Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Haresh Manyar | -- | 4375 | 2023-12-08 14:01:21 | | | |
| 2 | Fanny Huang | Meta information modification | 4375 | 2023-12-12 04:45:13 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Shah, S.R.; Mazumdar, N.J.; Centeno-Pedrazo, A.; Deka, D.; Artioli, N.; Manyar, H. Transition-Metal-Catalyzed Carboxylation. Encyclopedia. Available online: https://encyclopedia.pub/entry/52530 (accessed on 24 July 2026).
Shah SR, Mazumdar NJ, Centeno-Pedrazo A, Deka D, Artioli N, Manyar H. Transition-Metal-Catalyzed Carboxylation. Encyclopedia. Available at: https://encyclopedia.pub/entry/52530. Accessed July 24, 2026.
Shah, Sagarkumar Rajendrakumar, Nayan Jyoti Mazumdar, Ander Centeno-Pedrazo, Dhanapati Deka, Nancy Artioli, Haresh Manyar. "Transition-Metal-Catalyzed Carboxylation" Encyclopedia, https://encyclopedia.pub/entry/52530 (accessed July 24, 2026).
Shah, S.R., Mazumdar, N.J., Centeno-Pedrazo, A., Deka, D., Artioli, N., & Manyar, H. (2023, December 08). Transition-Metal-Catalyzed Carboxylation. In Encyclopedia. https://encyclopedia.pub/entry/52530
Shah, Sagarkumar Rajendrakumar, et al. "Transition-Metal-Catalyzed Carboxylation." Encyclopedia. Web. 08 December, 2023.
Copy Citation
Carbon dioxide is ideal for carboxylation reactions as a renewable and sustainable C1 feedstock and has significant recognition owing to its low cost, non-toxicity, and high abundance. To depreciate the environmental concentration of CO2, which causes the greenhouse gas effect, developing new catalytic protocols for organic synthesis in CO2 utilization is of great importance.
carboxylation
carbon dioxide
carboxylic acids
1. Introduction
The rapidly increasing greenhouse gas concentrations in the environment, resulting in the current climate crisis, are a cause of huge concern. Over the past few decades, carbon dioxide emissions have drastically expanded due to an increase in the usage of fossil fuels and deforestation, with a world annual average environmental concentration of 418.37 ppm in 2023 [1]. Figure 1 shows that, from 1959 to 2022, the level of CO2 concentration increased severely, with a 0.5% growth rate annually [2]. Controlling the amount of CO2 in the atmosphere is a global challenge that needs to be resolved with the utmost priority. There has been increasing pressure on manufacturing industries to produce efficient methods for reducing CO2 emissions [3]. Since it is an abundant, non-toxic, non-combustible, renewable supply of carbon that is highly functional and an eco-friendly chemical reagent, carbon dioxide is a very attractive C1 building block in organic synthesis.
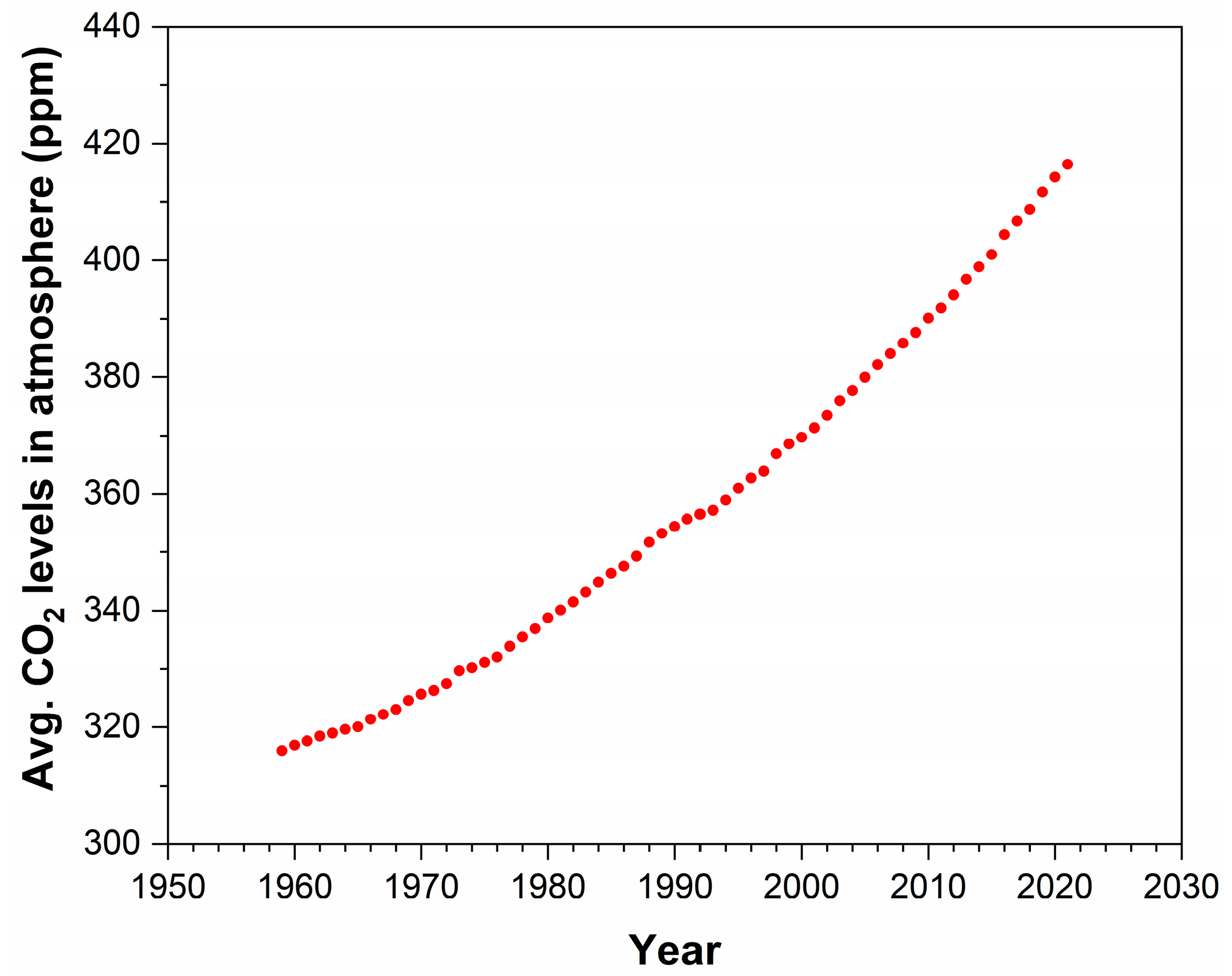
Figure 1. Concentration of CO2 from the years 1959 to 2022 in parts per million.
In a continuation of the interest in decarbonization, CO2 utilization, and environmental catalysis [4][5][6][7][8][9][10][11][12][13][14][15][16][17][18][19][20], researchers reviewed the recent progress made in carboxylation reactions for the synthesis of a variety of functionalized carboxylic acids and their derivatives. For the use of CO2 as a C1 feedstock in organic synthesis, it is very essential to capture CO2 in the C–C formation and produce value-added functional groups, such as carboxylic acids, lactams, carbonates, and lactones [21]. It is particularly challenging to employ due to its thermodynamic stability and kinetic inertness. Therefore, employing CO2 as a substrate faces numerous issues, such as high-pressure or high-temperature reaction conditions. Despite the substantial progress that has been made, there are still a lot of restrictions in this area regarding the scope of the substrate, the reaction system, and the activation techniques [22]. A variety of CO2 chemical transformations have been well developed in recent years [23]. Since the carboxylic acids are present in a wide variety of chemicals, such as agrochemicals, pharmaceuticals, biopharmaceuticals, cosmetics, and various polymers (Figure 2), it is particularly appealing to synthesize carboxylic acids with CO2 by forming C–C bonds [24][25]. Due to the importance of carboxylic acids and the benefits of promoting catalytic CO2 to produce carboxylic acid and its derivatives, much effort has been put into the synthesis of this motif using various reaction methods.
