Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Bertrand Liagre | -- | 3485 | 2023-12-04 09:35:23 | | | |
| 2 | Catherine Yang | Meta information modification | 3485 | 2023-12-05 01:50:15 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Limami, Y.; Pinon, A.; Wahnou, H.; Oudghiri, M.; Liagre, B.; Simon, A.; Duval, R.E. Ursolic Acid Targets Cancer Hallmarks. Encyclopedia. Available online: https://encyclopedia.pub/entry/52311 (accessed on 30 June 2026).
Limami Y, Pinon A, Wahnou H, Oudghiri M, Liagre B, Simon A, et al. Ursolic Acid Targets Cancer Hallmarks. Encyclopedia. Available at: https://encyclopedia.pub/entry/52311. Accessed June 30, 2026.
Limami, Youness, Aline Pinon, Hicham Wahnou, Mounia Oudghiri, Bertrand Liagre, Alain Simon, Raphaël Emmanuel Duval. "Ursolic Acid Targets Cancer Hallmarks" Encyclopedia, https://encyclopedia.pub/entry/52311 (accessed June 30, 2026).
Limami, Y., Pinon, A., Wahnou, H., Oudghiri, M., Liagre, B., Simon, A., & Duval, R.E. (2023, December 04). Ursolic Acid Targets Cancer Hallmarks. In Encyclopedia. https://encyclopedia.pub/entry/52311
Limami, Youness, et al. "Ursolic Acid Targets Cancer Hallmarks." Encyclopedia. Web. 04 December, 2023.
Copy Citation
Cancer is a multifactorial disease characterized by various hallmarks, including uncontrolled cell growth, evasion of apoptosis, sustained angiogenesis, tissue invasion, and metastasis, among others. Traditional cancer therapies often target specific hallmarks, leading to limited efficacy and the development of resistance. Thus, there is a growing need for alternative strategies that can address multiple hallmarks concomitantly. Ursolic acid (UA), a naturally occurring pentacyclic triterpenoid, has recently emerged as a promising candidate for multitargeted cancer therapy.
ursolic acid
cancer hallmarks
cell growth
apoptosis
1. Hallmark 1: Inhibition of Proliferative Signaling
Normal cells rely on tightly regulated cell cycle control mechanisms to ensure controlled proliferation and maintain the equilibrium of tissues. However, this precise control system is disrupted in cancer. Unlike normal cells, cancer cells exhibit a unique characteristic of releasing and responding to growth factors, particularly epidermal growth factor (EGF) and its receptor (EGFR) signaling [1]. By activating these growth factors, cancer cells acquire a heightened ability to stimulate their growth and division, independent of external signals. This phenomenon, known as self-sufficiency in cell proliferation, is primarily driven by three major signaling pathways, Akt, MAPK/ERK, and mTOR [2]. These pathways act as crucial drivers, fueling the uncontrolled growth and replication of cancer cells. Their activation promotes the survival and expansion of cancer cells, contributing to the relentless progression of the disease.
It has been revealed that UA exhibits multifaceted effects on various signaling pathways involved in cell cycle progression. UA has shown a capacity to inhibit the phosphorylation/activation of Akt [1][3], mTOR [4], Ras [5], and ERK [6] in a dose-dependent manner (Figure 1A). Furthermore, Yang et al. examined the effects of UA treatment on NSCLC and H1975 lung cancer cell lines that carry the EGFR T790M mutation [7]. This mutation is a major cause of resistance to EGFR tyrosine kinase inhibitors (EGFR-TKIs) such as erlotinib [8]. The results demonstrated that UA treatment effectively suppressed cell proliferation and motility while inducing apoptosis in these cells [7]. Furthermore, the study also extended its investigation to animal models by xenografting lung cancer cells that expressed the EGFR T790M mutation, providing additional evidence supporting the potential efficacy of UA in treating this specific subtype of NSCLC [7]. In a related context, UA markedly deterred the growth of xenograft tumors in colorectal cancer (CRC) models. The in vivo experiments revealed that UA not only ameliorated pathological features but also triggered apoptosis and arrested the cell cycle in xenograft CRC tissue [9]. These effects were attributed to the downregulation of the Wnt/β-catenin signaling cascade [9]. The comprehensive findings from both cellular and animal model studies underscore the potential of UA as a versatile therapeutic agent with promising anti-cancer properties, supporting its further exploration for the treatment of various cancer subtypes, including NSCLC and CRC.

Figure 1. UA targets cancer by (A) inhibiting of proliferative signaling, (B) inhibiting of growth suppressors, and (C) inhibiting of immune evasion.
2. Hallmark 2: Inhibition of Growth Suppressors
To bypass the growth-inhibitory signals originating from normal homeostatic processes, cancer cells exhibit a reduced responsiveness to external signals that normally impede their growth. These cells actively resist apoptotic control, which is crucial for tightly regulating cell death and curbing uncontrolled proliferation [2].
In normal conditions, apoptosis plays a pivotal role in eliminating cells that undergo excessive proliferation, thus maintaining a balanced cell population and removing aberrant or diseased cells [10]. On the other hand, autophagy serves as a cellular recycling system, responsible for eliminating abnormal proteins and cellular components, while promoting cellular regeneration. Cancer cells develop resistance to apoptotic signals, preventing programmed cell death and enhancing their survival [10]. Furthermore, cancer cells manipulate autophagy to enhance their growth potential and overcome nutrient scarcity, facilitating their survival and uncontrolled proliferation, even in challenging conditions [2].
This aberrant cellular behavior is primarily driven by Rb and p53 proteins, two prototypical tumor suppressors [4]. Interestingly, emerging research suggests that UA holds promise in targeting both proteins. Studies using human non-small cell lung cancer A549 cells showed the ability of UA to induce G1-phase arrest produced through the p53/p21-mediated and Cdks-inhibition pathway [11]. UA showed also an ability to increase the expression of E2F, a protein involved in the Rb signaling pathway (Figure 1B) [12].
