1 inhibits anterograde axonal transport of mitochondria by reducing Miro1 levels. Hum. Mol. Genet. 2017, 26, 4668–4679.
- Gautam, M.; Jara, J.H.; Kocak, N.; Rylaarsdam, L.E.; Dong, K.; Bigio, E.H.; Özdinler, P.H. Mitochondria, ER, and nuclear membrane defects reveal early mechanisms for upper motor neuron vulnerability with respect to TDP-43 pathology. Acta Neuropathol. 2019, 137, 47–69.
- Deng, J.; Wang, P.; Chen, X.; Cheng, H.; Liu, J.; Fushimi, K.; Zhu, L.; Wu, J.Y. FUS interacts with ATP synthase beta subunit and induces mitochondrial unfolded protein response in cellular and animal models. Proc. Natl. Acad. Sci. USA 2018, 115, E9678–E9686.
- Tsai, Y.L.; Coady, T.H.; Lu, L.; Zheng, D.; Alland, I.; Tian, B.; Shneider, N.A.; Manley, J.L. ALS/FTD-associated protein FUS induces mitochondrial dysfunction by preferentially sequestering respiratory chain complex mRNAs. Genes Dev. 2020, 34, 785–805.
- Shahidullah, M.; Le Marchand, S.J.; Fei, H.; Zhang, J.; Pandey, U.B.; Dalva, M.B.; Pasinelli, P.; Levitan, I.B. Defects in synapse structure and function precede motor neuron degeneration in Drosophila models of FUS-related ALS. J. Neurosci. 2013, 33, 19590–19598.
- Chung, M.J.; Suh, Y.L. Ultrastructural changes of mitochondria in the skeletal muscle of patients with amyotrophic lateral sclerosis. Ultrastruct. Pathol. 2002, 26, 3–7.
- Napoli, L.; Crugnola, V.; Lamperti, C.; Silani, V.; Di Mauro, S.; Bresolin, N.; Moggio, M. Ultrastructural mitochondrial abnormalities in patients with sporadic amyotrophic lateral sclerosis. Arch. Neurol. 2011, 68, 1612–1613.
- Naumann, M.; Pal, A.; Goswami, A.; Lojewski, X.; Japtok, J.; Vehlow, A.; Naujock, M.; Günther, R.; Jin, M.; Stanslowsky, N.; et al. Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation. Nat. Commun. 2018, 9, 335.
- Teyssou, E.; Chartier, L.; Roussel, D.; Perera, N.D.; Nemazanyy, I.; Langui, D.; Albert, M.; Larmonier, T.; Saker, S.; Salachas, F.; et al. The amyotrophic lateral sclerosis M114T PFN1 mutation deregulates alternative autophagy pathways and mitochondrial homeostasis. Int. J. Mol. Sci. 2022, 23, 5694.
- Loson, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667.
- Prudent, J.; Zunino, R.; Sugiura, A.; Mattie, S.; Shore, G.C.; McBride, H.M. MAPL sumoylation of Drp1 stabilizes an ER/mitochondrial platform required for cell death. Mol. Cell 2015, 59, 941–955.
- Frezza, C.; Cipolat, S.; de Brito, O.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 2006, 126, 177–189.
- Luo, G.; Yi, J.; Ma, C.; Xiao, Y.; Yi, F.; Yu, T.; Zhou, J. Defective mitochondrial dynamics is an early event in skeletal muscle of an amyotrophic lateral sclerosis mouse model. PLoS ONE 2013, 8, e82112.
- Joshi, A.U.; Saw, N.L.; Vogel, H.; Cunnigham, A.D.; Shamloo, M.; Mochly-Rosen, D. Inhibition of Drp1/Fis1 interaction slows progression of amyotrophic lateral sclerosis. EMBO Mol. Med. 2018, 10, e8166.
- Liu, W.; Yamashita, T.; Tian, F.; Morimoto, N.; Ikeda, Y.; Deguchi, K.; Abe, K. Mitochondrial fusion and fission proteins expression dynamically change in a murine model of amyotrophic lateral sclerosis. Curr. Neurovasc. Res. 2013, 10, 222–230.