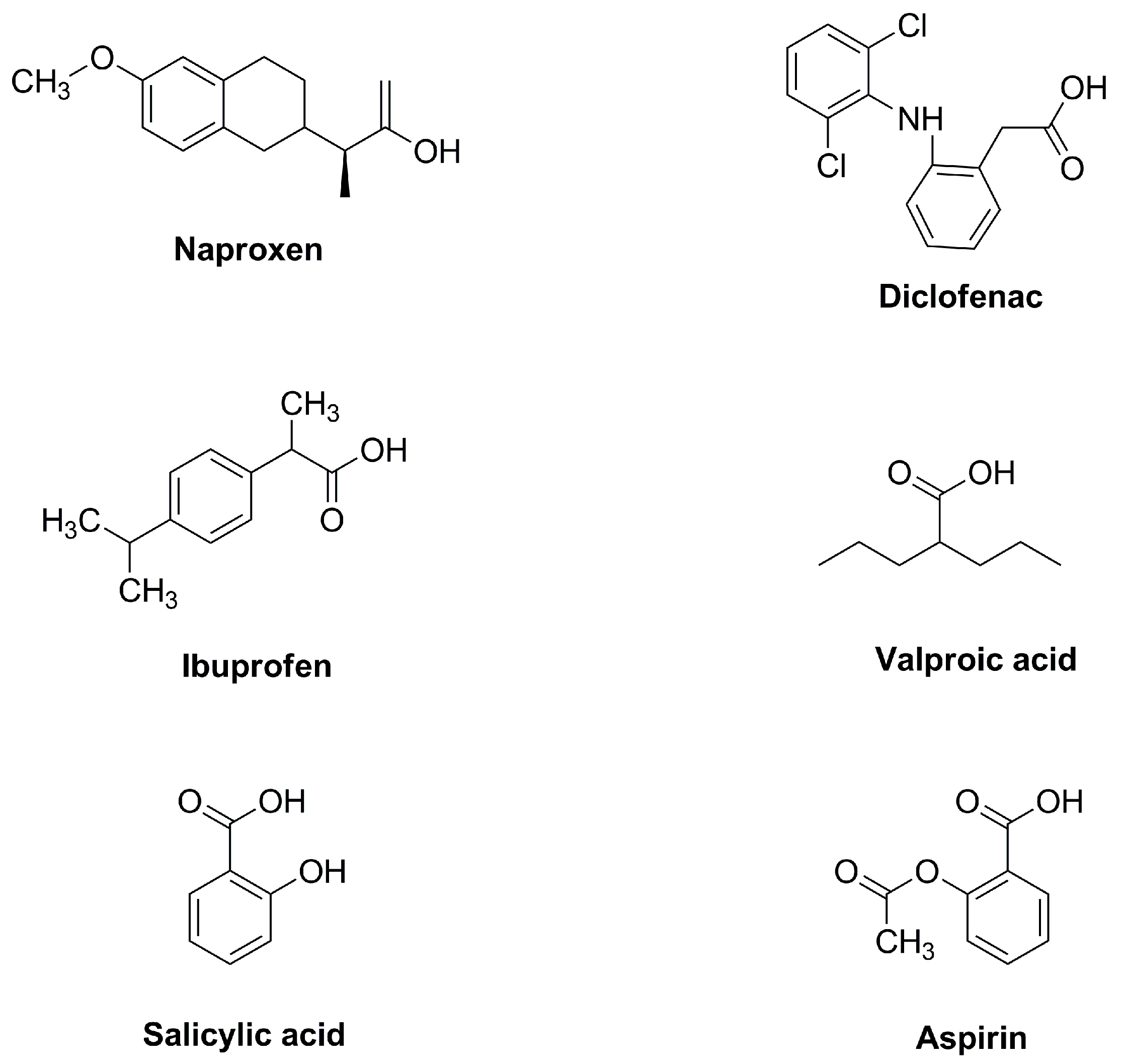
Figure 2. Carboxylic acids in pharmaceutical products.
Previously, strong nucleophilic reagents such as Grignard reagents were used for carboxylation (Scheme 1), in which CO2 acted as an electrophile. However, the main drawback with Grignard reagents is that they are air-sensitive and readily react with protic solvents and acidic functional groups, such as alcohol and amines [26]. Nevertheless, there are other well-known traditional synthetic pathways based on the hydrolysis of nitriles via acids, the oxidation of primary alcohol, and the carbonylation of organic halides with dangerous and poisonous carbon monoxide that can be used to synthesize carboxylic acid. The most appealing and simple way is the catalytic CO2 fixation process, which inserts CO2 as an electrophile into a metal–carbon bond to form a stable C–C bond under mild reaction conditions and exceptional chemo-site selectivity [27][28]. Diverse transition-metal-catalyzed carboxylation processes have been reported and well reviewed over the past ten years, as they enhance functional group compatibility with carboxylation reactions using CO2 as the C1 feedstock [29][30][31][32][33][34][35][36].
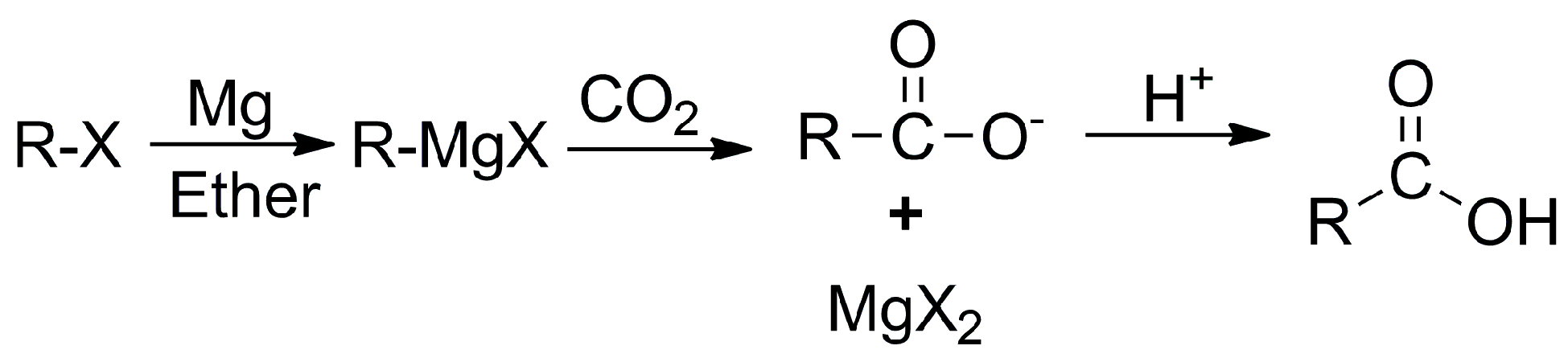
Scheme 1. Carboxylation with a Grignard reagent.
2. Transition-Metal-Catalyzed Carboxylation
2.1. Carboxylation of C–X Bonds (Halogens and Oxygen)
2.1.1. Pd-, Ni-, and Cu-Catalyzed Carboxylation of C-Halogens with CO2
The metal-catalyzed carboxylation of carbon nucleophiles with CO2 has developed into a potent substitute for the conventional synthetic methods for these compounds in recent years. The discovery that organometallic reagents can often be derived from organic halides through conventional metalation techniques has compelled chemists to explore the direct carboxylation of organic pseudohalides with CO2 [37]. Martin and his co-workers evolved the process for the reductive carboxylation of organic (pseudo) halides using CO2, and it still plays a significant role in this field [38][39]. From both theoretical and practical perspectives, the ideal method for producing carboxylic acids and their derivatives would involve catalytically inserting direct CO2 into aryl halides.