Nevertheless, p53 and Rb operate within a larger network with functional redundancy, demonstrating that multiple mechanisms regulate cell proliferation [13]. This complex interplay ensures robust control over cell growth even in the absence of p53 or Rb (Table 1).
Table 1. Cancer hallmarks, mechanisms of action, and the main pathways and markers involved [2][14][15].
| N° | Cancer Hallmark | Mechanisms of Action | Main Pathways/Markers | References |
|---|---|---|---|---|
| 1 | Sustaining proliferative signaling | Cellular proliferation | Cdk, Akt, MAPK/ERK, and mTOR | [14] |
| 2 | Evading growth suppressors | Tumor suppressors | Rb, p53 | [14] |
| 3 | Avoiding immune destruction | Immune checkpoints | PD1/PD-L1, TIM3, MMP2, and LAG3 | [2] |
| 4 | Tumor promoting inflammation | NF-κB signaling | NF-κB, IKK-β | [2] |
| Tumor-associated macrophages | CD68, CD163, iNOS | [2] | ||
| 5 | Genome instability and mutation | Chromosomal instability | PARP, BRCA, 53BP1, and cyclin-dependent kinase | [2] |
| 6 | Activating invasion and metastasis | Extracellular matrix (ECM) | Hyaluronan, Versican, Collagen IV | [14] |
| Adhesion molecules | CEACAM1, DCC, E-Cadherin | [14] | ||
| Secreted factors | Tenascin C, Fibrinogen, Periostin | [14] | ||
| 7 | Polymorphic microbiomes | Gut dysbiosis | Microbiota | [15] |
| 8 | Inducing angiogenesis | Angiogenesis | VEGF, FGF-β, PDGF | [14] |
| 9 | Resisting cell death | Apoptosis | Caspases, Bcl-2, and p53 | [14] |
| Autophagy | MAPK, ATG, and p62 | [14] | ||
| 10 | Enabling replicative immortality | Telomere regulation | TRF1/TRF2/POT1/TIN2/RAP1/TPP1 | [14] |
| p53 signaling | p53, MDM2, p14ARF/p19ARF, E2F-1 | [14] | ||
| 11 | Non-mutational epigenetic reprogramming | Epithelial-to-mesenchymal transition (EMT) | TFG-β, Wnt, SNAIL, Twist, Cadherins, Vimentin | [15] |
| Hypoxia | HIF1α/2α, HIF1β, CAIX, AP-1/c-jun, GLUT-1 | [2][15] | ||
| 12 | Deregulating cellular energetics | |||
| Glycolysis | Tomm20, V-ATPase, GAPDH | [2] | ||
| Mitochondrial metabolism | COX IV, VDAC1/Porin, ATPase β | [2] | ||
| 13 | Senescent Cells | Senescence-associated secretory phenotype | Senescence-associated β-galactosidase and uPAR | [15] |
| 14 | Unlocking phenotypic plasticity | Blocked differentiation | RAR α, HDAC, SOX10, α-ketoglutarate | [15] |
| Dedifferentiation plasticity | HOXA5, SMAD4 | [15] | ||
| Transdifferentiation | PTF1A, MIST1 | [15] |
3. Hallmark 3: Inhibition of Immune Evasion
The human immune system is a complex network of cells, tissues, and organs that plays a crucial role in defending the body against foreign pathogens and diseases [16]. However, its importance extends beyond infection control, as it also functions to identify and eliminate unhealthy and ailing cells, including cancer cells [16]. T cells and NK cells are crucial in the fight against cancer [8]. T cells recognize tumor antigens and orchestrate immune responses, while NK cells detect and eliminate cancer cells directly [16].
In cancer immunotherapy, targeting immune checkpoint molecules like PD1/PD-L1, TIM3, and LAG3 has revolutionized treatment approaches [2]. These molecules regulate the activity of T cells, and blocking them can unleash the immune system’s ability to recognize and attack cancer cells more effectively [16]. UA has shown promise in targeting some proteins involved in the immune evasion of cancer. Kang et al. showed that UA can exhibit anticancer activities by inhibiting MMP2 and PD-L1 expression through EGFR/JAK2/STAT3 signaling in NSCLC cells A549 and H460 (Figure 1C) [17][18].
4. Hallmark 4: Reducing Tumor Inflammation
The tumor microenvironment manipulates immune cells to support tumor survival instead of eliminating cancer cells. Immune cells in the TME secrete chemicals such as reactive oxygen species (ROS) that are actively mutagenic for nearby cancer cells [19]. Immune cells also release factors that aid in tumor growth and metastasis, rather than identifying and destroying cancer cells [2][20]. Key inflammatory mechanisms affected by the tumor are NF-κB, immune checkpoint signaling, and inflammasome signaling [2].
One potential approach to counteract the tumor-mediated hijacking of immune cells in the tumor microenvironment is the use of UA. Studies have demonstrated that UA possesses the ability to reduce the levels of inflammatory proteins such as TNF-α, NF-κB, and cyclooxygenase-2 (COX-2) in several cancer cell lines and other inflammatory models mainly by targeting JAK/STAT signaling pathway [21][22][23][24][25][26]. Furthermore, UA can also act by inhibiting NLRP3 inflammasome activation (Figure 2A) [27].

Figure 2. UA targets cancer by (A) reducing tumor inflammation, (B) regulating genome instability and mutation (where 1, 2, and 3 refer, respectively, to single-strand break, double-strand break, and DNA not repaired), (C) inhibiting cancer invasion metastasis, and (D) inhibiting angiogenesis.
By targeting these inflammatory pathways, UA can potentially alleviate the detrimental effects of chronic inflammation that can lead to carcinogenesis and promote cellular health.