- Onesto, E.; Colombrita, C.; Gumina, V.; Borghi, M.O.; Dusi, S.; Doretti, A.; Fagiolari, G.; Invernizzi, F.; Moggio, M.; Tiranti, V.; et al. Gene-specific mitochondria dysfunctions in human TARDBP and C9ORF72 fibroblasts. Acta Neuropathol. Commun. 2016, 4, 47.
- Jiang, Z.; Wang, W.; Perry, G.; Zhu, X.; Wang, X. Mitochondrial dynamic abnormalities in amyotrophic lateral sclerosis. Transl. Neurodegener. 2015, 4, 14.
- Palomo, G.M.; Granatiero, V.; Kawamata, H.; Konrad, C.; Kim, M.; Arreguin, A.J.; Zhao, D.; Milner, T.A.; Manfredi, G. Parkin is a disease modifier in the mutant SOD1 mouse model of ALS. EMBO Mol. Med. 2018, 10, e8888.
- Lopez-Gonzalez, R.; Lu, Y.; Gendron, T.F.; Karydas, A.; Tran, H.; Yang, D.; Petrucelli, L.; Miller, B.L.; Almeida, S.; Gao, F.B. Poly(GR) in C9ORF72-related ALS/FTD compromises mitochondrial function and increases oxidative stress and DNA damage in iPSC-derived motor neurons. Neuron 2016, 92, 383–391.
- Dafinca, R.; Scaber, J.; Ababneh, N.; Lalic, T.; Weir, G.; Christian, H.; Vowles, J.; Douglas, A.G.; Fletcher-Jones, A.; Browne, C.; et al. C9orf72 hexanucleotide expansions are associated with altered endoplasmic reticulum calcium homeostasis and stress granule formation in induced pluripotent stem cell-derived neurons from patients with amyotrophic lateral sclerosis and frontotemporal dementia. Stem Cells 2016, 34, 2063–2078.
- Davis, S.A.; Itaman, S.; Khalid-Janney, C.M.; Sherard, J.A.; Dowell, J.A.; Cairns, N.J.; Gitcho, M.A. TDP-43 interacts with mitochondrial proteins critical for mitophagy and mitochondrial dynamics. Neurosci. Lett. 2018, 678, 8–15.
- Ghiasi, P.; Hosseinkhani, S.; Noori, A.; Nafissi, S.; Khajeh, K. Mitochondrial complex I deficiency and ATP/ADP ratio in lymphocytes of amyotrophic lateral sclerosis patients. Neurol. Res. 2012, 34, 297–303.
- Magrane, J.; Sahawneh, M.A.; Przedborski, S.; Estevez, A.G.; Manfredi, G. Mitochondrial dynamics and bioenergetic dysfunction is associated with synaptic alterations in mutant SOD1 motor neurons. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 229–242.
- Carri, M.T.; Ferri, A.; Battistoni, A.; Famhy, L.; Gabbianelli, R.; Poccia, F.; Rotilio, G. Expression of a Cu, Zn superoxide dismutase typical of familial amyotrophic lateral sclerosis induces mitochondrial alteration and increase of cytosolic Ca2+ concentration in transfected neuroblastoma SH-SY5Y cells. FEBS Lett. 1997, 414, 365–368.
- Menzies, F.M.; Cookson, M.R.; Taylor, R.W.; Turnbull, D.M.; Chrzanowska-Lightowlers, Z.M.; Dong, L.; Figlewicz, D.A.; Shaw, P.J. Mitochondrial dysfunction in a cell culture model of familial amyotrophic lateral sclerosis. Brain 2002, 125, 1522–1533.
- Rizzardini, M.; Mangolini, A.; Lupi, M.; Ubezio, P.; Bendotti, C.; Cantoni, L. Low levels of ALS-linked Cu/Zn superoxide dismutase increase the production of reactive oxygen species and cause mitochondrial damage and death in motor neuron-like cells. J. Neurol. Sci. 2005, 232, 95–103.
- Ikawa, M.; Okazawa, H.; Tsujikawa, T.; Matsunaga, A.; Yamamura, O.; Mori, T.; Hamano, T.; Kiyono, Y.; Nakamoto, Y.; Yoneda, M. Increased oxidative stress is related to disease severity in the ALS motor cortex: A PET study. Neurology 2015, 84, 2033–2039.
