Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Madhvi Sharma | -- | 3898 | 2023-12-06 10:47:49 | | | |
| 2 | Fanny Huang | Meta information modification | 3898 | 2023-12-08 04:01:57 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Sharma, M.; Sidhu, A.K.; Samota, M.K.; Gupta, M.; Koli, P.; Choudhary, M. Types of Histone Post-Translational Modifications. Encyclopedia. Available online: https://encyclopedia.pub/entry/52426 (accessed on 26 July 2026).
Sharma M, Sidhu AK, Samota MK, Gupta M, Koli P, Choudhary M. Types of Histone Post-Translational Modifications. Encyclopedia. Available at: https://encyclopedia.pub/entry/52426. Accessed July 26, 2026.
Sharma, Madhvi, Amanpreet K. Sidhu, Mahesh Kumar Samota, Mamta Gupta, Pushpendra Koli, Mukesh Choudhary. "Types of Histone Post-Translational Modifications" Encyclopedia, https://encyclopedia.pub/entry/52426 (accessed July 26, 2026).
Sharma, M., Sidhu, A.K., Samota, M.K., Gupta, M., Koli, P., & Choudhary, M. (2023, December 06). Types of Histone Post-Translational Modifications. In Encyclopedia. https://encyclopedia.pub/entry/52426
Sharma, Madhvi, et al. "Types of Histone Post-Translational Modifications." Encyclopedia. Web. 06 December, 2023.
Copy Citation
Abiotic stresses profoundly alter plant growth and development, resulting in yield losses. Plants have evolved adaptive mechanisms to combat these challenges, triggering intricate molecular responses to maintain tissue hydration and temperature stability during stress. A pivotal player in this defense is histone modification, governing gene expression in response to diverse environmental cues. Post-translational modifications (PTMs) of histone tails, including acetylation, phosphorylation, methylation, ubiquitination, and sumoylation, regulate transcription, DNA processes, and stress-related traits.
histone
ubiquitination
methylation
stress tolerance
post-translational modifications
1. Introduction
Plants face formidable challenges when confronted with abiotic stresses, necessitating precise adjustments across various physiological pathways. These adaptive responses encompass critical functions, such as photosynthesis, antioxidant regulation, water uptake, ion homeostasis, and osmolyte synthesis [1][2][3][4][5][6]. In the face of environmental constraints, these morphophysiological adaptations are underpinned by an intricate network of post-translational modifications (PTMs), orchestrated by multifaceted molecular mechanisms [7].
Plant adaptation to abiotic stresses hinges on the dynamic regulation of gene expression, encompassing a multitude of genes controlled by an array of transcription factors (TFs) and chromatin-associated factors. While substantial attention has been devoted to elucidating the roles of TFs, enzymes catalyzing covalent histone modifications, and chromatin remodeling complexes, the contribution of histone chaperones remains less explored, and their significance in this context remains enigmatic. Notably, protein phosphorylation serves as a well-established mechanism for transmitting stress signals, whereas emerging modifications, like S-nitrosylation, are still in their infancy. In the realm of PTMs, ubiquitin and SUMO conjugations emerged as central regulatory processes in eukaryotes [8][9]. These modifications, however, exert distinct effects contingent upon the transcriptional or translational stage at which the targeted transcript or protein is situated. Consequently, the interplay of these diverse PTMs collectively dictates the ultimate impact on the associated cellular processes and phenotypic outcomes.
Chromatin regulation emerges as a pivotal player in governing gene expression, with DNA methylation, histone modifications, and other genome activities intricately intertwined with adaptive responses to environmental challenges in plants [10][11][12]. An array of epigenetic mechanisms, including DNA methylation, histone modifications, ATP-dependent chromatin remodeling, incorporation of histone variants, and regulation by noncoding RNA, orchestrate the structure and function of chromatin [13][14]. Specifically, methylation, acetylation, phosphorylation, ubiquitination, and sumoylation represent a subset of PTMs occurring on the N-terminal tails of histone proteins. These modifications, collectively referred to as the “histone code,” are pivotal in establishing and perpetuating epigenetic memory, profoundly influencing chromatin structure and gene expression [15][16].
Within the intricate world of chromatin, the configuration significantly influences genome expression, largely regulated by the interplay of DNA methylation machinery and histone chaperones, also known as nucleosome assembly/disassembly factors. They facilitate nucleosome assembly and disassembly, impacting replication-dependent and replication-independent processes and collaborate with free histones to prevent indiscriminate histone–DNA interactions. Hence, histone chaperones play a critical role in modulating histone availability and the incorporation into nucleosomes [17][18].
In summary, this multifaceted interplay of plant responses to abiotic stresses, encompassing diverse pathways, PTMs, and chromatin dynamics, underscores the intricate web of adaptations crucial for plant survival in challenging environments (Figure 1).
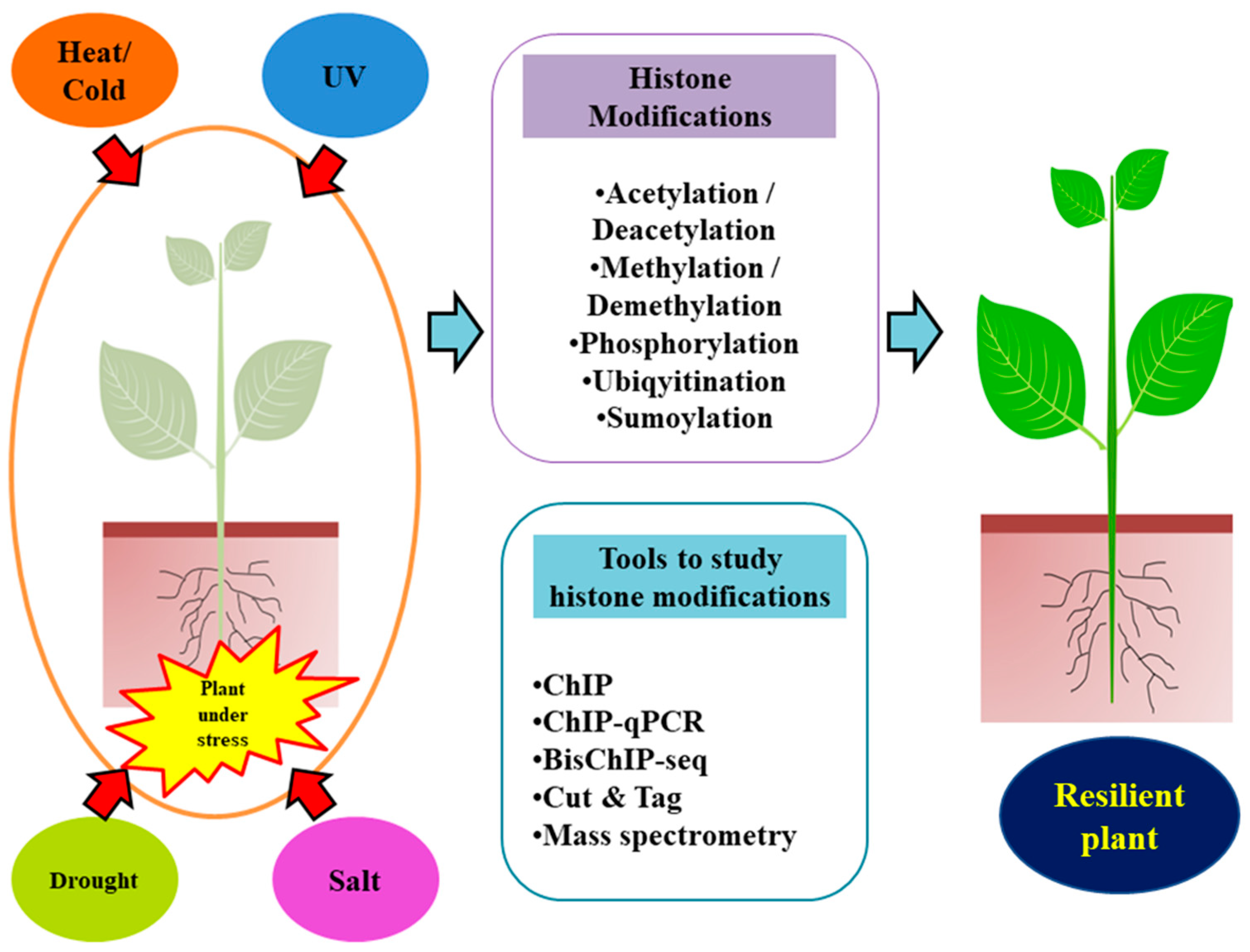
Figure 1. Plants are exposed to many abiotic stresses due to unpredictable climate changes, including cold and hot temperatures, drought, salinity, and ultraviolet radiation, all of which affect their productivity. Dynamic changes in histone modifications are important for the regulation of genes during environmental stress.