5. Hallmark 5: Genome Instability and Mutation
Cancer cells exhibit a high rate of proliferation, which increases the likelihood of genetic alterations and mutations affecting genes involved in cell division and tumor suppression. This genetic instability fosters the development of further cancerous adaptations within the cells. These changes can arise from direct DNA mutations or epigenetic modifications, which can disrupt protein expression levels and compromise genomic integrity [2][15]. Precision cancer therapies have been developed to target specific components of the cell cycle, such as checkpoint kinases (e.g., Chk1 and Chk2 proteins), as well as DNA damage repair enzymes like BRCA and 53BP1. These therapies aim to disrupt the molecular mechanisms that facilitate cancer cell proliferation and survival, offering potential avenues for more precise and effective treatments [15][28].
In the same context, it has been observed that UA exhibits the capacity to effectively inhibit the phosphorylation of Chk1, Chk2, and BRCA in a dose-dependent manner [29], as illustrated in Figure 2B. Furthermore, the administration of UA has effectively modulated the generation of both cellular and mitochondrial ROS. This, in turn, triggers a response in embryonic CSCs known as DNA damage response (DDR), strongly suggesting the potential for UA-induced cell death [29].
UA also weakens surveillance mechanisms by blocking 53BP1 foci formation induced by VRK1. This is achieved by inhibiting the catalytic activity of VRK1 through direct binding to its catalytic domain [28].
6. Hallmark 6: Activating Invasion and Metastasis
Tissue invasion refers to the process by which tumor cells expand into nearby tissues, while metastasis involves the migration of tumor cells from the primary tumor site to distant locations, where they establish secondary tumors [14][30]. One key mechanism involved in these processes is the well-documented epithelial-to-mesenchymal transition, which plays a crucial role in facilitating uninhibited cell division and enabling metabolic adaptations that promote cell survival under nutrient-deprived and stressful conditions [2][14][30].
These cancer mechanisms entail extensive modifications in cell–cell and cell–matrix interactions, as well as cellular transformations that promote invasion and migration. Specific targets involved in these processes include collagen and CEACAM1, which undergo alterations to facilitate the behavior of cancer cells [2][17][30]. These changes contribute to the invasive properties of cancer cells, allowing them to penetrate tissues and migrate to distant sites, ultimately leading to the establishment of secondary tumors [30].
In studies conducted on mesothelioma cells, it has been observed that UA exhibits the ability to impede the process of epithelial-to-mesenchymal transition (EMT). Specifically, UA activates E-cadherin, while concurrently inhibiting the expression of N-cadherin, vimentin, and Twist (Figure 2C) [3]. In addition, studies involving 4T1 tumor-bearing mice have shown that UA can inhibit lung metastasis [12]. Furthermore, UA also inhibits MMP2 an enzyme that facilitates the breakdown of the extracellular matrix, allowing cancer cells to invade surrounding tissues and enter blood vessels or lymphatics for metastatic dissemination [17].
7. Hallmark 7: Polymorphic Microbiomes
Variations in the microbial communities within our bodies, known as microbiomes, have been found to have a significant impact on cancer characteristics, development, and progression. Research suggests that polymorphic microbiomes can serve as a distinctive enabling factor that influences the acquisition of cancer hallmarks, potentially promoting or protecting against different types of cancer [31][32]. For instance, studies have revealed the presence of microbiomes with either cancer-promoting or cancer-protective properties, influencing the incidence and progression of colon cancer [32]. Furthermore, the composition of the gut microbiome has been shown to influence the immune system, affecting anti-tumor immunity and the response to immunotherapy in patients with melanoma [33][34]. Notably, the emerging concept of polymorphic microbiomes intersects with established hallmarks such as genome instability, mutations, and tumor-promoting inflammation [15]. This indicates that the composition of microbiomes can significantly contribute to the development and progression of cancer, acting alongside other well-known cancer hallmarks.
UA has shown potential in modulating the composition of the gut microbiota and improving the microenvironment within the digestive system [35]. Studies have demonstrated that UA can bring about changes in the gut microbiota by promoting the growth and enrichment of beneficial bacteria such as Bifidobacterium spp and Lactobacillus spp [35][36]. These alterations in the microbial composition contribute to a healthier gut environment. Moreover, the presence of an enhanced population of Bifidobacterium spp and Lactobacillus spp has been associated with immune regulatory effects [36]. Additionally, UA’s ability to promote the growth of these beneficial bacteria was correlated with its ability to correct the imbalance of Th17/Treg cells (Figure 2A) [36].
8. Hallmark 8: Reducing Angiogenesis
The establishment and growth of the vascular network play a pivotal role in metastasis as cancer cells require a sufficient supply of nutrients and oxygen, as well as a means of waste removal [37]. This crucial process is achieved through two mechanisms: angiogenesis, which involves the formation of new blood vessels, and lymphangiogenesis, which involves the formation of lymphatic vessels [37]. Abnormal signaling of specific growth factors, such as VEGF, FGF-β, and PDGF, significantly contribute to the promotion of tumor angiogenesis [2][14][37]. These growth factors play a significant role in stimulating the formation of blood vessels within tumors.
In vivo and in vitro studies conducted by Lin et al. using mice colorectal cancer (CRC) xenograft have shown that UA significantly reduced the intratumoral microvessel density [38]. Furthermore, by (i) suppressing the proliferation of endothelial cells, (ii) inhibiting their migration, and (iii) impeding the formation of capillary tubes, UA exhibits notable anti-angiogenic properties. Additionally, UA treatment significantly reduced the expression of VEGF-A and FGF-β in both CRC tumors and HT-29 cells (Figure 2D) [38].
9. Hallmarks 9 and 10: Resisting Cell Death and Enabling Replicative Immortality
Cancer cells possess mechanisms that enable them to resist cell death, a hallmark characteristic of cancer. This resistance is not solely due to a lack of response to external signals but rather arises from intrinsic mechanisms within the cancer cells. These mechanisms can be attributed to mutations that disrupt apoptotic signaling pathways or impair the detection of cellular damage [2][15].