- Chan, D.C. Mitochondrial dynamics and its involvement in disease. Annu. Rev. Pathol. 2020, 15, 235–259.
- Bakula, D.; Scheibye-Knudsen, M. Mitoph Aging: Mitophagy in aging and disease. Front. Cell Dev. Biol. 2020, 8, 239.
- Sasaki, S.; Iwata, M. Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2007, 66, 10–16.
- Wiedemann, F.R.; Manfredi, G.; Mawrin, C.; Beal, M.F.; Schon, E.A. Mitochondrial DNA and respiratory chain function in spinal cords of ALS patients. J. Neurochem. 2002, 80, 616–625.
- Li, Q.; Vande Velde, C.; Israelson, A.; Xie, J.; Bailey, A.O.; Dong, M.Q.; Chun, S.J.; Roy, T.; Winer, L.; Yates, J.R.; et al. ALS-linked mutant superoxide dismutase 1 (SOD1) alters mitochondrial protein composition and decreases protein import. Proc. Natl. Acad. Sci. USA 2010, 107, 21146–21151.
- Liu, J.; Lillo, C.; Jonsson, P.A.; Vande Velde, C.; Ward, C.M.; Miller, T.M.; Subramaniam, J.R.; Rothstein, J.D.; Marklund, S.; Andersen, P.M.; et al. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron 2004, 43, 5–17.
- Pasinelli, P.; Belford, M.E.; Lennon, N.; Bacskai, B.J.; Hyman, B.T.; Trotti, D.; Brown, R.H., Jr. Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron 2004, 43, 19–30.
- Kihira, T.; Okamoto, K.; Yoshida, S.; Kondo, T.; Iwai, K.; Wada, S.; Kajimoto, Y.; Kondo, T.; Kokubo, Y.; Kuzuhara, S. Environmental characteristics and oxidative stress of inhabitants and patients with amyotrophic lateral sclerosis in a high incidence area on the Kii Peninsula, Japan. Int. Med. 2013, 52, 1479–1486.
- Bernard-Marissal, N.; Medard, J.J.; Azzedine, H.; Chrast, R. Dysfunction in endoplasmic reticulum-mitochondria crosstalk underlies SIGMAR1 loss of function mediated motor neuron degeneration. Brain 2015, 138, 875–890.
- Ono, Y.; Tanaka, H.; Takata, M.; Nagahara, Y.; Noda, Y.; Tsuruma, K.; Shimazawa, M.; Hozumi, I.; Hara, H. SA4503, a sigma-1 receptor agonist, suppresses motor neuron damage in in vitro and in vivo amyotrophic lateral sclerosis models. Neurosci. Lett. 2014, 559, 174–178.
- Zhou, J.; Li, A.; Li, X.; Yi, J. Dysregulated mitochondrial Ca2+ and ROS signaling in skeletal muscle of ALS mouse model. Arch. Biochem. Biophys. 2019, 663, 249–258.
- Martin, L.J.; Gertz, B.; Pan, Y.; Price, A.C.; Molkentin, J.D.; Chang, Q. The mitochondrial permeability transition pore in motor neurons: Involvement in the pathobiology of ALS mice. Exp. Neurol. 2009, 218, 333–346.
- Belosludtsev, K.N.; Dubinin, M.V.; Belosludtseva, N.V.; Mironova, G.D. Mitochondrial Ca2+ transport: Mechanisms, molecular structures, and role in cells. Biochemistry 2019, 84, 593–607.
- Keogh, M.J.; Chinnery, P.F. Mitochondrial DNA mutations in neurodegeneration. Biochim. Biophys. Acta 2015, 1847, 1401–1411.
- Wiedemann, F.R.; Winkler, K.; Kuznetsov, A.V.; Bartels, C.; Vielhaber, S.; Feistner, H.; Kunz, W.S. Impairment of mitochondrial function in skeletal muscle of patients with amyotrophic lateral sclerosis. J. Neurol. Sci. 1998, 156, 65–72.