Unlike prior studies’ authors, Correa and Martin achieved high yields in the carboxylation of aryl bromides to corresponding aryl carboxylic acids using dimethyl acetamide and 1 M hexane as solvents, 5 mol.% of Pd(OAc2) (Palladium(II) acetate), and the bulkier phosphide ligand tBu-Xphos (biphenyl monophosphine ligand) under 10 atm pressure of CO2 and 100 °C, relatively mild reaction conditions. The authors also highlighted that there was no need for the synthesis of organometallic intermediates, and they also avoided the use of toxic CO for the preparation of benzoic acids. As a reductant, highly reactive pyrophoric diethylzinc (Et2Zn) was used, and via an oxidative addition reaction, zinc carboxylates were produced as an intermediate product; with an acidic build-up, carboxylic acid was generated, while a Pd (0) catalyst was generated via reductive elimination [40]. The need for high pressures, a pyrophoric reducing agent (Et2Zn), and the restriction to aryl bromide analogs was a significant constraint that needed to be surmounted even though this process served as a starting point for the reductive catalytic carboxylation of organic halides with CO2.
To overcome the above issues, Osakada and his team conducted CO2 insertion using nickel as a catalyst in the halo arene in 1994 [41]. They carboxylated bromoarene into benzoic acid, in which the monoaryl nickel complex Ni(Br)Ph(bpy) reacted with CO2 in a DMF solvent without any reductant, yielding a mixture of 55% benzoic acid and 21% biphenyl, which was simultaneously generated via acidosis [41]. Fuzihara and his coworkers discovered a much more efficient catalytic reaction using a less noble nickel catalyst in 2012 under a CO2 pressure of 1 atm and at room temperature. The nickel catalyst is very effective in the carboxylation of aryl chlorides, as well as vinyl chlorides, in the presence of Mn powder as the reductant, which is easy to handle [39]. In their experiment, air-stable Mn powder was combined with Et4NI (tetraethyl ammonium iodide) as a reducing agent, which was an essential component to obtain a high yield. The reaction was performed using a 5 mol.% NiCl2(PPh3) complex (Dichlorobis(triphenylphosphine)nickel(II)) under room temperature (25 °C) with 1 atm pressure for CO2 and 10 mol.% of PPh3 (triphenylphosphine), which provides a high yield of 4 butyl benzoic acid (95%). Similarly, in 2019, a novel Ni-catalytic carboxylation reaction was discovered in which the potential of using the Ni-catalyzed reductive carboxylation of N-hydroxy phthalimido (NHP) esters with 13CO2 or 14CO2 to synthesize isotopically labeled aliphatic and aromatic carboxylic acids was independently discovered by the Tortajada group, potentially accelerating the drug discovery process [42]. In 2021, Cerveri’s team independently disclosed an enantioselective nickel-catalyzed tandem heck-coupling/carboxylation synthetic sequence with CO2 that enables easy access to 2,3-dihydrobenzofuran-3-ylacetic acids and analogous compounds under benign conditions [43].
By analyzing the reaction mechanism, it can be seen that Ni(II) plays a vital role, and to keep the state of nickel (0, I, II) stable, the phosphate ligand plays a fundamental role. In each catalytic cycle, Ni(I) is present. The reaction commences with the reduction of Ni(II)to Ni(0) with Mn as the reducing agent (A). Aryl chloride enters the oxidative addition reaction to produce a Ni(I) intermediate (B). The most crucial step crops up when Ni(II) reduces to a Ni(I) intermediate with Mn as a reductant (C), which then reacts with CO2 and generates the carboxylatonickel complex (D). Finally, the reduction of these complexes via Mn from Ni(I) to Ni(0) produces carboxylates.
By offering more flexibility and ease of implementation, catalytic-reductive reactions contribute value from a standpoint of simplicity, dependability, and step economics. Reductive carboxylation techniques, on the other hand, are by their very nature constrained to substrates that swiftly undergo oxidative addition. According to Liu and his team, to overcome this issue, unactivated alkyl electrophiles with beta hydrogens should ideally be applied in this field [44]. In 2014, these authors reported a novel catalytic method for producing valuable carboxylic acids via the carboxylation of inactive primary alkyl bromides and sulfonates that contain beta hydrogens with CO2. This approach eliminates the use of air- or moisture-sensitive components and is distinguished by its excellent functional group compatibility, its mild conditions, the ready accessibility of its beginning materials, and its simplicity of execution [44].
In 2013, the Cu-catalyzed reductive carboxylation of aryl iodide was reported by Vu and his group using a copper iodide (CuI)/TMEDA (n, n, n, n-tetramethylethylinediamine) or DMEDA (1,2 dimethyl ethylenediamine) catalyst combined with diethylzinc ligand as a reductant [45]. Carboxylation took place with a low amount of copper iodide/TMEDA (3 mol.%) or DMEDA catalyst under 1 atm of CO2, using DMSO (Dimethyl sulfoxide) as a solvent or DMA (Dimethyl acetamide) for electron-poor aryl iodide and a temperature range of 25–70 °C. The reaction mechanism was very similar to the Ni-catalyzed carboxylation reaction.
In 2019, Yan and their team produced numerous valuable fluoro-acrylic acids and di-fluoro carboxylates, which feature in numerous pharmaceuticals but are otherwise challenging to obtain; they were synthesized successfully through the Cu-catalyzed highly selective conventional ipso-carboxylation of C–F bonds in fluorinated alkenes with CO2 using di boron as a source, LiOtBu as a base, and xantophos as a perfect ligand in a dimethylformamide (DMF) solvent under 80 °C and 1 atm of CO2 [46].
2.1.2. Carboxylation of C–O Bonds with CO2 Catalyzed via Ni(II)
According to Martin et al., in 2013, C(sp2)- and C(sp3)-O bonds were carboxylated using a Ni (II) catalyst with CO2 under mild conditions [44]. Even though the reaction conditions and mechanism were the same as for the C-halides carboxylation reaction, this technique holds tremendous potential for using ester derivatives as potent substitutes for organic halides due to the absence of air- or moisture-sensitive reagents and high selectivity. This approach is applicable to both aryl and benzyl C–O linkages; however, it requires prior activation of the C–O bonds as aryl or benzyl esters [44].
2.2. Carboxylation of Organometallic Compounds Employing Carbon Dioxide (CO2)
In organic synthesis, the direct carboxylation and production of carboxylic acids and their derivatives using organometallic compounds with CO2 have been extensively studied [47]. The direct use of CO2 as a green and sustainable feedstock and the avoidance of harsh reaction conditions are two main benefits of this protocol. However, this synthesis necessitates a substantial quantity of costly and weak organometallic reagents [47][48]. Due to the difficulty in the synthesis of related organometallic reagents, the use of this reaction method is still underdeveloped.
2.2.1. Nickel- and Palladium-Catalyzed Carboxylation of R-M (M = Sn, Zn) Reagents with CO2
Direct CO2 insertion using Grignard and organolithium reagents is not suitable for delicate functional groups, such as ketones, aldehydes, nitrile, some alcohols, and amines, as it is extremely reactive with these groups [49]. In the last two decades, alternative techniques have been developed that can fix CO2 directly into metal–carbon bonds catalytically with accurate chemo-selectivity, avoiding harsh reaction conditions. In 1997, Shi and coworkers made a groundbreaking discovery by introducing CO2 into metal–carbon bonds, resulting in the production of carboxylic acids [50]. This was the first instance of a transition-metal-catalyzed process involving less polarized metals. CO2 fixation into allyl stanannes takes place under 33 atm pressure and a 70 °C temperature using palladium as a catalyst with a phosphine ligand (8 mol.% of Pd(PPh3)4 or Pd(PBu3)4). Johansson and his coworkers made progress in 2006 by utilizing a pincer-palladium system (PCP) to achieve high selectivity in producing terminal alkene products [51]. This method allows for rapid CO2 fixation, resulting in the synthesis of alkenyl carboxylate. As part of the process, allyl stanannes were used to trans-metallize palladium. Afterward, CO2 was inserted into the Pd–C bond, resulting in the formation of carboxylates [52][53].