2. Epigenetic Memory and Chromatin Dynamics in Plant Stress Responses
Plants often experience unfavorable environmental conditions in growing habitats. Depending on the stress response, plants can retain information for a time after a previous stress (known as stress memory), so they can adapt more quickly to the same adversity in the future [19]. Epigenetic regulation is closely associated with the development of stress memories [19][20][21]. Research showed that stress treatment can alter the chromatin status of genes that respond to stress, and these changes persist after recovery and even in progeny [22][23][24]. Several factors play a role in regulating gene expression in eukaryotic cells, including the dynamic environment of chromatin. Epigenetic mechanisms, including covalent modifications to DNA and histone tails, are crucial for inducing favorable chromatin states that enable gene expression in response to stress and hence bestowing the plants with better adaptation. Several epigenetic factors cause chromatin modifications on exposure to various abiotic stresses in plants [25]. Each nucleosome contains a basic core histone octamer composed of four types of histone proteins, namely H2A, H2B, H3, and H4. In addition to these core histones, many histone variants are also reported. To date, only one variant is observed for H4, whereas several variants are encountered for H2A, H2B, and H3. These variants are believed to enhance the dynamics of nucleosome, diversity and play a crucial role in epigenetic genome regulation (Figure 2). The study of these variants can provide various clues in understanding the mechanism behind epigenetic genome regulation.
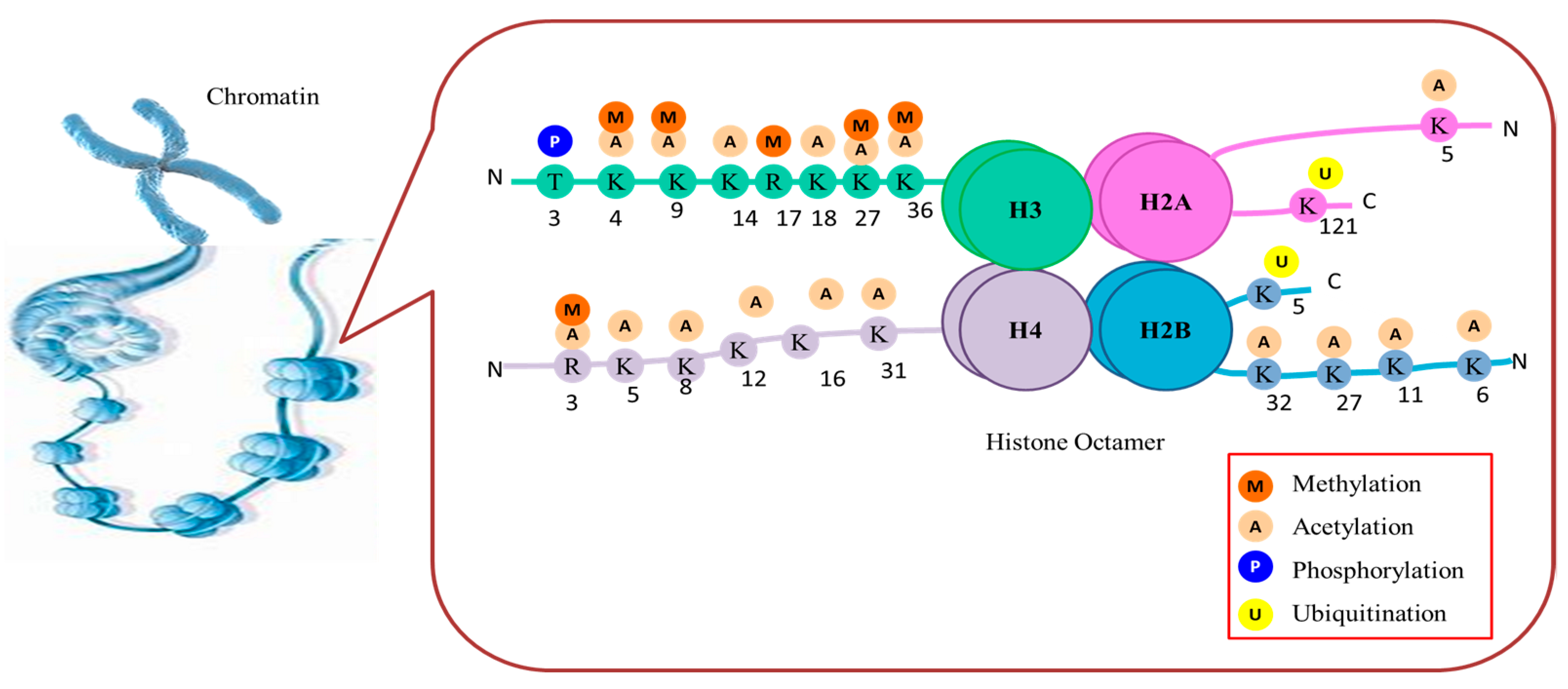
Figure 2. Histone modifications at different polypeptide amino acid residue locations and the group added by the different enzymes.
The majority of histone modifications take place at the N-terminal coil known as the histone tail rather than the globular C-terminal domain. Many basic amino acid residues, such as arginine and lysine, can be found in high concentrations in histones. The amino acid residues in histone tails can change chemically through acetylation, methylation, phosphorylation, and ubiquitination. The aforementioned modifications are believed to influence the functioning of genes located in proximity to core histones. Most histone-modifying enzymes are remarkably conserved across the plant realm, including well-researched and defined histone modifiers, such as histone methyltransferases (HMTs), histone demethylases (HDMs), histone deacetylases (HDACs), and histone acetyltransferases (HATs). Other less-studied enzymes include kinases, arginine deiminases, lysine- and arginine-specific methyltransferases, ubiquitinates, lysine and arginine-specific demethylases, and deubiquitinases. While there are studies on histone modifications and stress response in plants, this research seems to delve into the specific mechanisms and examples of how histone modifications influence stress tolerance in different plant species.
3. Molecular Sculptors: Types of Histone Post-Translational Modifications
3.1. Acetylation and Deacetylation
Histone acetylation involves a covalent alteration that enables the transfer of acetyl groups from acetyl CoA to the ε-amino group of lysine residues within histone molecules. This modification leads to the neutralization of lysine’s positive charge, subsequently diminishing the binding affinity between the modified histone and DNA [26][27]. Conversely, histone deacetylation is associated with a “closed” chromatin structure and the repression of gene activity [28]. Histone acetyltransferases and histone deacetylases are responsible for the reversible acetylation of histones (Table 1). At the outset, attention centered on identifying enzymes responsible for the introduction (“writers”) and removal (“erasers”) of these modifications [28]. Among these enzymatic agents, there are those that add chemical groups to histone tails or core domains, such as HATs, kinases, methyltransferases, and ubiquitinases. In contrast, there are enzymes that eliminate these modifications, including HDMs, phosphatases, deubiquitinases, and HDACs. Histone acetylation and deacetylation are critical regulators of plant stress tolerance. In Arabidopsis thaliana, a model plant, the HD2-type histone deacetylase HD2C has been found to negatively regulate drought tolerance. HD2C represses the expression of drought-responsive genes by deacetylating histones in their promoter regions, leading to reduced gene expression and impaired drought response in the plant [29]. Conversely, in Arabidopsis, certain genes positively regulate salt stress tolerance by enhancing the expression of salt-responsive genes. As an example, HD2C and HDA6 (histone deacetylase) work in tandem to govern the reaction to salt stress by controlling the expression of ABA-responsive genes including ABSCISIC ACID INSENSITIVE1 and ABSCISIC ACID INSENSITIVE2. The heightened expression of HDA705, a counterpart of Arabidopsis’ HDA6 or HDA7, led to diminished ABA levels and reduced salt stress tolerance in Arabidopsis seedlings [30]. Histone acetylation patterns also impact heat stress responses in wheat (Triticum aestivum). TaHAG1 is a gene that encodes a histone acetyltransferase that is orthologous to Arabidopsis AtHAG1/GCN5, and rice OsHAG702 promotes heat stress tolerance in wheat [31]. Moreover, histone acetylation can also modulate plant defense responses against pathogens. In Arabidopsis, the histone acetyltransferase HAG1 (HISTONE ACETYLTRANSFERASE OF THE GNAT FAMILY 1) is involved in activating defense genes against bacterial pathogens. HAG1 acetylates histones in the promoters of these genes, promoting their expression and enhancing the plant’s resistance to bacterial infection [32]. Furthermore, epigenetic memory and priming have been observed in maize (Zea mays), where exposure to brief drought stress induces changes in histone acetylation patterns, leading to improved drought tolerance upon subsequent stress [33][34].