- Wang, W.Y.; Pan, L.; Su, S.C.; Quinn, E.J.; Sasaki, M.; Jimenez, J.C.; Mackenzie, I.R.; Huang, E.J.; Tsai, L.H. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat. Neurosci. 2013, 16, 1383–1391.
- Ferrante, R.J.; Browne, S.E.; Shinobu, L.A.; Bowling, A.C.; Baik, M.J.; MacGarvey, U.; Kowall, N.W.; Brown, R.H., Jr.; Beal, M.F. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J. Neurochem. 1997, 69, 2064–2074.
- Qiu, H.; Lee, S.; Shang, Y.; Wang, W.Y.; Au, K.F.; Kamiya, S.; Barmada, S.J.; Finkbeiner, S.; Lui, H.; Carlton, C.E.; et al. ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J. Clin. Investig. 2014, 124, 981–999.
- Comi, G.P.; Bordoni, A.; Salani, S.; Franceschina, L.; Sciacco, M.; Prelle, A.; Fortunato, F.; Zeviani, M.; Napoli, L.; Bresolin, N.; et al. Cytochrome c oxidase subunit I microdeletion in a patient with motor neuron disease. Ann. Neurol. 1998, 43, 110–116.
- Finsterer, J. Mitochondriopathy mimicking amyotrophic lateral sclerosis. Neurologist 2003, 9, 45–48.
- Jimenez-Pacheco, A.; Franco, J.M.; Lopez, S.; Gomez-Zumaquero, J.M.; Magdalena Leal-Lasarte, M.; Caballero-Hernandez, D.E.; Cejudo-Guillén, M.; Pozo, D. Epigenetic mechanisms of gene regulation in amyotrophic lateral sclerosis. Adv. Exp. Med. Biol. 2017, 978, 255–275.
- Stoccoro, A.; Mosca, L.; Carnicelli, V.; Cavallari, U.; Lunetta, C.; Marocchi, A.; Migliore, L.; Coppedè, F. Mitochondrial DNA copy number and D-loop region methylation in carriers of amyotrophic lateral sclerosis gene mutations. Epigenomics 2018, 10, 1431–1443.
- Maekawa, M.; Sugano, K.; Ushiama, M.; Fukayama, N.; Nomoto, K.; Kashiwabara, H.; Fujita, S.; Kakizoe, T. Heterogeneity of DNA methylation status analyzed by bisulfite-PCR-SSCP and correlation with clinico-pathological characteristics in colorectal cancer. Clin. Chem. Lab. Med. 2001, 39, 121–128.
- Chestnut, B.A.; Chang, Q.; Price, A.; Lesuisse, C.; Wong, M.; Martin, L.J. Epigenetic regulation of motor neuron cell death through DNA methylation. J. Neurosci. 2011, 31, 16619–16636.
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega, A.L.; Pérez, S. PGC-1α, inflammation, and oxidative stress: An integrative view in metabolism. Oxid. Med. Cell Longev. 2020, 2020, 1452696.
- Handschin, C.; Rhee, J.; Lin, J.; Tarr, P.T.; Spiegelman, B.M. An autoregulatory loop controls peroxisome proliferatoractivated receptor gamma coactivator 1alpha expression in muscle. Proc. Natl. Acad. Sci. USA 2003, 100, 7111–7116.
- Zhu, L.; Wang, Q.; Zhang, L.; Fang, Z.; Zhao, F.; Lv, Z.; Gu, Z.; Zhang, J.; Wang, J.; Zen, K.; et al. Hypoxia induces PGC-1α expression and mitochondrial biogenesis in the myocardium of TOF patients. Cell Res. 2010, 20, 676–687.
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of mitochondrial biogenesis as a way for active longevity: Interaction between the Nrf2 and PGC-1α signaling pathways. Front. Genet. 2019, 10, 435.
- Thau, N.; Knippenberg, S.; Korner, S.; Rath, K.J.; Dengler, R.; Petri, S. Decreased mRNA expression of PGC-1α and PGC-1α regulated factors in the SOD1G93A ALS mouse model and in human sporadic ALS. J. Neuropathol. Exp. Neurol. 2012, 71, 1064–1074.