Apoptosis, also known as programmed cell death, exhibits distinct features including cell shrinkage, membrane blebbing, chromosome condensation (pyknosis), nuclear fragmentation (karyorrhexis), DNA fragmentation, and eventual engulfment of the cell by phagocytes [39]. On the other hand, autophagy, another cellular process, plays a vital role in enabling cells to survive in the face of various stressful conditions [40]. Cancer cells exploit autophagy to overcome nutrient limitations and facilitate tumor growth [41]. Moreover, autophagy can impact the tumor microenvironment by promoting angiogenesis, providing nutrients, and modulating inflammatory responses [2][10].
Cancer cells often exhibit alterations in key apoptotic signaling proteins such as caspases, Bcl-2 family, and p53. These proteins may be downregulated, mutated, or bypassed, contributing to the resistance of cancer cells to apoptosis. Similarly, the regulation of autophagy in cancer involves targeting specific pathways, including MAPK, ATG, and p62, which are involved in autophagy-related processes [2][10].
Notably, UA has been shown to elicit distinct effects on either mitochondrial or death receptors apoptotic pathways including the upregulation of the Fas receptor the cleavage of caspases-8, -3, -7, and -9, the regulation of Bcl-2 family proteins, and the activation of p53 leading to apoptotic cell death (Figure 3A) [6][21][42][43][44][45][46][47].
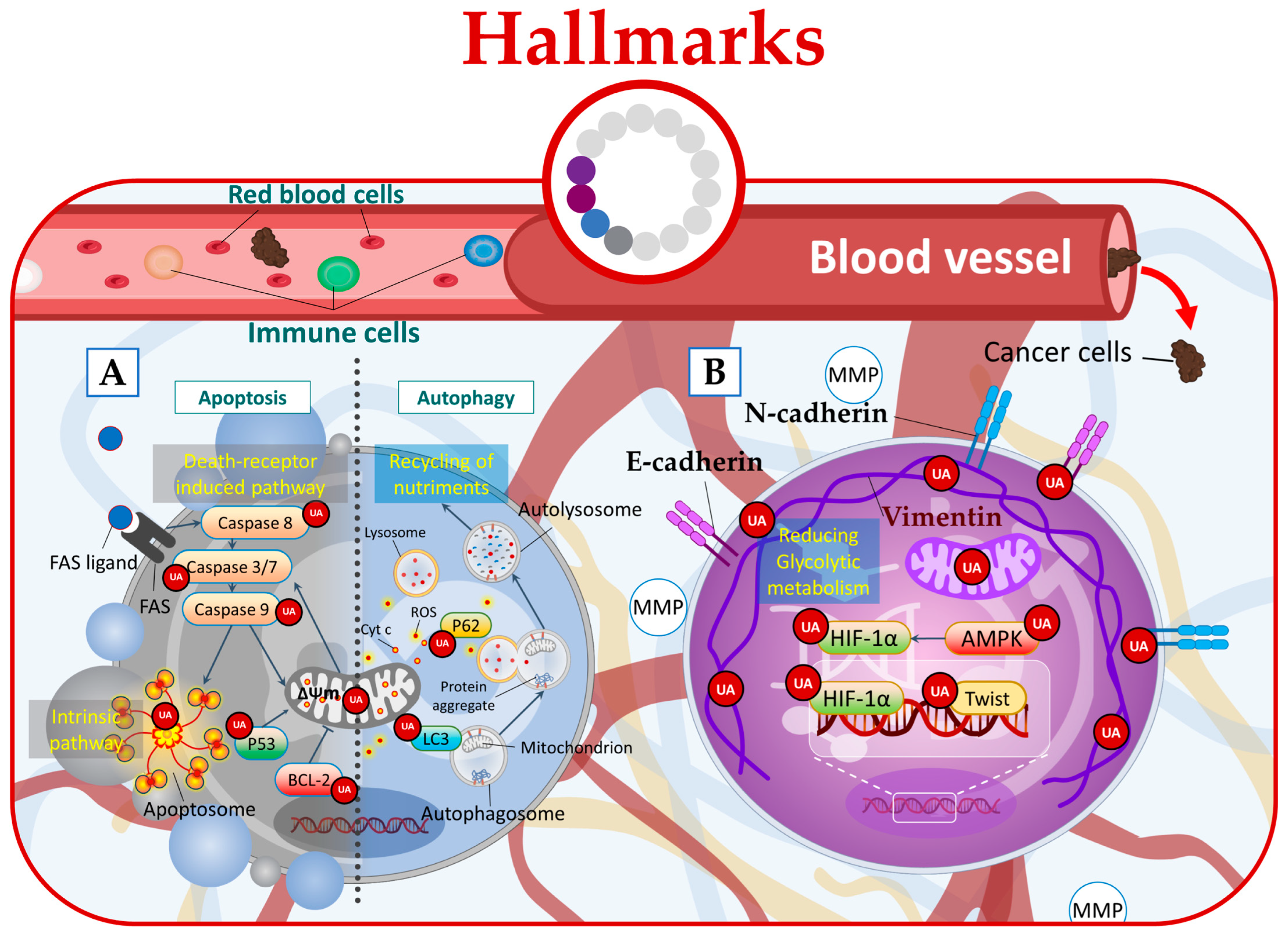
Figure 3. UA targets cancer by (A) inducing apoptosis and autophagy, (B) regulating microenvironmental factors, and disrupting cancer energy metabolism.
Furthermore, in phenotypically breast cancer cells, UA has demonstrated its anticancer potential by targeting glycolysis. By inhibiting Akt and inducing changes in the glycolytic pathway, UA leads to energy stress and activates AMPK (adenosine monophosphate-activated protein kinase), ultimately promoting a combined effect of autophagy and apoptosis at low micromolar concentrations (Figure 3A) [48].