Another essential element for the carboxylation process, which is facilitated via transition metals, is organozinc. Recent studies have indicated that nickel and palladium can serve as catalysts for coupling reactions between organozinc compounds and CO2 at room temperature [54], in which lithium chloride plays a vital role in achieving a high yield (70%); without it, only a 5% yield is achieved. Phosphine ligands (P(C–C6H11)3) were also essential for the reaction to give a desirable yield of carboxylic acid, in which electron-rich tricyclohexylphosphine increased the electron density of the oxygen by donating electrons, which enabled trans-metalation to occur. Further progress was achieved using diphenyl zinc, which provided a yield of 90% carboxylic acid [54]. At the same time, a method for carboxylating organozinc compounds in THF at 0 °C using palladium and nickel catalysts was developed by Yeung and Dong, in which the Aresta’s complex (Ni(η2-CO2)(PCy3)2) played a significant role [55]. In this process, Pd(OAc)2 was demonstrated to be an effective catalytic precursor to carbon dioxide activation. Aresta’s complex is used as a catalyst for the cross-coupling of organozinc reagents with CO2 under mild conditions. Palladium catalysts are superior to nickel catalysts when using aryl–zinc bromide, resulting in higher yields. During this study, the authors discovered that both aromatic and alkyl organozinc reagents can be employed with nickel catalysts, which achieved a good yield of carboxylic acids [55]. All these reactions share a consistent mechanism, which includes the initial introduction of nickel (0) to CO2 through oxidation addition, followed by transmetalation with the organozinc reagent, and ultimately leading to reductive elimination, resulting in the formation of the corresponding zinc carboxylate.
2.2.2. Copper (Cu)-, Palladium (Pd)-, and Rhodium (Rh)-Catalyzed Carboxylation of R B Reagents with CO2
The most popular catalyst for the deborylative carboxylation of organic boron compounds with CO2 is copper. The metal–carbon bond found in organocopper reagents is highly unusual and exhibits mild polarity. These reagents are capable of undergoing CO2 insertion under moderate conditions while being able to tolerate a wide range of functional groups [48]. Copper catalysts are highly encouraging for C–C bond formation via CO2 transformation, as copper catalysts can catalyze various C–H and C–halogen activation reactions, which involve Cu–C bond intermediates [56][57][58][59][60]. Iwasawa’s and Hou’s groups were able to carboxylate organoboronic esters using Cu and Rh through transition-metal catalysis using CO2 under ambient conditions. The use of nucleophilic organoboronic ester reagents has brought renewed attention to carboxylation reactions for their convenience and widespread application. In 2006, Iwasawa first reported on the use of a rhodium complex to catalyze organoboronic esters [61]. To achieve a high yield (75%) of benzoic acid in this reaction, it is essential to use a cesium fluoride (CsF) base and 1,3-bis(diphenylphosphino)propane (dppp) ligand. In the absence of them, only 12% benzoic acid will be produced. Following this significant finding, Iwasawa and Hou both separately developed a copper (I)-catalyzed carboxylation method for aryl boronic esters under mild conditions. This breakthrough discovery was a significant achievement and remarkable advancement in the field. Copper catalyst systems are superior to rhodium systems. Copper catalysts are cost-effective and readily available, and they exhibit a broader substrate scope, allowing for the synthesis of a diverse range of functionalized carboxylic acids. Iwasawa’s method, employing 5 mol.% of CuI, a 6 mol.% of bis-oxazoline ligand, excess CsF, and a DMF solvent, achieved remarkable carboxylate product yields from boronic esters. On the other hand, Hou’s group employed a copper(I) catalyst supported with an N-heterocyclic carbene ligand with 2 equiv. of strong base (t-BuOK) [62]. Despite the use of different additives, both reaction systems operated on the same mechanism.
When copper chloride reacts with t-BuOK, it produces a catalyst called t-BuOCu in a strong base system, which acts as an actual catalyst. After that, the organocopper complexes can be efficiently generated by undergoing a transmetalation process, followed by the insertion of CO2 into the Cu–C bond to produce copper carboxylates. The carboxylate salt is subsequently released by the base, which regenerates the catalyst. CsF played an essential role in this reaction mechanism by generating aryl fluoroborates [62]. Ohmiya’s team conducted a study in 2010 on the copper-catalyzed carboxylation of alkyl boranes with CO2, which encompassed a comparable reaction [63].
2.3. Utilizing CO2 to Carboxylate C–H Bonds
2.3.1. Carboxylation sp1 C–H Bonds with CO2
Cu-Catalyzed Carboxylation of sp1 C–H Bonds
In 1962, Allan Hay created a very successful technique for the oxidative homocoupling of terminal alkynes with Cu as a catalyst through C–H activation [64], in which copper chloride was the most suitable salt for the reaction with tertiary amines. Following that, Fukue and his group reported the catalytic carboxylation of terminal alkynes; they observed the insertion of CO2 in copper acetylide intermediates via a Cu complex [65]. To synthesize acidic ester, Fukue and Inoue used a mixture of terminal alkyne, 10 mol.% of bromo hexane, and 30 mol.% of K2CO3 at 100 °C for 4 h in the presence of copper iodide. Yu and their coworkers developed a method to achieve high yields of carboxylic acid under mild conditions using copper-NHC catalysis with CO2 and C-H activation as a solution to the harsh reaction conditions of Inoue and their research groups [66]. They reported that the CuCl-TMEDA (tetramethylethylenediamine) system is essential to achieving a good yield (93%). In 2014, the carboxylation of terminal alkynes with CO2 was examined by Yang and his team with the use of copper (1)-NHC complexes [67]. Through DFT calculations, they were able to determine the potential mechanism involved, as well as the role of NHC in the process [68]. In 2015, Trivedi and his group reported that a good yield of propiolic acids was obtained using a ferrocene-based bis-phosphene ligand and copper iodide as an effective catalyst for the carboxylation of terminal alkynes via C–H activation with CO2 under mild conditions, which was confirmed via single-crystal X-ray diffraction [69]. Bondarenko and their coworkers achieved the direct carboxylation of various terminal alkynes with CO2 by employing copper particles supported on readily accessible Al2O3 catalysts in the presence of calcium carbonate in 2017 [70]. In 2018, Gooßen and collaborators reported the salt-free carboxylation of terminal alkynes with CO2 using copper as a catalyst [71]. By activating the C–H bond with a copper(I) catalyst during this reaction, CO2 was introduced into alkynes using TMP (tetramethylpiperidine) as the base.