- Liang, H.; Ward, W.F.; Jang, Y.C.; Bhattacharya, A.; Bokov, A.F.; Jernigan, A.; Richardson, A.; Van Remmen, H. PGC-1α protects neurons and alters disease progression in an amyotrophic lateral sclerosis mouse model. Muscle Nerve 2011, 44, 947–956.
- Varghese, M.; Zhao, W.; Trageser, K.J.; Pasinetti, G.M. Peroxisome proliferator activator receptor gamma coactivator-1α overexpression in amyotrophic lateral sclerosis: A tale of two transgenics. Biomolecules 2020, 10, 760.
- Ladd, A.C.; Keeney, P.M.; Govind, M.M.; Bennett, J.P., Jr. Mitochondrial oxidative phosphorylation transcriptome alterations in human amyotrophic lateral sclerosis spinal cord and blood. Neuromol. Med. 2014, 16, 714–726.
- Yang, X.; Pan, W.; Xu, G.; Chen, L. Mitophagy: A crucial modulator in the pathogenesis of chronic diseases. Clin. Chim. Acta 2020, 502, 245–254.
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226.
- Duan, W.; Guo, M.; Yi, L.; Zhang, J.; Bi, Y.; Liu, Y.; Li, Y.; Li, Z.; Ma, Y.; Zhang, G.; et al. Deletion of Tbk1 disrupts autophagy and reproduces behavioral and locomotor symptoms of FTD-ALS in mice. Aging 2019, 11, 2457–2476.
- Kim, N.C.; Tresse, E.; Kolaitis, R.M.; Molliex, A.; Thomas, R.E.; Alami, N.H.; Wang, B.; Joshi, A.; Smith, R.B.; Ritson, G.P.; et al. VCP is essential for mitochondrial quality control by PINK1/Parkin and this function is impaired by VCP mutations. Neuron 2013, 78, 65–80.
- Gerbino, V.; Kaunga, E.; Ye, J.; Canzio, D.; O’Keeffe, S.; Rudnick, N.D.; Guarnieri, P.; Lutz, C.M.; Maniatis, T. The loss of TBK1 kinase activity in motor neurons or in all cell types differentially impacts ALS disease progression in SOD1 mice. Neuron 2020, 106, 789–805.
- Johnson, J.O.; Mandrioli, J.; Benatar, M.; Abramzon, Y.; Van Deerlin, V.M.; Trojanowski, J.Q.; Gibbs, J.R.; Brunetti, M.; Gronka, S.; Wuu, J.; et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 2010, 68, 857–864.
- Harding, O.; Evans, C.S.; Ye, J.; Cheung, J.; Maniatis, T.; Holzbaur, E.L.F. ALS- and FTD-associated missense mutations in TBK1 differentially disrupt mitophagy. Proc. Natl. Acad. Sci. USA 2021, 118, e2025053118.
- Sasaki, S. Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2011, 70, 349–359.
- Li, L.; Zhang, X.; Le, W. Altered macroautophagy in the spinal cord of SOD1 mutant mice. Autophagy 2008, 4, 290–293.
- Lagier-Tourenne, C.; Polymenidou, M.; Hutt, K.R.; Vu, A.Q.; Baughn, M.; Huelga, S.C.; Clutario, K.M.; Ling, S.-C.; Liang, T.Y.; Mazur, C. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-MRNAs. Nat. Neurosci. 2012, 15, 1488–1497.
- Madruga, E.; Maestro, I.; Martínez, A. Mitophagy modulation, a new player in the race against ALS. Int. J. Mol. Sci. 2021, 22, 740.
- Millecamps, S.; Julien, J.P. Axonal transport deficits and neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 161–176.
- Magrane, J.; Cortez, C.; Gan, W.B.; Manfredi, G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 2014, 23, 1413–1424.
- Anoar, S.; Woodling, N.S.; Niccoli, T. Mitochondria dysfunction in frontotemporal dementia/Amyotrophic Lateral Sclerosis: Lessons from Drosophila models. Front. Neurosci. 2021, 15, 786076.