Additionally, UA treatment was found to induce autophagy in Esophageal Squamous Cell Carcinoma (ESCC) cells. This was evidenced by the increased levels of LC3-II protein, which is a marker of autophagosome formation, and the concurrent decrease in p62 protein levels [49]. Moreover, UA increased cellular ROS leading to ROS-dependent autophagy (Figure 3A) [49].
10. Hallmark 11: Non-Mutational Epigenetic Reprogramming
Non-mutational epigenetic reprogramming can occur through microenvironmental factors such as hypoxia or EMT [50]. Hypoxia, for instance, can induce hypermethylation by suppressing the activity of TET demethylases. EMT is responsible for the reversible induction of cancer cell invasiveness at the periphery of solid tumors. Epigenetic regulatory heterogeneity is a significant aspect of non-mutational epigenetic reprogramming [15].
In this context, UA emerges as a potential therapeutic agent capable of targeting the hypoxic tumor microenvironment directly. It achieves this by downregulating HIF-1α (hypoxia-inducible factor-1α), an oxygen-dependent transcriptional activator [12], or through AMPK modulation [51]. By targeting these key regulators, UA has the potential to disrupt the hypoxic microenvironment of tumors. Additionally, UA exhibits the ability to target EMT by acting on crucial pathways and markers involved, including TGF-β, Wnt [52], SNAIL, Twist, cadherins, and vimentin (Figure 3B) [53].
11. Hallmark 12: Deregulating Cellular Energetics
Due to their rapid proliferation, cancer cells have heightened energy and nutrient demands, even in environments with limited oxygen supply [15]. This necessitates significant metabolic alterations in cancer cells to meet their metabolic needs. One prominent metabolic adaptation observed in cancer cells is the Warburg effect. This phenomenon involves a shift towards increased glycolysis, even in the presence of oxygen, leading to the diversion of pyruvate away from the Krebs cycle and towards lactate production [15][54]. This altered metabolic pathway allows cancer cells to generate energy quickly, although less efficiently than oxidative phosphorylation. In addition to the reliance on glycolysis, cancer cells often exhibit an increased utilization of glutamine to support their rapid proliferation [15].
Targeting the hypoxic tumor microenvironment involves focusing on key regulators such as HIF-1α and AMPK. HIF-1α plays a vital role in enabling cancer cells to adapt to hypoxia by promoting the expression of genes involved in glycolysis, angiogenesis, and cell survival. AMPK, on the other hand, is a metabolic sensor that switches to a tumor-promoting mode to protect against metabolic, oxidative, and genotoxic stress. Modulating these factors presents potential therapeutic avenues to disrupt the tumor microenvironment and interfere with cancer cell metabolism [15].
In this regard, UA demonstrates its potential by effectively targeting hypoxia through the HIF-1α pathway, leading to the downregulation of HIF-1α protein expression [12]. Furthermore, UA can disrupt the energy metabolism of cancer cells by directly acting on the glycolytic metabolism and mitochondrial respiration function [54]. Additionally, UA derivatives showed the ability to suppress cancer cell glucose metabolism by acting on 2-deoxy-D-glucose (2-DG) (Figure 3B) [50].
12. Hallmark 13: Senescent Cells
Cellular senescence is a process of irreversible cell cycle arrest that likely evolved as a protective mechanism to maintain tissue homeostasis [55]. It involves the cessation of cell division, accompanied by changes in cell morphology, metabolism, and the activation of senescence-associated secretory phenotype (SASP) [15][55]. Various internal and external factors, such as nutrient deprivation, DNA damage, organelle dysfunction, and disruptions in cellular signaling, can trigger senescence [55]. While senescent cells typically function as a defense against neoplasia, it is important to note that in certain cases, they can paradoxically promote tumor development and progression [56]. The secreted cytokines and growth factors from senescent cells, known as the SASP, can have either tumor-suppressive or oncogenic effects depending on the specific cellular context, cell type, and characteristics of the tumor [56].
Senescence-associated beta-galactosidase and uPAR (urokinase plasminogen activator receptor) are commonly used markers to identify senescent cells, with uPAR being a more recently described biomarker that exhibits broad induction during senescence.
In a cancer context, there is limited research investigating the impact of natural compounds on senescent cells. However, a study conducted by Han et al. shed light on this subject and revealed that UA effectively reduced senescence-associated beta-galactosidase (SA-β-gal) activity [57]. Furthermore, signaling pathways that regulate SASP, such as NF-κB, mTOR, and p38, or individual SASP factors that promote tumor progression [56] can also be targeted by UA (Figure 4A) [4][22][58].