Ag-Catalyzed Carboxylation of sp1 C–H Bonds with CO2
In 2011, Lu and their coworkers reported the ligand-free direct carboxylation of terminal alkynes with CO2 [68]. They used silver iodide at a stochiometric amount. In this reaction, silver propiolate acts as a nucleophile, and silver phenyl acetylide and silver phenylpropiolate were prepared and directly used as catalysts, which finally converted phenylacetylene to phenylpropanoid acid with a good yield. These authors found that silver propiolate was generated as an intermediate during the reaction, which then reacted with terminal alkynes and CS2CO3, which produced cesium propiolate. Upon acidification, the final product was synthesized as propiolic acid [72]. In 2012, a similar catalytic system to that described by Lu and their coworkers was reported by Gooßen and his group in which they used a very low quantity (500 ppm) of silver under mild conditions [73]. In 2014, Hong’s group discovered that carbon dioxide (CO2) produced through combustion can be utilized in organic syntheses with the same efficacy as hyper-pure CO2 gas [74]. The direct carboxylation of terminal alkynes is efficiently catalyzed via silver iodide, as it effectively combines the capture of ethanolamine with the utilization of CO2 [72]. In 2016, Li and Sun synthesized novel silver N-heterocyclic carbene complexes (NHC) using CO2 via the carboxylation of terminal alkynes. Through the utilization of various substrates, including alkyl alkynes and aryl alkynes with low electron densities, this system achieved a remarkable yield of propiolic acids. Additionally, the catalyst is highly reusable and can be employed multiple times without sacrificing its effectiveness.
2.3.2. Carboxylation of sp2 C–H Bonds with CO2
Cu- and Au-Catalyzed Carboxylation of sp2 C-H Bonds with CO2
In 2010, Nolan et al. reported a high yield via the regioselective carboxylation of oxazole and multi-substituted fluorobenzene, and it was successfully catalyzed using the copper complex 1,3-bis(2,6-diisopropylphenyl) imidazol-2-ylidene [Cu (IPr)OH] [75][76]. Extensive studies have been performed using copper complexes with varied ligands to facilitate the carboxylation of benzoxazoles. The [(IPr)CuCl] compound has proven to be a potent catalyst for the carboxylation of heterocycle with KOtBu [77]. According to these authors’ research, copper complexes with nitrogen-based ligands are not very effective unless the C–H bonds are acidic and able to activate them. This is why they do not work well with benzothiazole and benzofuran, which have less acidic C–H bonds. In 2012, Fukuzawa and Hou’s group discovered the use of 1,2,3-Triazol-5-ylidene copper(I) complexes (tzNHC-Cu) as a catalyst to carboxylate heteroaromatic groups like benzoxazole and benzothiazole via direct C–H carboxylation with CO2 [78]. This reaction was initiated by adding t-BuOK and 5 mol.% of tzNHC-Cu to a THF solvent under 1 atm CO2 pressure and 80 °C, in which, after 1 h, THF was removed via a vacuum and subsequently DMF was added, which achieved a good yield of benzoxylate carboxylic acid. The direct carboxylation of the C–H bond was successfully achieved by Nolan and his team using a [(IPr)AuOH] catalyst in 2010 [79]. The catalyst proved to be highly effective for the carboxylation of both the carbo- and heterocycle with a highly acidic regioselective C–H bond under mild conditions.
Carboxylation of sp2 C–H Bonds with CO2 Catalyzed by Pd and Rh
In a recent study, Iwasawa et al. reported the direct carboxylation of alkenyl C–H bonds with CO2 using Pd (II) as a catalyst [80]. According to their report, when 2-hydroxy styrene and 5% Pd (OAc)2 were treated with CS2CO3 and CO2 at 1-atmosphere pressure and 100 °C, the desired coumarins were produced with a high yield. In this study, coumarins were synthesized with high yields from a variety of substrates with either electron-donating or electron-withdrawing groups on the phenyl ring at the position of 2-hydroxystyrene. During the study, these authors found that this reaction produces the alkenyl palladium intermediate, which can be exploited for reversible nucleophilic carboxylation, and they used the single-crystal X-ray diffraction method to analyze it. The intermediate subsequently undergoes a reaction with another molecule of 2-hydroxystyrene 1 and a base, resulting in the formation of coumarin and the regeneration of the cyclometalated intermediate via the lactonization process. It is highly appealing and challenging in organic synthesis to catalyze nucleophilic carboxylation reactions via single-bond H-bond activation using carbon dioxide as the carbon source. As aryl rhodium(I) species are anticipated to have sufficient nucleophilicity for carboxylation and rhodium(I) complexes as well-known catalysts for C–H activation, Rh(I) was chosen as the catalyst for the carboxylation of aryl C–H bonds with CO2. The direct carboxylation of an inactive aryl C–H bond was achieved via Rh-catalyzed chelation-assisted C–H activation under atmospheric carbon dioxide pressure for the first time [81]. The carboxylation of aryl-substituted pyridine and pyrazoles was carried out in the DMA (N, N-dimethylacetamide) solvent at 70 °C in a CO2 atmosphere using 5 moles of [Rh(cyclooctene)Cl]2 and 12 moles of trimesitylphosphine (P(mes)3). By producing an essential intermediate methyl rhodium (I) species, AlMe2(OMe) was employed as a reducing agent, which was crucial during the catalytic cycle. The initial step involves the formation of aryl rhodium(I) from methyl rhodium(I), which is generated through the reaction between rhodium(I) chloride and a methyl aluminum reagent. This formation occurs through a chelation-assisted C–H bond activation, which is essentially an oxidative addition reaction. Following this, the elimination of methane takes place via a reductive elimination process. This step is succeeded by the generation of rhodium carboxylates, achieved through the nucleophilic carboxylation of aryl rhodium(I). The final stage of the process involves the transmetalation of the carboxylates with methyl aluminum reagents. This transmetalation also leads to the production of aluminum carboxylates and the release of methyl rhodium(I) species.
2.3.3. Carboxylation of sp3 C–H Bonds with CO2
Kikuchi et al. developed the silver-catalyzed carboxylation of several ketones containing alkyne derivatives with CO2 via C–C bond formation [82]. This reaction utilized mild reaction conditions with 10 atm pressure of CO2 and catalytic silver benzoate in the presence of 7-methyl-1,5,7-triazabicyclo [4.4.0] dec-5-ene (MTBD) to synthesize lactone derivatives with good to high yields. With this reaction system, it was possible to obtain the corresponding λ-lactone from aliphatic ketone derivatives at 50 °C under 2.0 MPa CO2 pressure. A lactone is synthesized in this reaction via the formation of a new C–C bond between sp3 carbon and CO2, which is stabilized via alkyne. The same group synthesized dihydroisobenzofuran derivatives with the reaction of o-alkynyl acetophenone derivatives and carbon dioxide via C–C bond formation using a silver catalyst [83]. Using a silver catalyst, the same group also synthesized dihydroisobenzofuran derivatives by combining o-alkynyl acetophenone derivatives with carbon dioxide.
2.4. Carboxylation of Ethylene with CO2 to Acrylates
It has been an unsolved problem and the most attractive reaction in catalysis research for more than thirty years to catalyze the synthesis of acrylates from the cheap and abundant building block of C1 carbon dioxide and alkenes. The synthesis of acrylic acids via the carboxylation of CO2 and alkene was first achieved by Hoberg and Minato [84]. Limbach et al. reported the CO2 coupling reaction with ethylene to acrylates by means of cyclometalation, followed by the elimination of beta-hydride [85]. A crucial step in the nickel-mediated acrylate synthesis from CO2 and ethylene was the formation of nickelalatones. The reaction was meticulously demonstrated in three distinct stages: (1) the formation of nickelalactone at room temperature using a Ni(dtbpe) (dtbpe = di-tert-butylphosphino ethane) ligand that reacts with C2H4 and CO2 with pressure of 2 bar and 6 bar subsequently in THF; (2) the utilization of potent base alkoxides in PhCl to effectively break the nickelalactone ring, which is challenging task, resulting in the formation of π -complexes featuring 2-coordinated acrylate; (3) the exchanging of ligands with ethylene resulting in the release of Ni(dtbpe)(CH2CH2) and an acrylate product under room temperature. A catalytic turnover of more than 10 was attained once the catalytic cycle was put together. A simple aqueous extraction was used to separate the Na acrylate formed during the reaction. Extensive studies have been conducted by Hoffman et al. on the carboxylation of ethylene to acrylates using CO2 mediated with nickel [86]. The main challenge encountered is the breakdown of the nickelalactone structure. They developed a new approach to producing lactones. However, all the existing methods for synthesizing acrylic acid complexes require too much activation energy and cannot function at normal temperatures.