Table 1. Histone modifications occurring under different abiotic stresses in plants. Special attention is given to the proteins acting as substrates for modifications, encompassing both histone and nonhistone proteins.
| Modification Type | Regulator Name | Crop | Stress Type | References |
|---|---|---|---|---|
| Acetylation Acetyltransferase | GCN5, AtHAC1 | Arabidopsis and Poplar | Heat, salinity, and drought (Chimeric dCas9 HAT) | [35][36][37] |
| Acetylation Acetyltransferase | HAT, AREB | Poplar | Drought | [36] |
| Deacetylation (Deactylase) | HDAC, IDS1 | Rice | Salinity | [38] |
| Deactylation (Deactylase) | HDAC, MYB96 | Arabidopsis | Drought | [39] |
| Deactylation Deacetylase |
HDA9, HDA15, HDA705, BdHD1, HD2C | Arabidopsis, Rice, and Brachypodium | Drought, salinity cold, and heat | [28][39][40][41][42] |
| H3K9 acetylation | HAT, GCN5, ZmEXPANSIN-B2 | Maize | Salinity | [43] |
| H3 hyperacetylation | HAT genes, OsHAT genes | Rice | Drought | [44][45] |
| Deactylation (Deactylase) | HDA9, CYP707A1, CYP707A2 | Arabidopsis | Drought | [46] |
| Deactylation (Deactylase) | BdHD1, WRKY24 | Purple False Brome or Stiff Brome | Drought | [47] |
| Acetylation | AtHAC1 | Arabidopsis | Heat | [48] |
| Acetylation | MYST, ELP3, GCN5 | Barley | Drought | [49] |
| Acetylation | OsHAC703, OsHAG703, OsHAF701, OsHAM70 |
Rice | Drought | [50] |
| Deacetylation (Deactylase) | 84KHDA903 | Tobacco | Drought | [51] |
| Deacetylation (Deactylase) | HD2C, HSFA3, HSFC1, HSP10 | Arabidopsis | Heat | [42] |
| Acetylation | GCN5, PtrNAC006, | Black Cottonwood Tree | Drought | [52] |
| Recruiter | MYB96, IDS1, AREB1 | Arabidopsis, Rice, and Poplar | Drought and salinity | [38][39][52] |
| Methylation Methyltransferase | ATX1, ATX4/5 | Arabidopsis | Drought | [53][54] |
| Demethylation Demethylase |
JMJ17 | Arabidopsis | Drought | [55] |
| Trimethylation | HMT | Arabidopsis | Gamma irradiation | [56] |
| Ubiquitination Ubiquitinase | HUB1/2, AtHUB2, OsHUB2 |
Arabidopsis, Cotton, and Rice | Salinity and drought | [57][58][59][60][61] |
| Phosphorylation Kinase | MLK1/2 | Arabidopsis | Drought and salinity | [62][63][64] |
| Ubiquitinase and deubiquitinase | H2B | Rice | Drought | [61] |
| Sumoylation | SUMO E3 ligase (AtSIZ1, OsSIZ1) | Arabidopsis and Rice | Heat | [65][66][67] |
| Ubiquitination | SNAC1 gene | Wheat | Salt and drought | [68] |
3.2. Methylation and Demethylation
The equilibrium of a specific covalent histone modification’s steady-state level is governed by a delicate interplay between enzymes that facilitate its addition and those that facilitate its removal. Protein arginine methyl transferases (PRMTs) and histone lysine methyl transferases (KMTs) mark lysine and arginine with methyl groups, respectively. Histone lysine methylation occurs primarily at Lys4, Lys9, Lys27, and Lys36 of H3 in Arabidopsis [69][70][71]. Overall, histone methylation at H3K9 and H3K27 is connected with gene silencing, whereas methylation at H3K4 and H3K36 is tied to gene activation. Based on the number of methyl groups added to histone molecules, methylation is classified as mono-, di-, or trimethylation, and gene expression varies depending on the level of modification [26]. For instance, in Arabidopsis, the trimethylation of Lys27 (H3K27me3) leads to gene expression repression, while the trimethylation of Lys4 (H3K4me3) leads to the activation of gene transcription [72]. These methylation marks can be eliminated by histone demethylases (HDMs) with the assistance of various cofactors in plants, including lysine-specific demethylase 1 (LSD1) and the Jumonji C domain-containing protein (JMJ) [73][74][75] (as shown in Table 1). Methylation occurs on lysine and/or arginine amino acids within histones, altering their interaction with reader proteins and consequently influencing chromatin structure, which in turn determines whether transcription is activated or repressed. In Arabidopsis, repressive histone methylation modifications, such as H4R3me2, H3K9me2/3, and H3K27me3, are observed, whereas active histone methylation modifications, such as H4R3me2, H3K4me3, and H3K36me2/3, are evident [76][77]. Unlike acetylation, which damages the electrostatic properties of histone proteins, histone methylation preserves the electron charge of lysine. The histone methylation mark’s mode of action (Tran’s effects) is presumably coordinated through hydrophobicity; however, this assertion is not absolute, and other hypotheses have been put forth. Moreover, a variety of histone H3 lysine residue methylation holds significance in plants, encompassing repressive dimethylation at Lys9 (H3K9me2) and trimethylation at Lys27 (H3K27me3), along with permissive trimethylation at Lys4 (H3K4me3) and Lys36 (H3K36me3) [78]. Additionally, plants exhibit two arginine methylation sites (H3R17 and H4R3) and five lysine methylation sites (H3K4, H3K9, H3K27, H3K36, and H4R20), each potentially holding a distinctive role in the orchestration of transcriptional regulation [76]. For instance, Polycomb Repressive Complex 2 (PRC2) having an HMT unit mediates the histone modification H3K27me3, which was reported to be associated with gene repression in eukaryotes [79][80]. The identification of these PRC2 complexes originally occurred in Drosophila as Hox gene regulators, subsequently revealing homologous PRC2 subunits within plants and animals [81][82].
Histone demethylation involves the removal of methyl groups from specific lysine or arginine residues on histone proteins. This modification can have a profound impact on chromatin structure and, subsequently, on the transcriptional regulation of genes involved in plant abiotic stress tolerance. Histone demethylation can either activate or suppress the transcription of stress-related genes [83]. The specific effect depends on the histone residue being demethylated and the enzyme responsible for the demethylation. For example, the removal of methyl groups from histone H3 lysine 4 (H3K4) is associated with gene activation, while the demethylation of histone H3 lysine 9 (H3K9) or histone H3 lysine 27 (H3K27) is linked to gene repression [84]. The demethylation of histone H3K4 and histone H3 lysine 36 (H3K36) near the promoter regions of stress-responsive genes can lead to their activation [85]. This allows plants to mount a rapid response to abiotic stress conditions by increasing the expression of genes involved in stress tolerance, such as those encoding heat shock proteins, antioxidant enzymes, and osmoprotectants.
3.3. Phosphorylation
Phosphorylation, the process of adding a phosphate group (PO43−) to a molecule, is orchestrated by specific protein kinases, while phosphatases facilitate phosphate group removal [86]. Within this complex landscape, histones, the proteins around which DNA is wound in chromatin, are subject to dynamic phosphorylation events that primarily target threonine (Thr), serine (Ser), and tyrosine (Tyr) residues [87]. Histone phosphorylation often responds to signals, such as DNA damage, extracellular cues, or cell division progression. In the context of histone modifications, phosphorylation on histone H3 is of particular interest, with prominent sites including Ser 10, Ser 28 (H3S10ph and H3S28ph), Thr 3, and Thr 11 (H3T3ph and H3T11ph) [88]. The importance of these modifications is underscored by observations in Arabidopsis where a mutant deficient in closely related Ser/Thr protein kinases (At3g03940 and At5g18190) displayed heightened sensitivity to osmotic and salt stress, along with dwarfism. Intriguingly, this mutant exhibited a significantly reduced level of phosphorylated histone H3 at Thr 3 (H3T3ph). Genome-wide assessments unveiled an elevation in H3T3ph at Thr 3 within pericentromeric regions of Arabidopsis thaliana under osmotic stress conditions [89][90][91].