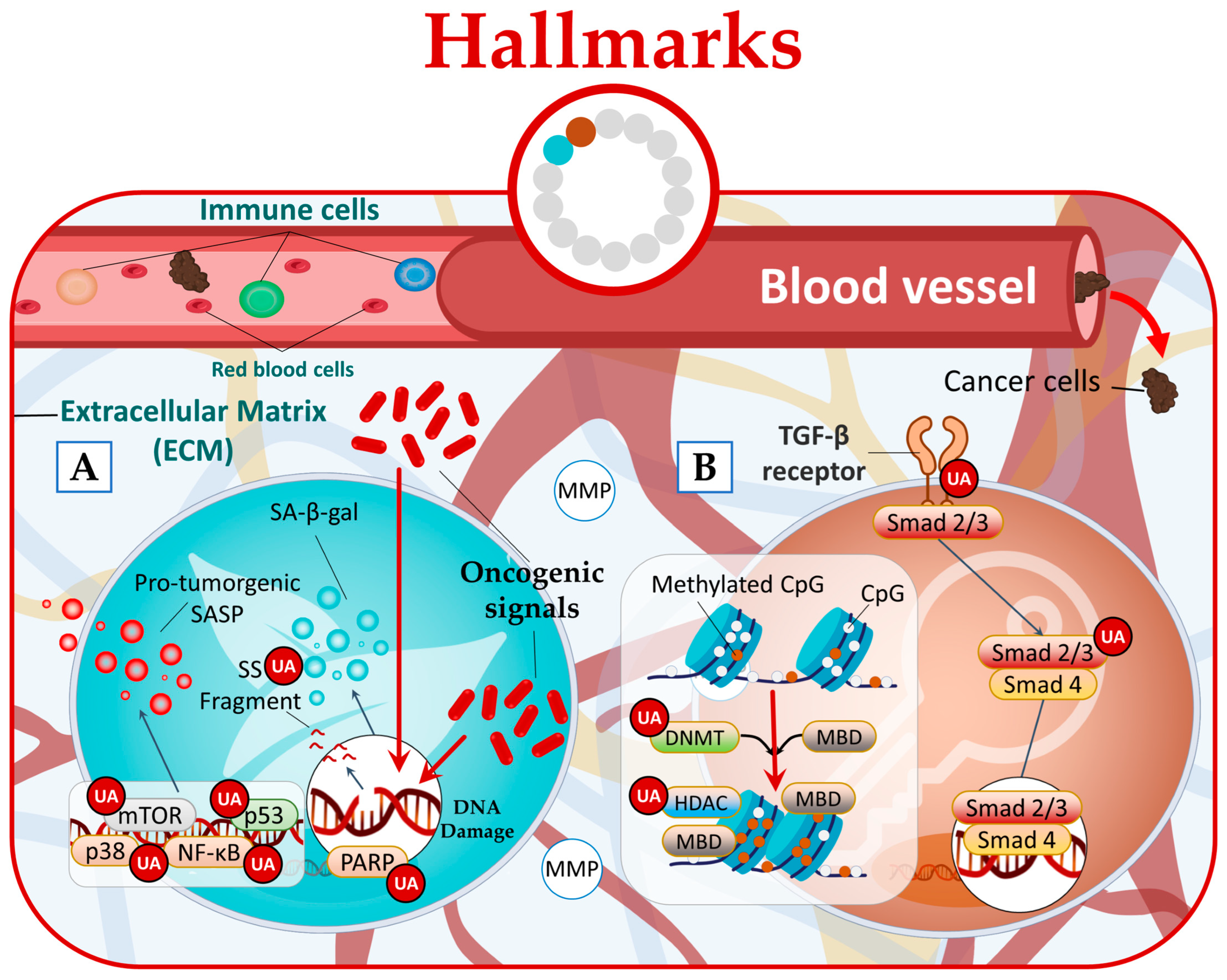
Figure 4. UA targets cancer by (A) reducing senescence state and (B) disrupting phenotypic plasticity.
13. Hallmark 14: Unlocking Phenotypic Plasticity
Cancer cells possess a unique ability to exhibit phenotypic plasticity, a characteristic that is typically restricted in normal cells. This plasticity plays a critical role in cancer initiation and progression by enabling disrupted differentiation states. It is considered a significant component of cancer pathogenesis [15]. It proposed that phenotypic plasticity represents a distinct capability within cancer, separate from the established core hallmarks.
Within this context, they identify three primary types of phenotypic plasticity. The first type involves the dedifferentiation of mature cells, which involves the reversion of these cells back to progenitor states. In colon cancer, for instance, the loss of transcription factors HOXA5 and SMAD4 is observed in advanced colon carcinomas, whereas they are highly expressed in differentiating epithelial cells. The second type encompasses blocked differentiation, where cancer cells are trapped in progenitor or stem cell states. This phenomenon is observed in various cancers such as acute promyelocytic leukemia (involving retinoic acid α nuclear receptor—RARα), acute myeloid leukemia, melanoma, and pancreatic cancer. The third type, known as transdifferentiation, occurs in pancreatic ductal adenocarcinoma (PDAC), where pancreatic acinar cells transform into a ductal cell phenotype. This process is regulated by transcription factors PTF1A and MIST1 [15].
Research studies have provided evidence that UA can disrupt all three forms of phenotypic plasticity. Specifically, UA has been found to effectively inhibit hyperproliferation of human dermal fibroblasts by regulating the Smad2/3 pathway [59]. Furthermore, UA has been found to possess the ability to downregulate the expression of epigenetic-modifying enzymes. This includes the DNA methyltransferases (DNMTs) DNMT1 and DNMT3a, as well as the histone deacetylases (HDACs) HDAC1, HDAC2, HDAC3, and HDAC8 belonging to class I, and HDAC6 and HDAC7 belonging to class II (Figure 4B) [60][61][62].
References
- Cheng, N.; Chytil, A.; Shyr, Y.; Joly, A.; Moses, H.L. Transforming growth factor-beta signaling-deficient fibroblasts enhance hepatocyte growth factor signaling in mammary carcinoma cells to promote scattering and invasion. Mol. Cancer Res. 2008, 6, 1521–1533.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Sohn, E.J.; Won, G.; Lee, J.; Yoon, S.W.; Lee, I.; Kim, H.J.; Kim, S.-H. Blockage of epithelial to mesenchymal transition and upregulation of let 7b are critically involved in ursolic acid induced apoptosis in malignant mesothelioma cell. Int. J. Biol. Sci. 2016, 12, 1279.
- Castrejón-Jiménez, N.S.; Leyva-Paredes, K.; Baltierra-Uribe, S.L.; Castillo-Cruz, J.; Campillo-Navarro, M.; Hernández-Pérez, A.D.; Luna-Angulo, A.B.; Chacón-Salinas, R.; Coral-Vázquez, R.M.; Estrada-García, I. Ursolic and oleanolic acids induce mitophagy in A549 human lung cancer cells. Molecules 2019, 24, 3444.
- Guo, W.; Xu, B.; Wang, X.; Zheng, B.; Du, J.; Liu, S. The Analysis of the Anti-Tumor Mechanism of Ursolic Acid Using Connectively Map Approach in Breast Cancer Cells Line MCF-7. Cancer Manag. Res. 2020, 12, 3469–3476.