- So, E.; Mitchell, J.C.; Memmi, C.; Chennell, G.; Vizcay-Barrena, G.; Allison, L.; Shaw, C.E.; Vance, C. Mitochondrial abnormalities and disruption of the neuromuscular junction precede the clinical phenotype and motor neuron loss in hFUSWT transgenic mice. Hum. Mol. Genet. 2018, 27, 463–474.
- Du, H.; Huo, Z.; Chen, Y.; Zhao, Z.; Meng, F.; Wang, X.; Liu, S.; Zhang, H.; Zhou, F.; Liu, J.; et al. Induced pluripotent stem cells and their applications in amyotrophic lateral sclerosis. Cells 2023, 12, 971.
- Tradewell, M.L.; Yu, Z.; Tibshirani, M.; Boulanger, M.C.; Durham, H.D.; Richard, S. Arginine methylation by PRMT1 regulates nuclear-cytoplasmic localization and toxicity of FUS/TLS harbouring ALS-linked mutations. Hum. Mol. Genet. 2012, 21, 136–149.
- Fang, T.; Al Khleifat, A.; Meurgey, J.H.; Jones, A.; Leigh, P.N.; Bensimon, G.; Al-Chalabi, A. Stage at which riluzole treatment prolongs survival in patients with amyotrophic lateral sclerosis: A retrospective analysis of data from a dose-ranging study. Lancet Neurol. 2018, 17, 416–422.
- Gao, M.; Zhu, L.; Chang, J.; Cao, T.; Song, L.; Wen, C.; Chen, Y.; Zhuo, Y.; Chen, F. Safety and efficacy of edaravone in patients with amyotrophic lateral sclerosis: A systematic review and meta-analysis. Clin. Drug Investig. 2023, 43, 1–11.
- Wei Zhao; Merina Varghese; Prashant Vempati; Anastasiya Dzhun; Alice Cheng; Jun Wang; Dale Lange; Amanda Bilski; Irene Faravelli; Giulio Maria Pasinetti; Caprylic Triglyceride as a Novel Therapeutic Approach to Effectively Improve the Performance and Attenuate the Symptoms Due to the Motor Neuron Loss in ALS Disease. PLOS ONE. 2012, 7, e49191.
- Petra Kaufmann; John L.P. Thompson; Gilberto Levy; Richard Buchsbaum; Jeremy Shefner; Lisa S. Krivickas; Jonathan Katz; Yvonne Rollins; Richard J. Barohn; Carlayne E. Jackson; Ezgi Tiryaki; Catherine Lomen‐Hoerth; Carmel Armon; Rup Tandan; Stacy A. Rudnicki; Kourosh Rezania; Robert Sufit; Alan Pestronk; Steven P. Novella; Terry Heiman‐Patterson; Edward J. Kasarskis; Erik P. Pioro; Jacqueline Montes; Rachel Arbing; Darleen Vecchio; Alexandra Barsdorf; Hiroshi Mitsumoto; Bruce Levin; Phase II trial of CoQ10 for ALS finds insufficient evidence to justify phase III. Ann. Neurol.. 2009, 66, 235-244.
- Tetsuro Matsuhashi; Takeya Sato; Shin-Ichiro Kanno; Takehiro Suzuki; Akihiro Matsuo; Yuki Oba; Motoi Kikusato; Emi Ogasawara; Tai Kudo; Kosuke Suzuki; Osamu Ohara; Hiroko Shimbo; Fumika Nanto; Hiroaki Yamaguchi; Daisuke Saigusa; Yasuno Mukaiyama; Akiko Watabe; Koichi Kikuchi; Hisato Shima; Eikan Mishima; Yasutoshi Akiyama; Yoshitsugu Oikawa; Ho Hsin-Jung; Yukako Akiyama; Chitose Suzuki; Mitsugu Uematsu; Masaki Ogata; Naonori Kumagai; Masaaki Toyomizu; Atsushi Hozawa; Nariyasu Mano; Yuji Owada; Setsuya Aiba; Teruyuki Yanagisawa; Yoshihisa Tomioka; Shigeo Kure; Sadayoshi Ito; Kazuto Nakada; Ken-Ichiro Hayashi; Hitoshi Osaka; Takaaki Abe; Mitochonic Acid 5 (MA-5) Facilitates ATP Synthase Oligomerization and Cell Survival in Various Mitochondrial Diseases. EBioMedicine. 2017, 20, 27-38.