This phosphorylation of histone H3 plays a crucial role in various cellular processes, including chromosome segregation, chromatin condensation, and transcriptional regulation [92]. Additionally, histone H2AX phosphorylation at Ser 129, known as γH2AX, is a pivotal player in the DNA damage response and repair. Rapid phosphorylation of H2AX occurs at sites of double-strand DNA breaks, catalyzed by PI3K kinases. This modification represents one of the earliest and most discernible post-translational signals triggered by DNA damage [93][94][95]. The Arabidopsis genome houses an extensive array of over 1000 protein kinases, including calcium-dependent protein kinases (CPKs), mitogen-activated protein kinases (MAPKs), receptor-like kinases (RLKs), and sucrose nonfermenting-related kinases (SnRKs). Alongside these, it hosts approximately 150 protein phosphatases, encompassing type 1 (PP1) and type 2A phosphatases, the protein tyrosine phosphatase family, and the metal-dependent protein phosphatase family [96]. Specific MAPKs, such as MPK3, MPK4, and MPK6, have been identified as key players in the phosphorylation of HSFA4A at Ser-309. This intricate regulatory mechanism serves to modulate the activity of the heat-activated factor HSFA4A. Elevated temperatures and increased salinity both trigger HSFA4A activation, and their combined action influences the accessibility of HSFA4A-binding sites within the promoters of target genes, like ZAT12, HSP17.6A, and WRKY30. This finely tuned orchestration governs the plant’s response to abiotic stresses [97]. Histone phosphorylation, influenced by the activity of MAPKs and the action of transcription factors, like HSFA4A, can further modulate gene expression. It can make the chromatin structure more permissive or restrictive for the transcription of the genes involved in heat stress response. Overall, the connection among MAPKs, HSFA4A, and histone phosphorylation lies in the signaling pathway activated by heat stress in plants. MAPKs are involved in the early signaling events of heat stress response, phosphorylating HSFA4A, which, in turn, activates the transcription of stress-responsive genes. Histone phosphorylation can then act as an additional layer of regulation, ensuring that the right genes are expressed in response to heat stress, contributing to the plant’s adaption and survival under adverse conditions.
Furthermore, rising temperatures induce the nuclear translocation of the BR-regulated transcription factor, brassinazole-resistant 1 (BZR1). In the nucleus, BZR1 binds to the promoter of PIF4 (phytochrome-interacting factor), leading to cell elongation [98][99]. Notably, histone H2A phosphorylation at Ser 95 in Arabidopsis, catalyzed by MuTP9-like kinases, such as MLK4 and MLK3, has been shown to promote flowering time and enhance the deposition of H2A.Z [92].
In summary, histone phosphorylation is a dynamic and finely regulated process involving a delicate balance between kinases and phosphatases. These kinases engage diverse targets to orchestrate distinct temperature-signaling pathways, thereby governing the plant’s responses to a range of temperatures from elevated to exceedingly high. This intricate regulatory network underscores the pivotal role of histone phosphorylation in plant stress tolerance.
3.4. Ubiquitination
The enzymatic process involving the transfer of one or more ubiquitin monomers to the protein substrate is termed ubiquitination (or ubiquitylation) [100]. Monoubiquitination brings about alterations in the subcellular localization, biochemical properties, or molecular functions of target proteins. In contrast, polyubiquitination serves as a signal for proteasome-mediated degradation [101]. This modification occurs specifically on lysine residues 119 of H2A and 120 of H2B. The ubiquitination process primarily encompasses three stages: ubiquitin protein activation, ubiquitin conjugation, and ubiquitin ligation. These steps necessitate the addition of ubiquitin to the target protein and are executed by their respective ubiquitin-activating enzymes (E1s), ubiquitin-conjugating enzymes (E2s), and ubiquitin ligases (E3s) [102]. Among the ubiquitination enzymes, the most abundant are E3 ubiquitin ligases. In rice and arabidopsis, a drought stress tolerance cascade involving 3 ubiquitin ligase OsPUB67 and its target protein OsDIS1 and OsRZP34 has been well explored. OsPUB67 positively regulates drought tolerance by promoting improved scavenging of ROS and closure of stomata under drought, whereas OsDIS1 and OsRZP34 are negative regulators that open the stomata under drought stress. OsPUB67 ubiquitinates the targets OsRZFP34 and OsDIS1 for proteolysis-mediated degradation, leading to an increased level of stomatal closure [103][104][105].
In a study conducted by Tripathi et al. [106], researchers delved into the physiological role of OsNAPL6, a putative rice NAP superfamily histone chaperone responsive to stress. This nuclear-localized histone chaperone possesses the ability to form nucleosome-like structures. Through a combination of overexpression and knockdown strategies, they unveiled a positive connection between OsNAPL6 expression levels and the plant’s ability to adapt to diverse abiotic stresses. Their investigation, involving comparative transcriptome profiling and promoter recruitment analyses, highlighted OsNAPL6′s role in stress response by influencing the expression of various genes associated with diverse functions. Many ubiquitin ligases were discovered in stress-related mutants, accounting for regulatory roles in abiotic stress tolerance, particularly drought tolerance, in Arabidopsis and crop species [107]. For example, a small regulatory protein (E3 ubiquitin ligase RING FINGER 1) imparts drought tolerance in durum wheat [1]. Furthermore, H2A (H2Aub) and H2B (H2Bub) monoubiquitination affect transcription in eukaryotes both actively and repressively. In Arabidopsis, H2AK121 monoubiquitination occurs independently of H3K27me3, but it does not cooperate with PRC2, which is required to maintain H3K27me3 [108][109].
In addition to DNA methylation, histone H3 heterochromatic methylation is required for H2B deubiquitination. H2B monoubiquitination, in more detail, activates transcription via the presence of H3K4me3 [110]. The experimental study in Arabidopsis observed a direct link between H2B monoubiquitination and plant immunity because they found that pathogen infection increases the H2B monoubiquitination at R-gene SNC1 [111]. Likewise, in tomatoes, the presence of the histone H2B monoubiquitination enzymes HUB1, HUB2, SlHUB1, and SlHUB2 has been identified as a contributor to resistance against B. cinerea. Their role is likely centered on maintaining a balance between the signaling pathways governed by SA and JA/ethylene. The expression of the gene governing the SA (salicylic acid)-mediated signaling pathway was significantly upregulated, while the expression of genes in the JA (Jasmonic acid)/ethylene pathway were critically downregulated. This interaction establishes a crucial connection between plant immunity and the process of ubiquitination [112]. According to research on wheat’s histone modification, TaHUB2 (the second histone H2B monoubiquitination enzyme) interacts with TaH2B in vernalization pathways and may be necessary for wheat heading [113][114]. TaHUB2 serves as a ubiquitin RING-type E3 ligase. In Arabidopsis, the HUB1 (histone monoubiquitination 1, gene encoding E3 ligase) mediated H2Bub1 (histone H2B monoubiquitination) has been proven as a mechanism regulating auxin biosynthesis [115]. A recent investigation revealed that GhUbox8, an E3 ligase of the U-box-type, collaboratively modulates histone monoubiquitination of H2A and H2B in conjunction with GhUBC2L, an E2 enzyme. This concerted action governs the expression of genes associated with cell cycle progression and organ development [116]. This discovery reinforces the significance of histone monoubiquitination in orchestrating the regulation of organ size within the context of cotton.
3.5. Sumoylation
The Small Ubiquitin-like Modifier (SUMO) protein family engages in a process of attaching to and detaching from various proteins within the cell, thereby modulating the functionality of these proteins. In the research conducted by Shiio et al. [117], it was demonstrated that SUMO can modify H4, leading to the recruitment of HDAC and HP1 proteins. This recruitment subsequently results in the suppression of transcriptional activity through competitive interactions with other active marks, such as methylation, acetylation, and ubiquitination. In Arabidopsis, there are instances of SUMOylated chromatin modifiers and components, such as HDA19, H2B, and GCN5, and the deubiquitinating enzyme UBP26, which functions to remove ubiquitin attached to H2B. As an illustration, exposure to heat stress (37 °C for 30 min) induces a reduction in H2B SUMOylation while simultaneously increasing SUMOylation in GCN5 HAT [118]. This phenomenon plays a pivotal role in modulating DNA methylation patterns during heat stress within Arabidopsis, as the SUMOylation of histone acetylases/deacetylases facilitates the conversion of euchromatic regions into heterochromatic ones [119]. In the Arabidopsis context, SUMOylation associated with chromatin serves as a pivotal switch, regulating the transcriptional balance between plant development and the response to heat stress. These SUMO-mediated changes in chromatin signals lead to the upregulation of heat-responsive genes and the downregulation of growth-related genes. Notably, the inactivation of the SUMO ligase gene SIZ1 resulted in reduced SUMO signals on chromatin and a corresponding attenuation of rapid transcriptional responses to heat stress [120][121].
Through a comprehensive approach encompassing proteomic and interactome analyses, a total of 350 SUMO targets and SUMO-interacting proteins were unveiled in Arabidopsis. This exploration extended to those entities that exhibited accumulation subsequent to subjecting plants to conditions of heat and oxidative stress, employing three distinct research methodologies [118][122]. The majority of SUMO substrates control nuclear activities, such as DNA methylation, DNA repair, RNA processing, chromatin remodeling, and gene transcription [123][124]. Genes, such as COP1 and SIZ1, serve as typical examples of proteins that dynamically regulate high-temperature-induced growth responses via SUMOylation or ubiquitination. Alternatively, SUMO can be subjected to controlled proteolysis via the generation of poly-SUMO chains mediated by SUMO ligases like PIAL1/2. This process is paralleled by the regulation of SUMO through the activity of SUMO ligases, such as PIAL1/2, which orchestrate the assembly of poly-SUMO chains. This polymeric configuration serves as a docking site for STUbLs, facilitating the conjugation of polyubiquitin chains to both SUMO and the target protein. Consequently, this orchestrated interaction primes the 26S proteasome for targeted degradation. This intricate interplay elevates the scope of transcriptional control to a greater level [125].