- Wu, C.C.; Cheng, C.H.; Lee, Y.H.; Chang, I.L.; Chen, H.Y.; Hsieh, C.P.; Chueh, P.J. Ursolic Acid Triggers Apoptosis in Human Osteosarcoma Cells via Caspase Activation and the ERK1/2 MAPK Pathway. J. Agric. Food Chem. 2016, 64, 4220–4226.
- Yang, K.; Chen, Y.; Zhou, J.; Ma, L.; Shan, Y.; Cheng, X.; Wang, Y.; Zhang, Z.; Ji, X.; Chen, L. Ursolic acid promotes apoptosis and mediates transcriptional suppression of CT45A2 gene expression in non-small-cell lung carcinoma harbouring EGFR T790M mutations. Br. J. Pharmacol. 2019, 176, 4609–4624.
- Nagano, T.; Tachihara, M.; Nishimura, Y. Mechanism of Resistance to Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors and a Potential Treatment Strategy. Cells 2018, 7, 212.
- Zhao, H.; Tang, S.; Tao, Q.; Ming, T.; Lei, J.; Liang, Y.; Peng, Y.; Wang, M.; Liu, M.; Yang, H.; et al. Ursolic Acid Suppresses Colorectal Cancer by Down-Regulation of Wnt/β-Catenin Signaling Pathway Activity. J. Agric. Food Chem. 2023, 71, 3981–3993.
- Mariño, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94.
- Hsu, Y.-L.; Kuo, P.-L.; Lin, C.-C. Proliferative inhibition, cell-cycle dysregulation, and induction of apoptosis by ursolic acid in human non-small cell lung cancer A549 cells. Life Sci. 2004, 75, 2303–2316.
- Gao, J.L.; Shui, Y.M.; Jiang, W.; Huang, E.Y.; Shou, Q.Y.; Ji, X.; He, B.C.; Lv, G.Y.; He, T.C. Hypoxia pathway and hypoxia-mediated extensive extramedullary hematopoiesis are involved in ursolic acid’s anti-metastatic effect in 4T1 tumor bearing mice. Oncotarget 2016, 7, 71802–71816.
- Amin, A.R.M.R.; Karpowicz, P.A.; Carey, T.E.; Arbiser, J.; Nahta, R.; Chen, Z.G.; Dong, J.-T.; Kucuk, O.; Khan, G.N.; Huang, G.S.; et al. Evasion of anti-growth signaling: A key step in tumorigenesis and potential target for treatment and prophylaxis by natural compounds. Semin. Cancer Biol. 2015, 35, S55–S77.
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70.
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46.
- Dutta, S.; Ganguly, A.; Chatterjee, K.; Spada, S.; Mukherjee, S. Targets of Immune Escape Mechanisms in Cancer: Basis for Development and Evolution of Cancer Immune Checkpoint Inhibitors. Biology 2023, 12, 218.
- Kang, D.Y.; Sp, N.; Lee, J.-M.; Jang, K.-J. Antitumor Effects of Ursolic Acid through Mediating the Inhibition of STAT3/PD-L1 Signaling in Non-Small Cell Lung Cancer Cells. Biomedicines 2021, 9, 297.
- Peng, K.; Zhang, Y.; Liu, D.; Chen, J. MMP2 is a immunotherapy related biomarker and correlated with cancer-associated fibroblasts infiltrate in melanoma. Cancer Cell Int. 2023, 23, 26.
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899.
- Hiam-Galvez, K.J.; Allen, B.M.; Spitzer, M.H. Systemic immunity in cancer. Nat. Rev. Cancer 2021, 21, 345–359.
- Hassan, L.; Pinon, A.; Limami, Y.; Seeman, J.; Fidanzi-Dugas, C.; Martin, F.; Badran, B.; Simon, A.; Liagre, B. Resistance to ursolic acid-induced apoptosis through involvement of melanogenesis and COX-2/PGE2 pathways in human M4Beu melanoma cancer cells. Exp. Cell Res. 2016, 345, 60–69.
- Bose, S.; Banerjee, S.; Mondal, A.; Chakraborty, U.; Pumarol, J.; Croley, C.R.; Bishayee, A. Targeting the JAK/STAT Signaling Pathway Using Phytocompounds for Cancer Prevention and Therapy. Cells 2020, 9, 1451.
- Cargnin, S.T.; Gnoatto, S.B. Ursolic acid from apple pomace and traditional plants: A valuable triterpenoid with functional properties. Food Chem. 2017, 220, 477–489.
- Zhou, J.X.; Wink, M. Evidence for Anti-Inflammatory Activity of Isoliquiritigenin, 18β Glycyrrhetinic Acid, Ursolic Acid, and the Traditional Chinese Medicine Plants Glycyrrhiza glabra and Eriobotrya japonica, at the Molecular Level. Medicines 2019, 6, 55.
- Zhang, Y.; Huang, L.; Shi, H.; Chen, H.; Tao, J.; Shen, R.; Wang, T. Ursolic acid enhances the therapeutic effects of oxaliplatin in colorectal cancer by inhibition of drug resistance. Cancer Sci. 2018, 109, 94–102.
- Yu, F.Y.; Tu, Y.; Deng, Y.; Guo, C.; Ning, J.; Zhu, Y.; Lv, X.; Ye, H. MiR-4500 is epigenetically downregulated in colorectal cancer and functions as a novel tumor suppressor by regulating HMGA2. Cancer Biol. Ther. 2016, 17, 1149–1157.
- Chen, Z.; Liu, Q.; Zhu, Z.; Xiang, F.; Zhang, M.; Wu, R.; Kang, X. Ursolic Acid Protects Against Proliferation and Inflammatory Response in LPS-Treated Gastric Tumour Model and Cells by Inhibiting NLRP3 Inflammasome Activation. Cancer Manag. Res. 2020, 12, 8413–8424.