References
- Ritchie, H.; Roser, M.; Rosado, P. CO2 and Greenhouse Gas Emissions. Our World Data 2020. Available online: https://ourworldindata.org/co2-and-greenhouse-gas-emissions (accessed on 24 October 2023).
- Ritchie, H.; Roser, M.; Rosado, P. Energy. Our World Data 2022. Available online: https://ourworldindata.org/energy (accessed on 24 October 2023).
- Riduan, S.N.; Zhang, Y. Recent Developments in Carbon Dioxide Utilization under Mild Conditions. Dalton Trans. 2010, 39, 3347–3357.
- Delarmelina, M.; Deshmukh, G.; Goguet, A.; Catlow, C.R.A.; Manyar, H. Role of Sulfation of Zirconia Catalysts in Vapor Phase Ketonization of Acetic Acid. J. Phys. Chem. C 2021, 125, 27578–27595.
- Ethiraj, J.; Wagh, D.; Manyar, H. Advances in Upgrading Biomass to Biofuels and Oxygenated Fuel Additives Using Metal Oxide Catalysts. Energy Fuels 2022, 36, 1189–1204.
- Keogh, J.; Deshmukh, G.; Manyar, H. Green Synthesis of Glycerol Carbonate via Transesterification of Glycerol Using Mechanochemically Prepared Sodium Aluminate Catalysts. Fuel 2022, 310, 122484.
- Ralphs, K.; Collins, G.; Manyar, H.; James, S.L.; Hardacre, C. Selective Hydrogenation of Stearic Acid Using Mechanochemically Prepared Titania-Supported Pt and Pt–Re Bimetallic Catalysts. ACS Sustain. Chem. Eng. 2022, 10, 6934–6941.
- Keogh, J.; Jeffrey, C.; Tiwari, M.S.; Manyar, H. Kinetic Analysis of Glycerol Esterification Using Tin Exchanged Tungstophosphoric Acid on K-10. Ind. Eng. Chem. Res. 2022, 62, 19095–19103.
- Tiwari, M.S.; Wagh, D.; Dicks, J.S.; Keogh, J.; Ansaldi, M.; Ranade, V.V.; Manyar, H.G. Solvent Free Upgrading of 5-Hydroxymethylfurfural (HMF) with Levulinic Acid to HMF Levulinate Using Tin Exchanged Tungstophosphoric Acid Supported on K-10 Catalyst. ACS Org. Inorg. Au 2023, 3, 27–34.
- Manyar, H. Synthesis of a Novel Redox Material UDCaT-3: An Efficient and Versatile Catalyst for Selective Oxidation, Hydroxylation and Hydrogenation Reactions. Adv. Synth. Catal. 2008, 350, 2286.
- Skillen, N.; Ralphs, K.; Craig, D.; McCalmont, S.; Muzio, A.F.V.; O’Rourke, C.; Manyar, H.; Robertson, P. Photocatalytic Reforming of Glycerol to H2 in a Thin Film Pt-TiO2 Recirculating Photo Reactor. J. Chem. Technol. Biotechnol. 2020, 95, 2619–2627.
- Jakubek, T.; Ralphs, K.; Kotarba, A.; Manyar, H. Nanostructured Potassium-Manganese Oxides Decorated with Pd Nanoparticles as Efficient Catalysts for Low-Temperature Soot Oxidation. Catal. Lett. 2019, 149, 100–106.
- Salisu, J.; Gao, N.; Quan, C.; Yanik, J.; Artioli, N. Co-Gasification of Rice Husk and Plastic in the Presence of CaO Using a Novel ANN Model-Incorporated Aspen plus Simulation. J. Energy Inst. 2023, 108, 101239.
- Quan, C.; Zhang, G.; Gao, N.; Su, S.; Artioli, N.; Feng, D. Behavior Study of Migration and Transformation of Heavy Metals during Oily Sludge Pyrolysis. Energy Fuels 2022, 36, 8311–8322.
- Byrne, E.L.; O’Donnell, R.; Gilmore, M.; Artioli, N.; Holbrey, J.D.; Swadźba-Kwaśny, M. Hydrophobic Functional Liquids Based on Trioctylphosphine Oxide (TOPO) and Carboxylic Acids. Phys. Chem. Chem. Phys. 2020, 22, 24744–24763.
- Castoldi, L.; Matarrese, R.; Kubiak, L.; Daturi, M.; Artioli, N.; Pompa, S.; Lietti, L. In-Depth Insights into N2O Formation over Rh- and Pt-Based LNT Catalysts. Catal. Today 2019, 320, 141–151.
- Yilleng, M.T.; Gimba, E.C.; Ndukwe, G.I.; Bugaje, I.M.; Rooney, D.W.; Manyar, H.G. Batch to Continuous Photocatalytic Degradation of Phenol Using TiO2 and Au-Pd Nanoparticles Supported on TiO2. J. Environ. Chem. Eng. 2018, 6, 6382–6389.
- O’Donnell, R.; Ralphs, K.; Grolleau, M.; Manyar, H.; Artioli, N. Doping Manganese Oxides with Ceria and Ceria Zirconia Using a One-Pot Sol–Gel Method for Low Temperature Diesel Oxidation Catalysts. Top. Catal. 2020, 63, 351–362.
- Coney, C.; Hardacre, C.; Morgan, K.; Artioli, N.; York, A.P.E.; Millington, P.; Kolpin, A.; Goguet, A. Investigation of the oxygen storage capacity behaviour of three way catalysts using spatio-temporal analysis. Appl. Catal. B Environ. 2019, 258, 117918.
- Lietti, L.; Righini, L.; Castoldi, L.; Artioli, N.; Forzatti, P. Labeled 15NO study on N2 and N2O formation over Pt-Ba/Al2O3 NSR catalysts. Top. Catal. 2013, 56, 7–13.
- He, M.; Sun, Y.; Han, B. Green Carbon Science: Scientific Basis for Integrating Carbon Resource Processing, Utilization, and Recycling. Angew. Chem. Int. Ed. 2013, 52, 9620–9633.
- Ye, J.-H.; Ju, T.; Huang, H.; Liao, L.-L.; Yu, D.-G. Radical Carboxylative Cyclizations and Carboxylations with CO2. Acc. Chem. Res. 2021, 54, 2518–2531.
- Sakakura, T.; Choi, J.-C.; Yasuda, H. Transformation of Carbon Dioxide. Chem. Rev. 2007, 107, 2365–2387.
- Maag, H. Prodrugs of Carboxylic Acids. In Prodrugs: Challenges and Rewards Part 1; Stella, V.J., Borchardt, R.T., Hageman, M.J., Oliyai, R., Maag, H., Tilley, J.W., Eds.; Biotechnology: Pharmaceutical Aspects; Springer: New York, NY, USA, 2007; pp. 703–729. ISBN 9780387497853.
- Ajvazi, N.; Stavber, S. Alcohols in direct carbon-carbon and carbon-heteroatom bond-forming reactions: Recent advances. Arkivoc 2018, part ii, 288–329.
- Valera Lauridsen, J.M.; Cho, S.Y.; Bae, H.Y.; Lee, J.-W. CO2 (De)Activation in Carboxylation Reactions: A Case Study Using Grignard Reagents and Nucleophilic Bases. Organometallics 2020, 39, 1652–1657.
- Aresta, M.; Dibenedetto, A. Key Issues in Carbon Dioxide Utilization as a Building Block for Molecular Organic Compounds in the Chemical Industry. In CO2 Conversion and Utilization; Song, C., Gaffney, A.F., Fujimoto, K., Eds.; American Chemical Society: Washington, DC, USA, 2002; Volume 809, pp. 54–70. ISBN 9780841237476.