References
- Guerra, D.; Crosatti, C.; Khoshro, H.H.; Mastrangelo, A.M.; Mica, E.; Mazzucotelli, E. Post-transcriptional and post-translational regulations of drought and heat response in plants: A spider’s web of mechanisms. Front. Plant Sci. 2015, 6, 57.
- Gupta, N.K.; Shavrukov, Y.; Singhal, R.K.; Borisjuk, N. (Eds.) Multiple Abiotic Stress Tolerances in Higher Plants: Addressing the Growing Challenges; CRC Press: Boca Raton, FL, USA, 2023.
- Ahammed, G.J.; Li, X.; Liu, A.; Chen, S. Brassinosteroids in plant tolerance to abiotic stress. J. Plant Growth Regul. 2020, 39, 1451–1464.
- Jerome Jeyakumar, J.M.; Ali, A.; Wang, W.M.; Thiruvengadam, M. Characterizing the role of the miR156-SPL Network in plant development and stress response. Plants 2020, 9, 1206.
- Lohani, N.; Jain, D.; Singh, M.B.; Bhalla, P.L. Engineering multiple abiotic stress tolerance in canola, Brassica napus. Front. Plant Sci. 2020, 11, 3.
- Ghosh, U.K.; Islam, M.N.; Siddiqui, M.N.; Khan, M.A.R. Understanding the roles of osmolytes for acclimatizing plants to changing environment: A review of potential mechanism. Plant Signal. Behav. 2021, 16, 1913306.
- Ramazi, S.; Zahiri, J. Posttranslational modifications in proteins: Resources, tools and prediction methods. Database 2021, 2021, baab012.
- Ghimire, S.; Tang, X.; Zhang, N.; Liu, W.; Si, H. SUMO and SUMOylation in plant abiotic stress. Plant Growth Regul. 2020, 91, 317–325.
- Ran, H.; Li, C.; Zhang, M.; Zhong, J.; Wang, H. Neglected PTM in Animal Adipogenesis: E3-mediated Ubiquitination. Gene 2023, 878, 147574.
- Rehman, M.; Tanti, B. Understanding epigenetic modifications in response to abiotic stresses in plants. Biocatal. Agric. Biotechnol. 2020, 27, 101673.
- Akhter, Z.; Bi, Z.; Ali, K.; Sun, C.; Fiaz, S.; Haider, F.U.; Bai, J. In Response to Abiotic Stress, DNA Methylation Confers Epigenetic Changes in Plants. Plants 2021, 10, 1096.
- Vyse, K.; Faivre, L.; Romich, M.; Pagter, M.; Schubert, D.; Hincha, D.K.; Zuther, E. Transcriptional and post-transcriptional regulation and transcriptional memory of chromatin regulators in response to low temperature. Front. Plant Sci. 2020, 11, 39.
- Grisan, V. Regulation of the Transcription Cycle by Co-Ordinate Interaction of Atp-Dependent Chromatin Remodeling and Histone Post-Translational Modifications. Doctoral Dissertation, University of Birmingham, Birmingham, UK, 2019.
- Luo, M.; Ríos, G.; Sarnowski, T.J.; Zhang, S.; Mantri, N.; Charron, J.B.; Libault, M. New insights into mechanisms of epigenetic modifiers in plant growth and development. Front. Plant Sci. 2020, 10, 1661.
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080.
- Ali, F.; Dar, J.S.; Magray, A.R.; Ganai, B.A.; Chishti, M.Z. Posttranslational Modifications of Proteins and Their Role in Biological Processes and Associated Diseases. In Protein Modificomics; Academic Press: Cambridge, MA, USA, 2019; pp. 1–35.
- Duc, C.; Benoit, M.; Le Goff, S.; Simon, L.; Poulet, A.; Cotterell, S.; Tatout, C.; Probst, A.V. The histone chaperone complex HIR maintains nucleosome occupancy and counterbalances impaired histone deposition in CAF-1 complex mutants. Plant J. 2015, 81, 707–722.
- Pardal, A.J.; Fernandes-Duarte, F.; Bowman, A.J. The histone chaperoning pathway: From ribosome to nucleosome. Essays Biochem. 2019, 63, 29–43.
- Chaudhry, U.K.; Gökçe, Z.N.; Gökçe, A.F. Salt stress and plant molecular responses. In Plant Defense Mechanisms; Intech Open: London, UK, 2022; p. 105.
- Lamke, J.; Baurle, I. Epigenetic and chromatin-based mechanisms in environmental stress adaptation and stress memory in plants. Genome Biol. 2017, 18, 124.
- Friedrich, T.; Faivre, L.; Baurle, I.; Schubert, D. Chromatin-based mechanisms of temperature memory in plants. Plant Cell Environ. 2019, 42, 762–770.
- Avramova, Z. Transcriptional ‘memory’ of a stress: Transient chromatin and memory (epigenetic) marks at stress-response genes. Plant J. 2015, 83, 149–159.
- Hilker, M.; Schwachtje, J.; Baier, M.; Balazadeh, S.; Baurle, I.; Geiselhardt, S.; Hincha, D.K.; Kunze, R.; Mueller-Roeber, B.; Rillig, M.C.; et al. Priming and memory of stress responses in organisms lacking a nervous system. Biol. Rev. 2016, 91, 1118–1133.
- Yang, H.; Berry, S.; Olsson, T.S.G.; Hartley, M.; Howard, M.; Dean, C. Distinct Phases of Polycomb Silencing to Hold Epigenetic Memory of Cold in Arabidopsis. Science 2017, 357, 1142–1145.
- Chang, Y.N.; Zhu, C.; Jiang, J.; Zhang, H.; Zhu, J.K.; Duan, C.G. Epigenetic Regulation in Plant Abiotic Stress Responses. J. Integr. Plant Biol. 2020, 62, 563–580.
- Zhao, T.; Zhan, Z.; Jiang, D. Histone Modifications and Their Regulatory Roles in Plant Development and Environmental Memory. J. Genet. Genom. 2019; in press.
- Csizmok, V.; Forman-Kay, J.D. Complex Regulatory Mechanisms Mediated by the Interplay of Multiple Post-Translational Modifications. Curr. Opin. Struct. Biol. 2018, 48, 58–67.
- Park, J.; Lim, C.J.; Shen, M.; Park, H.J.; Cha, J.Y.; Iniesto, E.; Rubio, V.; Mengiste, T.; Zhu, J.K.; Bressan, R.A.; et al. Epigenetic Switch from Repressive to Permissive Chromatin in Response to Cold Stress. Proc. Natl. Acad. Sci. USA 2018, 115, E5400–E5409.
- Tahir, M.S.; Karagiannis, J.; Tian, L. HD2A and HD2C Co-Regulate Drought Stress Response by Modulating Stomatal Closure and Root Growth in Arabidopsis. Front. Plant Sci. 2022, 13, 1062722.
- Luo, M.; Wang, Y.Y.; Liu, X.; Yang, S.; Lu, Q.; Cui, Y.; Wu, K. HD2C interacts with HDA6 and is involved in ABA and salt stress response in Arabidopsis. J. Exp. Bot. 2012, 63, 3297–3306.
- Lin, J.; Song, N.; Liu, D.; Liu, X.; Chu, W.; Li, J.; Chang, S.; Liu, Z.; Chen, Y.; Yang, Q.; et al. Histone Acetyltransferase TaHAG1 Interacts with TaNACL to Promote Heat Stress Tolerance in Wheat. Plant Biotechnol. J. 2022, 20, 1645–1647.
- Rymen, B.; Kawamura, A.; Lambolez, A.; Inagaki, S.; Takebayashi, A.; Iwase, A.; Sakamoto, Y.; Sako, K.; Favero, D.S.; Ikeuchi, M.; et al. Histone Acetylation Orchestrates Wound-Induced Transcriptional Activation and Cellular Reprogramming in Arabidopsis. Commun. Biol. 2019, 2, 404.
- Kang, H.; Fan, T.; Wu, J.; Zhu, Y.; Shen, W.H. Histone Modification and Chromatin Remodeling in Plant Response to Pathogens. Front. Plant Sci. 2022, 13, 986940.
- Forestan, C.; Farinati, S.; Zambelli, F.; Pavesi, G.; Rossi, V.; Varotto, S. Epigenetic Signatures of Stress Adaptation and Flowering Regulation in Response to Extended Drought and Recovery in Zea mays. Plant Cell Environ. 2020, 43, 55–75.