- Nirma D. Perera; Rebecca K. Sheean; Chew L. Lau; Yea Seul Shin; Philip M. Beart; Malcolm K. Horne; Bradley J. Turner; Rilmenidine promotes MTOR-independent autophagy in the mutant SOD1 mouse model of amyotrophic lateral sclerosis without slowing disease progression. Autophagy. 2017, 14, 534-551.
- Ernesto Miquel; Adriana Cassina; Laura Martínez-Palma; José M. Souza; Carmen Bolatto; Sebastián Rodríguez-Bottero; Angela Logan; Robin A.J. Smith; Michael P. Murphy; Luis Barbeito; Rafael Radi; Patricia Cassina; Neuroprotective effects of the mitochondria-targeted antioxidant MitoQ in a model of inherited amyotrophic lateral sclerosis. Free. Radic. Biol. Med.. 2014, 70, 204-213.
- Thierry Bordet; Bruno Buisson; Magali Michaud; Cyrille Drouot; Pascale Galéa; Pierre Delaage; Natalia P. Akentieva; Alex S. Evers; Douglas F. Covey; Mariano A. Ostuni; Jean-Jacques Lacapère; Charbel Massaad; Michael Schumacher; Esther-Marie Steidl; Delphine Maux; Michel Delaage; Christopher E. Henderson; Rebecca M. Pruss; Identification and Characterization of Cholest-4-en-3-one, Oxime (TRO19622), a Novel Drug Candidate for Amyotrophic Lateral Sclerosis. J. Pharmacol. Exp. Ther.. 2007, 322, 709-720.
- Patricia Cassina; Adriana Cassina; Mariana Pehar; Raquel Castellanos; Mandi Gandelman; Andrés de León; Kristine M. Robinson; Ronald P. Mason; Joseph S. Beckman; Luis Barbeito; Rafael Radi; Mitochondrial Dysfunction in SOD1G93A-Bearing Astrocytes Promotes Motor Neuron Degeneration: Prevention by Mitochondrial-Targeted Antioxidants. J. Neurosci.. 2008, 28, 4115-4122.
- Weixing Zhao; Zhipeng Xu; Jiangbei Cao; Qiang Fu; Yishuang Wu; Xiaoying Zhang; Yue Long; Xuan Zhang; Yitian Yang; Yunfeng Li; Weidong Mi; Elamipretide (SS-31) improves mitochondrial dysfunction, synaptic and memory impairment induced by lipopolysaccharide in mice. J. Neuroinflammation. 2019, 16, 1-19.
- Perera, N.D.; Sheean, R.K.; Lau, C.L.; Shin, Y.S.; Beart, P.M.; Horne, M.K.; Turner, B.J. Rilmenidine promotes MTOR-independent autophagy in the mutant SOD1 mouse model of amyotrophic lateral sclerosis without slowing disease progression. Autophagy 2018, 14, 534–551.
- Magrì, A.; Lipari, C.L.R.; Risiglione, P.; Zimbone, S.; Guarino, F.; Caccamo, A.; Messina, A. ERK1/2-dependent TSPO overactivation associates with the loss of mitophagy and mitochondrial respiration in ALS. Cell Death Dis. 2023, 14, 122.
- Golko-Perez, S.; Amit, T.; Bar-Am, O.; Youdim, M.B.; Weinreb, O. A novel iron chelator-radical scavenger ameliorates motor dysfunction and improves life span and mitochondrial biogenesis in SOD1G93A ALS mice. Neurotox. Res. 2017, 31, 230–244.
- Li, X.; Dong, L.; Li, A.; Yi, J.; Brotto, M.; Zhou, J. Butyrate ameliorates mitochondrial respiratory capacity of the motor-neuron-like cell line NSC34-G93A, a cellular model for ALS. Biomolecules 2022, 12, 333.