- Wu, X.-F.; Fang, X.; Wu, L.; Jackstell, R.; Neumann, H.; Beller, M. Transition-Metal-Catalyzed Carbonylation Reactions of Olefins and Alkynes: A Personal Account. Acc. Chem. Res. 2014, 47, 1041–1053.
- Huang, K.; Sun, C.-L.; Shi, Z.-J. Transition-Metal-Catalyzed C–C Bond Formation through the Fixation of Carbon Dioxide. Chem. Soc. Rev. 2011, 40, 2435–2452.
- Tsuji, Y.; Fujihara, T. Carbon Dioxide as a Carbon Source in Organic Transformation: Carbon–Carbon Bond Forming Reactions by Transition-Metal Catalysts. Chem. Commun. 2012, 48, 9956–9964.
- Duong, H.A.; Huleatt, P.B.; Tan, Q.-W.; Shuying, E.L. Regioselective Copper-Catalyzed Carboxylation of Allylboronates with Carbon Dioxide. Org. Lett. 2013, 15, 4034–4037.
- Yu, D.; Teong, S.P.; Zhang, Y. Transition Metal Complex Catalyzed Carboxylation Reactions with CO2. Coord. Chem. Rev. 2015, 293–294, 279–291.
- Luan, Y.-X.; Ye, M. Transition Metal-Mediated or Catalyzed Hydrocarboxylation of Olefins with CO2. Tetrahedron Lett. 2018, 59, 853–861.
- Yi, Y.; Hang, W.; Xi, C. Recent Advance of Transition-Metal-Catalyzed Tandem Carboxylation Reaction of Unsaturated Hydrocarbons with Organometallic Reagents and CO2. Youji Huaxue 2021, 41, 80.
- Ran, C.-K.; Liao, L.-L.; Gao, T.-Y.; Gui, Y.-Y.; Yu, D.-G. Recent Progress and Challenges in Carboxylation with CO2. Curr. Opin. Green. Sustain. Chem. 2021, 32, 100525.
- Liu, X.-F.; Zhang, K.; Tao, L.; Lu, X.-B.; Zhang, W.-Z. Recent Advances in Electrochemical Carboxylation Reactions Using Carbon Dioxide. Green. Chem. Eng. 2022, 3, 125–137.
- Seyferth, D. Alkyl and Aryl Derivatives of the Alkali Metals: Useful Synthetic Reagents as Strong Bases and Potent Nucleophiles. 1. Conversion of Organic Halides to Organoalkali-Metal Compounds. Organometallics 2006, 25, 2–24.
- Kingston, C.; Wallace, M.A.; Allentoff, A.J.; deGruyter, J.N.; Chen, J.S.; Gong, S.X.; Bonacorsi, S.; Baran, P.S. Direct Carbon Isotope Exchange through Decarboxylative Carboxylation. J. Am. Chem. Soc. 2019, 141, 774–779.
- Fujihara, T.; Nogi, K.; Xu, T.; Terao, J.; Tsuji, Y. Nickel-Catalyzed Carboxylation of Aryl and Vinyl Chlorides Employing Carbon Dioxide. J. Am. Chem. Soc. 2012, 134, 9106–9109.
- Correa, A.; Martín, R. Palladium-Catalyzed Direct Carboxylation of Aryl Bromides with Carbon Dioxide. J. Am. Chem. Soc. 2009, 131, 15974–15975.
- Osakada, K.; Sato, R.; Yamamoto, T. Nickel-Complex-Promoted Carboxylation of Haloarenes Involving Insertion of CO2 into NiII-C Bonds. Organometallics 1994, 13, 4645–4647.
- Tortajada, A.; Duan, Y.; Sahoo, B.; Cong, F.; Toupalas, G.; Sallustrau, A.; Loreau, O.; Audisio, D.; Martin, R. Catalytic Decarboxylation/Carboxylation Platform for Accessing Isotopically Labeled Carboxylic Acids. ACS Catal. 2019, 9, 5897–5901.
- Cerveri, A.; Giovanelli, R.; Sella, D.; Pedrazzani, R.; Monari, M.; Nieto Faza, O.; López, C.S.; Bandini, M. Enantioselective CO2 Fixation Via a Heck-Coupling/Carboxylation Cascade Catalyzed by Nickel. Chem.–A Eur. J. 2021, 27, 7657–7662.
- Correa, A.; León, T.; Martin, R. Ni-Catalyzed Carboxylation of C(Sp2)– and C(Sp3)–O Bonds with CO2. J. Am. Chem. Soc. 2014, 136, 1062–1069.
- Tran-Vu, H.; Daugulis, O. Copper-Catalyzed Carboxylation of Aryl Iodides with Carbon Dioxide. ACS Catal. 2013, 3, 2417–2420.
- Yan, S.-S.; Wu, D.-S.; Ye, J.-H.; Gong, L.; Zeng, X.; Ran, C.-K.; Gui, Y.-Y.; Li, J.; Yu, D.-G. Copper-Catalyzed Carboxylation of C–F Bonds with CO2. ACS Catal. 2019, 9, 6987–6992.
- Bew, S.P. Carboxylic Acids. In Comprehensive Organic Functional Group Transformations, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2005; pp. 19–125. ISBN 9780080446554.
- Ebert, G.W.; Juda, W.L.; Kosakowski, R.H.; Ma, B.; Dong, L.; Cummings, K.E.; Phelps, M.V.B.; Mostafa, A.E.; Luo, J. Carboxylation and Esterification of Functionalized Arylcopper Reagents. J. Org. Chem. 2005, 70, 4314–4317.
- Hoffmann, R.W. Protecting-Group-Free Synthesis. Synthesis 2006, 2006, 3531–3541.
- Shi, M.; Nicholas, K.M. Palladium-Catalyzed Carboxylation of Allyl Stannanes. J. Am. Chem. Soc. 1997, 119, 5057–5058.
- Johansson, R.; Wendt, O.F. Insertion of CO2 into a Palladiumallyl Bond and a Pd(ii) Catalysed Carboxylation of Allyl Stannanes. Dalton Trans. 2007, 4, 488–492.
- Wu, J.; Hazari, N. Palladium Catalyzed Carboxylation of Allylstannanes and Boranes Using CO2. Chem. Commun. 2011, 47, 1069–1071.
- Hruszkewycz, D.P.; Wu, J.; Hazari, N.; Incarvito, C.D. Palladium(I)-Bridging Allyl Dimers for the Catalytic Functionalization of CO2. J. Am. Chem. Soc. 2011, 133, 3280–3283.
- Ochiai, H.; Jang, M.; Hirano, K.; Yorimitsu, H.; Oshima, K. Nickel-Catalyzed Carboxylation of Organozinc Reagents with CO2. Org. Lett. 2008, 10, 2681–2683.
- Yeung, C.S.; Dong, V.M. Beyond Aresta’s Complex: Ni- and Pd-Catalyzed Organozinc Coupling with CO2. J. Am. Chem. Soc. 2008, 130, 7826–7827.
- Shibasaki, M.; Kanai, M. Asymmetric Synthesis of Tertiary Alcohols and α-Tertiary Amines via Cu-Catalyzed C−C Bond Formation to Ketones and Ketimines. Chem. Rev. 2008, 108, 2853–2873.
- Evano, G.; Blanchard, N.; Toumi, M. Copper-Mediated Coupling Reactions and Their Applications in Natural Products and Designed Biomolecules Synthesis. Chem. Rev. 2008, 108, 3054–3131.
- Deutsch, C.; Krause, N.; Lipshutz, B.H. CuH-Catalyzed Reactions. Chem. Rev. 2008, 108, 2916–2927.