- Zhong, R.; Cui, D.; Ye, Z.H. A group of Populus trichocarpa DUF231 proteins exhibit differential O-acetyltransferase activities toward xylan. PLoS ONE 2018, 13, e0194532.
- Li, S.; Lin, Y.J.; Wang, P.; Zhang, B.; Li, M.; Chen, S.; Shi, R.; Tunlaya-Anukit, S.; Liu, X.; Wang, Z.; et al. The AREB1 Transcription Factor Influences Histone Acetylation to Regulate Drought Responses and Tolerance in Populus trichocarpa. Plant Cell. 2019, 31, 663–686.
- Roca Paixão, J.F.; Gillet, F.X.; Ribeiro, T.P.; Bournaud, C.; Lourenço-Tessutti, I.T.; Noriega, D.D.; Melo, B.P.; de Almeida-Engler, J.; Grossi-de-Sa, M.F. Improved Drought Stress Tolerance in Arabidopsis by CRISPR/dCas9 Fusion with a Histone Acetyl Transferase. Sci. Rep. 2019, 9, 8080.
- Cheng, X.; Zhang, S.; Tao, W.; Zhang, X.; Liu, J.; Sun, J.; Zhang, H.; Pu, L.; Huang, R.; Chen, T. INDETERMINATE SPIKELET1 Recruits Histone Deacetylase and a Transcriptional Repression Complex to Regulate Rice Salt Tolerance. Plant Physiol. 2018, 178, 824–837.
- Lee, H.G.; Seo, P.J. MYB96 Recruits the HDA15 Protein to Suppress Negative Regulators of ABA Signaling in Arabidopsis. Nat. Commun. 2019, 10, 1713.
- Zheng, Y.; Fornelli, L.; Compton, P.D.; Sharma, S.; Canterbury, J.; Mullen, C.; Kelleher, N.L. Unabridged Analysis of Human Histone H3 by Differential Top-Down Mass Spectrometry Reveals Hypermethylated Proteoforms from MMSET/NSD2 Overexpression. Mol. Cell Proteom. 2016, 15, 776–790.
- Song, Y.; Wang, R.; Li, L.W.; Liu, X.; Wang, Y.F.; Wang, Q.X.; Zhang, Q. Long Non-coding RNA HOTAIR Mediates the Switching of Histone H3 Lysine 27 Acetylation to Methylation to Promote Epithelial-to-Mesenchymal Transition in Gastric Cancer. Int. J. Oncol. 2019, 54, 77–86.
- Buszewicz, D.; Archacki, R.; Palusiński, A.; Kotliński, M.; Fogtman, A.; Iwanicka-Nowicka, R.; Sosnowska, K.; Kuciński, J.; Pupel, P.; Olędzki, J.; et al. HD2C Histone Deacetylase and a SWI/SNF Chromatin Remodeling Complex Interact and Both Are Involved in Mediating the Heat Stress Response in Arabidopsis. Plant Cell Environ. 2016, 39, 2108–2122.
- Li, H.; Yan, S.; Zhao, L.; Tan, J.; Zhang, Q.; Gao, F.; Wang, P.; Hou, H.; Li, L. Histone Acetylation Associated Up-Regulation of the Cell Wall Related Genes Is Involved in Salt Stress Induced Maize Root Swelling. BMC Plant Biol. 2014, 14, 105.
- Eom, S.H.; Hyun, T.K. Histone Acetyltransferases (HATs) in Chinese Cabbage: Insights from Histone H3 Acetylation and Expression Profiling of HATs in Response to Abiotic Stresses. J. Am. Soc. Hortic. Sci. 2018, 143, 296–303.
- Fang, H.; Liu, X.; Thorn, G.; Duan, J.; Tian, L. Expression Analysis of Histone Acetyltransferases in Rice under Drought Stress. Biochem. Biophys. Res. Commun. 2014, 443, 400–405.
- Baek, D.; Shin, G.; Kim, M.C.; Shen, M.; Lee, S.Y.; Yun, D.J. Histone Deacetylase HDA9 with ABI4 Contributes to Abscisic Acid Homeostasis in Drought Stress Response. Front. Plant Sci. 2020, 11, 143.
- Song, J.; Henry, H.A.L.; Tian, L. Brachypodium Histone Deacetylase BdHD1 Positively Regulates ABA and Drought Stress Responses. Plant Sci. 2019, 283, 355–365.
- Ivanova, T.; Dincheva, I.; Badjakov, I.; Iantcheva, A. Transcriptional and Metabolic Profiling of Arabidopsis thaliana Transgenic Plants Expressing Histone Acetyltransferase HAC1 upon the Application of Abiotic Stress—Salt and Low Temperature. Metabolites 2023, 13, 994.
- Papaefthimiou, D.; Likotrafiti, E.; Kapazoglou, A.; Bladenopoulos, K.; Tsaftaris, A. Epigenetic Chromatin Modifiers in Barley: III. Isolation and Characterization of the Barley GNAT-MYST Family of Histone Acetyltransferases and Responses to Exogenous ABA. Plant Physiol. Biochem. 2010, 48, 98–107.
- Billah, M.; Aktar, S.; Brestic, M.; Zivcak, M.; Khaldun, A.B.M.; Uddin, M.S.; Hossain, A. Progressive Genomic Approaches to Explore Drought- and Salt-Induced Oxidative Stress Responses in Plants under Changing Climate. Plants 2021, 10, 1910.
- Ma, X.; Zhang, B.; Liu, C.; Tong, B.; Guan, T.; Xia, D. Expression of a Populus Histone Deacetylase Gene 84KHDA903 in Tobacco Enhances Drought Tolerance. Plant Sci. 2017, 265, 1–11.
- Li, S.; He, X.; Gao, Y.; Zhou, C.; Chiang, V.L.; Li, W. Histone acetylation changes in plant response to drought stress. Genes 2021, 12, 1409.
- Ding, J.; Shen, J.; Mao, H.; Xie, W.; Li, X.; Zhang, Q. RNA-Directed DNA Methylation Is Involved in Regulating Photoperiod-Sensitive Male Sterility in Rice. Mol. Plant. 2012, 5, 1210–1216.
- Liu, J.; Shi, Y.; Yang, S. Insights into the Regulation of C-Repeat Binding Factors in Plant Cold Signaling. J. Integr. Plant Biol. 2018, 60, 780–795.
- Huang, S.; Zhang, A.; Jin, J.B.; Zhao, B.; Wang, T.J.; Wu, Y.; Xu, Z.Y. Arabidopsis Histone H3K4 Demethylase JMJ 17 Functions in Dehydration Stress Response. N. Phytol. 2019, 223, 1372–1387.
- Mozgova, I.; Mikulski, P.; Pecinka, A.; Farrona, S. Epigenetic Mechanisms of Abiotic Stress Response and Memory in Plants. In Epigenetics in Plants of Agronomic Importance: Fundamentals and Applications: Transcriptional Regulation and Chromatin Remodelling in Plants; Springer: Berlin/Heidelberg, Germany, 2019; pp. 1–64.
- Zhou, S.; Chen, Q.; Sun, Y.; Li, Y. Histone H2B Monoubiquitination Regulates Salt Stress-Induced Microtubule Depolymerization in Arabidopsis. Plant Cell Environ. 2017, 40, 1512–1530.
- Zhou, Y.; Romero-Campero, F.J.; Gómez-Zambrano, Á.; Turck, F.; Calonje, M. H2A Mono ubiquitination in Arabidopsis thaliana Is Generally Independent of LHP1 and PRC2 Activity. Genome Biol. 2017, 18, 6.
- Zarreen, F.; Karim, M.J.; Chakraborty, S. The Diverse Roles of Histone H2B Mono ubiquitination in the Life of Plants. J. Exp. Bot. 2022, 73, 3854–3865.
- Chen, K.; Tang, W.S.; Zhou, Y.B.; Xu, Z.S.; Chen, J.; Ma, Y.Z.; Li, H.Y. Overexpression of GmUBC9 Gene Enhances Plant Drought Resistance and Affects Flowering Time via Histone H2B Mono ubiquitination. Front. Plant Sci. 2020, 11, 555794.
- Ma, S.; Tang, N.; Li, X.; Xie, Y.; Xiang, D.; Fu, J.; Shen, J.; Yang, J.; Tu, H.; Li, X.; et al. Reversible Histone H2B Monoubiquitination Fine-Tunes Abscisic Acid Signaling and Drought Response in Rice. Mol. Plant. 2019, 12, 263–277.
- Wang, Z.; Casas-Mollano, J.A.; Xu, J.; Riethoven, J.-J.M.; Zhang, C.; Cerutti, H. Osmotic Stress Induces Phosphorylation of Histone H3 at Threonine 3 in Pericentromeric Regions of Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2015, 112, 8487–8492.