- Li, X.; Chen, C.; Zhan, X.; Li, B.; Zhang, Z.; Li, S.; Xie, Y.; Song, X.; Shen, Y.; Liu, J.; et al. R13 preserves motor performance in SOD1G93A mice by improving mitochondrial function. Theranostics 2021, 11, 7294–7307.
- Obrador, E.; Salvador-Palmer, R.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.L.; Estrela, J.M. The link between oxidative stress, redox status, bioenergetics and mitochondria in the pathophysiology of ALS. Int. J. Mol. Sci. 2021, 22, 6352.
- Kaufmann, P.; Thompson, J.L.; Levy, G.; Buchsbaum, R.; Shefner, J.; Krivickas, L.S.; Katz, J.; Rollins, Y.; Barohn, R.J.; Jackson, C.E.; et al. Phase II trial of CoQ10 for ALS finds insufficient evidence to justify phase III. Ann. Neurol. 2009, 66, 235–244.
- Matsuhashi, T.; Sato, T.; Kanno, S.I.; Suzuki, T.; Matsuo, A.; Oba, Y.; Kikusato, M.; Ogasawara, E.; Kudo, T.; Suzuki, K.; et al. Mitochonic acid 5 (MA-5) facilitates ATP synthase oligomerization and cell survival in various mitochondrial diseases. eBioMedicine 2017, 20, 27–38.
- Coughlan, K.S.; Halang, L.; Woods, I.; Prehn, J.H. A high-fat jelly diet restores bioenergetic balance and extends lifespan in the presence of motor dysfunction and lumbar spinal cord motor neuron loss in TDP-43A315T mutant C57BL6/J mice. Dis. Model. Mech. 2016, 9, 1029–1037.
- Steyn, F.J.; Li, R.; Kirk, S.E.; Tefera, T.W.; Xie, T.Y.; Tracey, T.J.; Kelk, D.; Wimberger, E.; Garton, F.C.; Roberts, L.; et al. Altered skeletal muscle glucose-fatty acid flux in amyotrophic lateral sclerosis. Brain Commun. 2020, 2, fcaa154.
- Zhao, Z.; Lange, D.J.; Voustianiouk, A.; MacGrogan, D.; Ho, L.; Suh, J.; Humala, N.; Thiyagarajan, M.; Wang, J.; Pasinetti, G.M. A ketogenic diet as a potential novel therapeutic intervention in amyotrophic lateral sclerosis. BMC Neurosci. 2006, 7, 29.
- Zhao, W.; Varghese, M.; Vempati, P.; Dzhun, A.; Cheng, A.; Wang, J.; Lange, D.; Bilski, A.; Faravelli, I.; Pasinetti, G.M. Caprylic triglyceride as a novel therapeutic approach to effectively improve the performance and attenuate the symptoms due to the motor neuron loss in ALS disease. PLoS ONE 2012, 7, e49191.
- Jain, S.S.; Paglialunga, S.; Vigna, C.; Ludzki, A.; Herbst, E.A.; Lally, J.S.; Schrauwen, P.; Hoeks, J.; Tupling, A.R.; Bonen, A.; et al. High-fat diet-induced mitochondrial biogenesis is regulated by mitochondrial-derived reactive oxygen species activation of CaMKII. Diabetes 2014, 63, 1907–1913.
- Bolognesi, M.L. Harnessing polypharmacology with medicinal chemistry. ACS Med. Chem. Lett. 2019, 10, 273–275.
- Andrea Magrì; Cristiana Lucia Rita Lipari; Pierpaolo Risiglione; Stefania Zimbone; Francesca Guarino; Antonella Caccamo; Angela Messina; ERK1/2-dependent TSPO overactivation associates with the loss of mitophagy and mitochondrial respiration in ALS. Cell Death Dis.. 2023, 14, 1-10.
- Weixing Zhao; Zhipeng Xu; Jiangbei Cao; Qiang Fu; Yishuang Wu; Xiaoying Zhang; Yue Long; Xuan Zhang; Yitian Yang; Yunfeng Li; Weidong Mi; Elamipretide (SS-31) improves mitochondrial dysfunction, synaptic and memory impairment induced by lipopolysaccharide in mice. J. Neuroinflammation. 2019, 16, 1-19.