- Wang, Y.; Liu, J.; Zheng, G. Designing Copper-Based Catalysts for Efficient Carbon Dioxide Electroreduction. Adv. Mater. 2021, 33, 2005798.
- Nitopi, S.; Bertheussen, E.; Scott, S.B.; Liu, X.; Engstfeld, A.K.; Horch, S.; Seger, B.; Stephens, I.E.L.; Chan, K.; Hahn, C.; et al. Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte. Chem. Rev. 2019, 119, 7610–7672.
- Ukai, K.; Aoki, M.; Takaya, J.; Iwasawa, N. Rhodium(I)-Catalyzed Carboxylation of Aryl- and Alkenylboronic Esters with CO2. J. Am. Chem. Soc. 2006, 128, 8706–8707.
- Ohishi, T.; Nishiura, M.; Hou, Z. Carboxylation of Organoboronic Esters Catalyzed by N-Heterocyclic Carbene Copper(I) Complexes. Angew. Chem. Int. Ed. 2008, 47, 5792–5795.
- Ohmiya, H.; Tanabe, M.; Sawamura, M. Copper-Catalyzed Carboxylation of Alkylboranes with Carbon Dioxide: Formal Reductive Carboxylation of Terminal Alkenes. Org. Lett. 2011, 13, 1086–1088.
- Hay, A.S. Oxidative Coupling of Acetylenes. II. J. Org. Chem. 1962, 27, 3320–3321.
- Fukue, Y.; Oi, S.; Inoue, Y. Direct Synthesis of Alkyl 2-Alkynoates from Alk-1-Ynes, CO2, and Bromoalkanes Catalysed by Copper(I) or Silver(I) Salt. J. Chem. Soc. Chem. Commun. 1994, 18, 2091.
- Zhang, W.-Z.; Li, W.-J.; Zhang, X.; Zhou, H.; Lu, X.-B. Cu(I)-Catalyzed Carboxylative Coupling of Terminal Alkynes, Allylic Chlorides, and CO2. Org. Lett. 2010, 12, 4748–4751.
- Yu, D.; Zhang, Y. Copper- and Copper– N. -Heterocyclic Carbene-Catalyzed C–H Activating Carboxylation of Terminal Alkynes with CO2 at Ambient Conditions. Proc. Natl. Acad. Sci. USA 2010, 107, 20184–20189.
- Yang, L.; Yuan, Y.; Wang, H.; Zhang, N.; Hong, S. Theoretical Insights into Copper(i)–NHC-Catalyzed C–H Carboxylation of Terminal Alkynes with CO2: The Reaction Mechanisms and the Roles of NHC. RSC Adv. 2014, 4, 32457–32466.
- Trivedi, M.; Singh, G.; Kumar, A.; Rath, N.P. 1,1′-Bis(Di-Tert-Butylphosphino)Ferrocene Copper(i) Complex Catalyzed C–H Activation and Carboxylation of Terminal Alkynes. Dalton Trans. 2015, 44, 20874–20882.
- Bondarenko, G.N.; Dvurechenskaya, E.G.; Magommedov, E.S.; Beletskaya, I.P. Copper(0) Nanoparticles Supported on Al2O3 as Catalyst for Carboxylation of Terminal Alkynes. Catal. Lett. 2017, 147, 2570–2580.
- Wendling, T.; Risto, E.; Krause, T.; Gooßen, L.J. Salt-Free Strategy for the Insertion of CO2 into C−H Bonds: Catalytic Hydroxymethylation of Alkynes. Chem.–A Eur. J. 2018, 24, 6019–6024.
- Zhang, X.; Zhang, W.-Z.; Ren, X.; Zhang, L.-L.; Lu, X.-B. Ligand-Free Ag(I)-Catalyzed Carboxylation of Terminal Alkynes with CO2. Org. Lett. 2011, 13, 2402–2405.
- Arndt, M.; Risto, E.; Krause, T.; Gooßen, L.J. C–H Carboxylation of Terminal Alkynes Catalyzed by Low Loadings of Silver(I)/DMSO at Ambient CO2 Pressure. ChemCatChem 2012, 4, 484–487.
- Kim, S.H.; Kim, K.H.; Hong, S.H. Carbon Dioxide Capture and Use: Organic Synthesis Using Carbon Dioxide from Exhaust Gas. Angew. Chem. Int. Ed. 2014, 53, 771–774.
- Gaillard, S.; Cazin, C.S.J.; Nolan, S.P. N-Heterocyclic Carbene Gold(I) and Copper(I) Complexes in C–H Bond Activation. Acc. Chem. Res. 2012, 45, 778–787.
- Boogaerts, I.I.F.; Fortman, G.C.; Furst, M.R.L.; Cazin, C.S.J.; Nolan, S.P. Carboxylation of N–H/C–H Bonds Using N-Heterocyclic Carbene Copper(I) Complexes. Angew. Chem. Int. Ed. 2010, 49, 8674–8677.
- Zhang, L.; Cheng, J.; Ohishi, T.; Hou, Z. Copper-Catalyzed Direct Carboxylation of C–H Bonds with Carbon Dioxide. Angew. Chem. Int. Ed. 2010, 49, 8670–8673.
- Inomata, H.; Ogata, K.; Fukuzawa, S.; Hou, Z. Direct C–H Carboxylation with Carbon Dioxide Using 1,2,3-Triazol-5-Ylidene Copper(I) Complexes. Org. Lett. 2012, 14, 3986–3989.
- Boogaerts, I.I.F.; Nolan, S.P. Carboxylation of C−H Bonds Using N.-Heterocyclic Carbene Gold(I) Complexes. J. Am. Chem. Soc. 2010, 132, 8858–8859.
- Sasano, K.; Takaya, J.; Iwasawa, N. Palladium(II)-Catalyzed Direct Carboxylation of Alkenyl C–H Bonds with CO2. J. Am. Chem. Soc. 2013, 135, 10954–10957.
- Mizuno, H.; Takaya, J.; Iwasawa, N. Rhodium(I)-Catalyzed Direct Carboxylation of Arenes with CO2 via Chelation-Assisted C−H Bond Activation. J. Am. Chem. Soc. 2011, 133, 1251–1253.
- Kikuchi, S.; Sekine, K.; Ishida, T.; Yamada, T. C–C Bond Formation with Carbon Dioxide Promoted by a Silver Catalyst. Angew. Chem. Int. Ed. 2012, 51, 6989–6992.
- Sekine, K.; Takayanagi, A.; Kikuchi, S.; Yamada, T. Silver-Catalyzed C–C Bond Formation with Carbon Dioxide: Significant Synthesis of Dihydroisobenzofurans. Chem. Commun. 2013, 49, 11320–11322.
- Hoberg, H.; Minato, M. Palladium-Catalysed Dimerization of Isoprene with Carbon Dioxide in the Presence of Organotin Ethoxide and 1,8-DiazabicycloUndec-7-Ene (DBU). J. Organomet. Chem. 1991, 406, C25–C281991.
- Lejkowski, M.L.; Lindner, R.; Kageyama, T.; Bódizs, G.É.; Plessow, P.N.; Müller, I.B.; Schäfer, A.; Rominger, F.; Hofmann, P.; Futter, C.; et al. The First Catalytic Synthesis of an Acrylate from CO2 and an Alkene—A Rational Approach. Chem.–A Eur. J. 2012, 18, 14017–14025.
- Plessow, P.N.; Schäfer, A.; Limbach, M.; Hofmann, P. Acrylate Formation from CO2 and Ethylene Mediated by Nickel Complexes: A Theoretical Study. Organometallics 2014, 33, 3657–3668.
More
Information
Subjects:
Chemistry, Applied
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
973
Revisions:
2 times
(View History)
Update Date:
12 Dec 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No