- Wang, Y.; Cai, S.; Yin, L.; Shi, K.; Xia, X.; Zhou, Y.; Yu, J.; Zhou, J. Tomato HsfA1a Plays a Critical Role in Plant Drought Tolerance by Activating ATG Genes and Inducing Autophagy. Autophagy 2015, 11, 2033–2047.
- Ueda, M.; Seki, M. Histone Modifications Form Epigenetic Regulatory Networks to Regulate Abiotic Stress Response. Plant Physiol. 2020, 182, 15–26.
- Cai, H.; Wang, H.; Zhou, L.; Li, B.; Zhang, S.; He, Y.; Xu, Y. Time-Series Transcriptomic Analysis of Contrasting Rice Materials under Heat Stress Reveals a Faster Response in the Tolerant Cultivar. Int. J. Mol. Sci. 2023, 24, 9408.
- Mishra, N.; Srivastava, A.P.; Esmaeili, N.; Hu, W.; Shen, G. Overexpression of the Rice Gene OsSIZ1 in Arabidopsis Improves Drought-, Heat-, and Salt-Tolerance Simultaneously. PLoS ONE 2018, 13, e0201716.
- Zhang, S.S.; Yang, H.; Ding, L.; Song, Z.T.; Ma, H.; Chang, F.; Liu, J.X. Tissue-Specific Transcriptomics Reveals an Important Role of the Unfolded Protein Response in Maintaining Fertility upon Heat Stress in Arabidopsis. Plant Cell. 2017, 29, 1007–1023.
- Saad, A.S.I.; Li, X.; Li, H.P.; Huang, T.; Gao, C.S.; Guo, M.W.; Liao, Y.C. A Rice Stress-Responsive NAC Gene Enhances Tolerance of Transgenic Wheat to Drought and Salt Stresses. Plant Sci. 2013, 203, 33–40.
- Liang, Q.; Geng, Q.; Jiang, L.; Liang, M.; Li, L.; Zhang, C.; Wang, W. Protein Methylome Analysis in Arabidopsis Reveals Regulation in RNA-Related Processes. J. Proteom. 2020, 213, 103601.
- Liu, Y.; Liu, K.; Yin, L.; Yu, Y.; Qi, J.; Shen, W.H.; Zhu, J.; Zhang, Y.; Dong, A. H3K4me2 Functions as a Repressive Epigenetic Mark in Plants. Epigenetics Chromatin. 2019, 12, 1–14.
- Fiorucci, A.S.; Bourbousse, C.; Concia, L.; Rougée, M.; Deton-Cabanillas, A.F.; Zabulon, G.; Layat, E.; Latrasse, D.; Kim, S.K.; Chaumont, N.; et al. Arabidopsis S2Lb Links AtCOMPASS-like and SDG2 Activity in H3K4me3 Independently from Histone H2B Monoubiquitination. Genome Biol. 2019, 20, 1–21.
- Zheng, B.; Chen, X. Dynamics of Histone H3 Lysine 27 Trimethylation in Plant Development. Curr. Opin. Plant Biol. 2011, 14, 123–129.
- Xiao, J.; Lee, U.S.; Wagner, D. Tug of War: Adding and Removing Histone Lysine Methylation in Arabidopsis. Curr. Opin. Plant Biol. 2016, 34, 41–53.
- Cheng, K.; Xu, Y.; Yang, C.; Ouellette, L.; Niu, L.; Zhou, X.; Chu, L.; Zhuang, F.; Liu, J.; Wu, H.; et al. Histone Tales: Lysine Methylation, a Protagonist in Arabidopsis Development. J. Exp. Bot. 2020, 71, 793–807.
- Wang, T.S.; Cheng, J.K.; Lei, Q.Y.; Wang, Y.P. A Switch for Transcriptional Activation and Repression: Histone Arginine Methylation. In The DNA, RNA, and Histone Methylomes; Springer: Berlin/Heidelberg, Germany, 2019; pp. 521–541.
- Liu, W.; Tanasa, B.; Tyurina, O.V.; Zhou, T.Y.; Gassmann, R.; Liu, W.T.; Ohgi, K.A.; Benner, C.; Garcia-Bassets, I.; Aggarwal, A.K.; et al. PHF8 Mediates Histone H4 Lysine 20 Demethylation Events Involved in Cell Cycle Progression. Nature 2010, 466, 508–512.
- Wang, H.; Wang, H.; Shao, H.; Tang, X. Recent Advances in Utilizing Transcription Factors to Improve Plant Abiotic Stress Tolerance by Transgenic Technology. Front. Plant Sci. 2016, 7, 67.
- Berger, S.L. The Complex Language of Chromatin Regulation during Transcription. Nature 2007, 447, 407–412.
- Simon, J.A.; Kingston, R.E. Mechanisms of Polycomb Gene Silencing: Knowns and Unknowns. Nat. Rev. Mol. Cell Biol. 2009, 10, 697–708.
- Margueron, R.; Reinberg, D. The Polycomb Complex PRC2 and Its Mark in Life. Nature 2011, 469, 343–349.
- Vijayanathan, M.; Trejo-Arellano, M.G.; Mozgová, I. Polycomb Repressive Complex 2 in Eukaryotes—An Evolutionary Perspective. Epigenomes 2022, 6, 3.
- Baile, F.; Gómez-Zambrano, A.; Calonje, M. Roles of Polycomb Complexes in Regulating Gene Expression and Chromatin Structure in Plants. Plant Commun. 2021, 3, 100267.
- Liu, Y.; Wang, J.; Liu, B.; Xu, Z.Y. Dynamic regulation of DNA methylation and histone modifications in response to abiotic stresses in plants. J. Integr. Plant Biol. 2022, 64, 2252–2274.
- Pandey, G.; Sharma, N.; Pankaj Sahu, P.; Prasad, M. Chromatin-based epigenetic regulation of plant abiotic stress response. Curr. Genom. 2016, 17, 490–498.
- Cui, X.; Zheng, Y.; Lu, Y.; Issakidis-Bourguet, E.; Zhou, D.X. Metabolic Control of Histone Demethylase Activity Involved in Plant Response to High Temperature. Plant Physiol. 2021, 185, 1813–1828.
- Lama, P. Identification and Functional Characterization of GmMYB176-Specific Protein Kinases in Soybean; The University of Western Ontario: London, ON, Canada, 2016.
- Yuan, L.; Liu, X.; Luo, M.; Yang, S.; Wu, K. Involvement of Histone Modifications in Plant Abiotic Stress Responses. J. Integr. Plant Biol. 2013, 55, 892–901.
- Houben, A.; Demidov, D.; Caperta, A.D.; Karimi, R.; Agueci, F.; Vlasenko, L. Phosphorylation of Histone H3 in Plants-A Dynamic Affair. Biochim. Biophys. Acta (BBA)—Gene Struct. Expr. 2007, 1769, 308–315.
- Ramakrishnan, M.; Satish, L.; Kalendar, R.; Narayanan, M.; Kandasamy, S.; Sharma, A.; Emamverdian, A.; Wei, Q.; Zhou, M. The dynamism of transposon methylation for plant development and stress adaptation. Int. J. Mol. Sci. 2021, 22, 11387.
- Sawicka, A.; Seiser, C. Sensing Core Histone Phosphorylation—A Matter of Perfect Timing. Biochim. Biophys. Acta (BBA)—Gene Regul. Mech. 2014, 1839, 711–718.
- Wang, F.; Higgins, J.M.G. Histone Modifications and Mitosis: Countermarks, Landmarks, and Bookmarks. Trends Cell Biol. 2013, 23, 175–184.
- Su, Y.; Wang, S.; Zhang, F.; Zheng, H.; Liu, Y.; Huang, T.; Ding, Y. Phosphorylation of Histone H2A at Serine 95: A Plant-Specific Mark Involved in Flowering Time Regulation and H2A.Z Deposition. Plant Cell 2017, 29, 2197–2213.
- Stadler, J.; Richly, H. Regulation of DNA Repair Mechanisms: How the Chromatin Environment Regulates the DNA Damage Response. Int. J. Mol. Sci. 2017, 18, 1715.
- Dubrez, L.; Causse, S.; Bonan, N.B.; Dumétier, B.; Garrido, C. Heat-Shock Proteins: Chaperoning DNA Repair. Oncogene 2020, 39, 516–529.
- Aleksandrov, R.; Hristova, R.; Stoynov, S.; Gospodinov, A. The Chromatin Response to Double-Strand DNA Breaks and Their Repair. Cells 2020, 9, 1853.
- Kerk, D.; Templeton, G.; Moorhead, G.B. Evolutionary Radiation Pattern of Novel Protein Phosphatases Revealed by Analysis of Protein Data from the Completely Sequenced Genomes of Humans, Green Algae, and Higher Plants. Plant Physiol. 2008, 146, 351–367.
- Andrási, N.; Rigó, G.; Zsigmond, L.; Pérez-Salamó, I.; Papdi, C.; Klement, E.; Pettkó-Szandtner, A.; Baba, A.I.; Ayaydin, F.; Dasari, R. The Mitogen-Activated Protein Kinase 4-Phosphorylated Heat Shock Factor A4A Regulates Responses to Combined Salt and Heat Stresses. J. Exp. Bot. 2019, 70, 4903–4918.
- Ibáñez, C.; Delker, C.; Martinez, C.; Bürstenbinder, K.; Janitza, P.; Lippmann, R.; Ludwig, W.; Sun, H.; James, G.V.; Klecker, M. Brassinosteroids Dominate Hormonal Regulation of Plant Thermomorphogenesis via BZR1. Curr. Biol. 2018, 28, 303–310.e3.
- Huang, T.; Zhang, H.; Zhou, Y.; Su, Y.; Zheng, H.; Ding, Y. Phosphorylation of Histone H2A at Serine 95 Is Essential for Flowering Time and Development in Arabidopsis. Front. Plant Sci. 2021, 12, 761008.
- Swatek, K.N.; Komander, D. Ubiquitin Modifications. Cell Res. 2016, 26, 399–422.
- Oss-Ronen, L.; Sarusi, T.; Cohen, I. Histone Mono-Ubiquitination in Transcriptional Regulation and Its Mark on Life: Emerging Roles in Tissue Development and Disease. Cells 2022, 11, 2404.
- Yu, F.; Wu, Y.; Xie, Q. Ubiquitin-Proteasome System in ABA Signaling: From Perception to Action. Mol. Plant. 2016, 9, 21–33.
- Qin, Q.; Wang, Y.; Huang, L.; Du, F.; Zhao, X.; Li, Z.; Wang, W.; Fu, B. A U-Box E3 Ubiquitin Ligase OsPUB67 Is Positively Involved in Drought Tolerance in Rice. Plant Mol. Biol. 2020, 102, 89–107.
- Ning, Y.; Jantasuriyarat, C.; Zhao, Q.; Zhang, H.; Chen, S.; Liu, J.; Liu, L.; Tang, S.; Park, C.H.; Wang, X.; et al. The SINA E3 Ligase OsDIS1 Negatively Regulates Drought Response in Rice. Plant Physiol. 2011, 157, 242–255.
- Hsu, K.H.; Liu, C.C.; Wu, S.J.; Kuo, Y.Y.; Lu, C.A.; Wu, C.R.; Lian, P.J.; Hong, C.Y.; Ke, Y.T.; Huang, J.H.; et al. Expression of a Gene Encoding a Rice RING Zinc-Finger Protein, OsRZFP34, Enhances Stomata Opening. Plant Mol. Biol. 2014, 86, 125–137.
- Tripathi, A.K.; Pareek, A.; Singla-Pareek, S.L. A NAP-family histone chaperone functions in abiotic stress response and adaptation. Plant Physiol. 2016, 171, 2854–2868.
- Lyzenga, W.J.; Booth, J.K.; Stone, S.L. The Arabidopsis RING-Type E3 Ligase XBAT32 Mediates the Proteasomal Degradation of the Ethylene Biosynthetic Enzyme, 1-Aminocyclopropane-1-Carboxylate Synthase 7. Plant J. 2012, 71, 23–34.
- Bratzel, F.; López-Torrejón, G.; Koch, M.; Del Pozo, J.C.; Calonje, M. Keeping Cell Identity in Arabidopsis Requires PRC1 RING-Finger Homologs That Catalyze H2A Monoubiquitination. Curr. Biol. 2010, 20, 1853–1862.
- Zhou, M.; Paša-Tolić, L.; Stenoien, D.L. Profiling of Histone Post-Translational Modifications in Mouse Brain with High-Resolution Top-Down Mass Spectrometry. J. Proteome Res. 2017, 16, 599–608.
- Geng, F.; Wenzel, S.; Tansey, W.P. Ubiquitin and Proteasomes in Transcription. Annu. Rev. Biochem. 2012, 81, 177–201.
- Zou, B.; Yang, D.L.; Shi, Z.; Dong, H.; Hua, J. Monoubiquitination of Histone 2B at the Disease Resistance Gene Locus Regulates Its Expression and Impacts Immune Responses in Arabidopsis. Plant Physiol. 2014, 165, 309–318.
- Zhang, Y.; Li, D.; Zhang, H.; Hong, Y.; Huang, L.; Liu, S.; Li, X.; Ouyang, Z.; Song, F. Tomato Histone H2B Monoubiquitination Enzymes SlHUB1 and SlHUB2 Contribute to Disease Resistance against Botrytis cinerea through Modulating the Balance between SA- and JA/ET-Mediated Signaling Pathways. BMC Plant Biol. 2015, 15, 252.
- Kim, J.H.; Lim, S.D.; Jang, C.S. Oryza sativa Drought-, Heat-, and Salt-Induced RING Finger Protein 1 (OsDHSRP1) Negatively Regulates Abiotic Stress-Responsive Gene Expression. Plant Mol. Biol. 2020, 103, 235–252.
- Kim, J.H.; Khan, I.U.; Kim, M.S.; Seo, Y.W. Functional Characterization of Wheat Histone H2B Monoubiquitination Enzyme TaHUB2 in Response to Vernalization in Keumkang (Triticum aestivum L.). J. Plant Interact. 2021, 16, 93–103.
- Zhang, Y.; Lai, X.; Yang, S.; Ren, H.; Yuan, J.; Jin, H.; Shi, C.; Lai, Z.; Xia, G. Functional Analysis of Tomato CHIP Ubiquitin E3 Ligase in Heat Tolerance. Sci. Rep. 2021, 11, 1713.
- Wang, P.; Guo, K.; Su, Q.; Deng, J.; Zhang, X.; Tu, L. Histone Ubiquitination Controls Organ Size in Cotton (Gossypium hirsutum). Plant J. 2022, 110, 1005–1020.
- Shiio, Y.; Eisenman, R.N. Histone Sumoylation Is Associated with Transcriptional Repression. Proc. Natl. Acad. Sci. USA 2003, 100, 13225–13230.
- Miller, M.J.; Barrett-Wilt, G.A.; Hua, Z.; Vierstra, R.D. Proteomic Analyses Identify a Diverse Array of Nuclear Processes Affected by Small Ubiquitin-like Modifier Conjugation in Arabidopsis. Proc. Natl. Acad. Sci. USA 2010, 107, 16512–16517.
- To, T.K.; Kim, J.M.; Matsui, A.; Kurihara, Y.; Morosawa, T.; Ishida, J.; Tanaka, M.; Endo, T.; Kakutani, T.; Toyoda, T.; et al. Arabidopsis HDA6 Regulates Locus-Directed Heterochromatin Silencing in Cooperation with MET1. PLoS Genet. 2011, 7, e1002055.
- Han, D.; Chen, C.; Xia, S.; Liu, J.; Shu, J.; Nguyen, V.; Lai, J.; Cui, Y.; Yang, C. Chromatin-Associated SUMOylation Controls the Transcriptional Switch between Plant Development and Heat-Stress Responses. Plant Commun. 2020, 2, 100091.
- Han, G.; Qiao, Z.; Li, Y.; Yang, Z.; Wang, C.; Zhang, Y.; Liu, L.; Wang, B. RING Zinc Finger Proteins in Plant Abiotic Stress Tolerance. Front. Plant Sci. 2022, 13, 877011.
- Budhiraja, R.; Hermkes, R.; Müller, S.; Schmidt, J.; Colby, T.; Panigrahi, K.; Coupland, G.; Bachmair, A. Substrates Related to Chromatin and to RNA-Dependent Processes Are Modified by Arabidopsis SUMO Isoforms That Differ in a Conserved Residue with Influence on Desumoylation. Plant Physiol. 2009, 149, 1529–1540.
- Augustine, R.C.; Vierstra, R.D. SUMOylation: Re-wiring the Plant Nucleus during Stress and Development. Curr. Opin. Plant Biol. 2018, 45, 143–154.
- Roy, D.; Sadanandom, A. SUMO Mediated Regulation of Transcription Factors as a Mechanism for Transducing Environmental Cues into Cellular Signaling in Plants. Cell. Mol. Life Sci. 2021, 78, 2641–2664.
- Wawrzyn’ska, A.; Sirko, A. Proteasomal Degradation of Proteins Is Important for the Proper Transcriptional Response to Sulfur Deficiency Conditions in Plants. Plant Cell Physiol. 2020, 61, 1548–1564.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
2 times
(View History)
Update Date:
08 Dec 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No