- Kim, S.H.; Ryu, H.G.; Lee, J.; Shin, J.; Harikishore, A.; Jung, H.Y.; Kim, Y.S.; Lyu, H.N.; Oh, E.; Baek, N.I.; et al. Ursolic acid exerts anti-cancer activity by suppressing vaccinia-related kinase 1-mediated damage repair in lung cancer cells. Sci. Rep. 2015, 5, 14570.
- Inoue, M.; Honma, Y.; Urano, T.; Suzumiya, J. Japanese apricot extract (MK615) potentiates bendamustine-induced apoptosis via impairment of the DNA damage response in lymphoma cells. Oncol. Lett. 2017, 14, 792–800.
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. Ther. 2020, 5, 28.
- Jain, T.; Sharma, P.; Are, A.C.; Vickers, S.M.; Dudeja, V. New Insights Into the Cancer–Microbiome–Immune Axis: Decrypting a Decade of Discoveries. Front. Immunol. 2021, 12, 622064.
- Sadrekarimi, H.; Gardanova, Z.R.; Bakhshesh, M.; Ebrahimzadeh, F.; Yaseri, A.F.; Thangavelu, L.; Hasanpoor, Z.; Zadeh, F.A.; Kahrizi, M.S. Emerging role of human microbiome in cancer development and response to therapy: Special focus on intestinal microflora. J. Transl. Med. 2022, 20, 301.
- Dzutsev, A.; Badger, J.H.; Perez-Chanona, E.; Roy, S.; Salcedo, R.; Smith, C.K.; Trinchieri, G. Microbes and Cancer. Annu. Rev. Immunol. 2017, 35, 199–228.
- Okumura, S.; Konishi, Y.; Narukawa, M.; Sugiura, Y.; Yoshimoto, S.; Arai, Y.; Sato, S.; Yoshida, Y.; Tsuji, S.; Uemura, K.; et al. Gut bacteria identified in colorectal cancer patients promote tumourigenesis via butyrate secretion. Nat. Commun. 2021, 12, 5674.
- Sheng, Q.; Li, F.; Chen, G.; Li, J.; Li, J.; Wang, Y.; Lu, Y.; Li, Q.; Li, M.; Chai, K. Ursolic Acid Regulates Intestinal Microbiota and Inflammatory Cell Infiltration to Prevent Ulcerative Colitis. J. Immunol. Res. 2021, 2021, 6679316.
- Chen, W.; Yu, Y.; Liu, Y.; Song, C.; Chen, H.; Tang, C.; Song, Y.; Zhang, X. Ursolic acid regulates gut microbiota and corrects the imbalance of Th17/Treg cells in T1DM rats. PLoS ONE 2022, 17, e0277061.
- Saman, H.; Raza, S.S.; Uddin, S.; Rasul, K. Inducing Angiogenesis, a Key Step in Cancer Vascularization, and Treatment Approaches. Cancers 2020, 12, 1172.
- Lin, J.; Chen, Y.; Wei, L.; Hong, Z.; Sferra, T.J.; Peng, J. Ursolic acid inhibits colorectal cancer angiogenesis through suppression of multiple signaling pathways. Int. J. Oncol. 2013, 43, 1666–1674.
- Arandjelovic, S.; Ravichandran, K.S. Phagocytosis of apoptotic cells in homeostasis. Nat. Immunol. 2015, 16, 907–917.
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364.
- Pandey, A.; Yadav, P.; Shukla, S. Unfolding the role of autophagy in the cancer metabolism. Biochem. Biophys. Rep. 2021, 28, 101158.
- Harmand, P.O.; Duval, R.; Liagre, B.; Jayat-Vignoles, C.; Beneytout, J.L.; Delage, C.; Simon, A. Ursolic acid induces apoptosis through caspase-3 activation and cell cycle arrest in HaCat cells. Int. J. Oncol. 2003, 23, 105–112.
- Manu, K.A.; Kuttan, G. Ursolic acid induces apoptosis by activating p53 and caspase-3 gene expressions and suppressing NF-kappaB mediated activation of bcl-2 in B16F-10 melanoma cells. Int. Immunopharmacol. 2008, 8, 974–981.
- Duval, R.E.; Harmand, P.O.; Jayat-Vignoles, C.; Cook-Moreau, J.; Pinon, A.; Delage, C.; Simon, A. Differential involvement of mitochondria during ursolic acid-induced apoptotic process in HaCaT and M4Beu cells. Oncol. Rep. 2008, 19, 145–149.
- Harmand, P.O.; Duval, R.; Delage, C.; Simon, A. Ursolic acid induces apoptosis through mitochondrial intrinsic pathway and caspase-3 activation in M4Beu melanoma cells. Int. J. Cancer 2005, 114, 1–11.
- Limami, Y.; Pinon, A.; Leger, D.Y.; Mousseau, Y.; Cook-Moreau, J.; Beneytout, J.L.; Delage, C.; Liagre, B.; Simon, A. HT-29 colorectal cancer cells undergoing apoptosis overexpress COX-2 to delay ursolic acid-induced cell death. Biochimie 2011, 93, 749–757.
- Pinon, A.; Limami, Y.; Micallef, L.; Cook-Moreau, J.; Liagre, B.; Delage, C.; Duval, R.E.; Simon, A. A novel form of melanoma apoptosis resistance: Melanogenesis up-regulation in apoptotic B16-F0 cells delays ursolic acid-triggered cell death. Exp. Cell Res. 2011, 317, 1669–1676.
- Lewinska, A.; Adamczyk-Grochala, J.; Kwasniewicz, E.; Deregowska, A.; Wnuk, M. Ursolic acid-mediated changes in glycolytic pathway promote cytotoxic autophagy and apoptosis in phenotypically different breast cancer cells. Apoptosis 2017, 22, 800–815.
- Lee, N.R.; Meng, R.Y.; Rah, S.Y.; Jin, H.; Ray, N.; Kim, S.H.; Park, B.H.; Kim, S.M. Reactive Oxygen Species-Mediated Autophagy by Ursolic Acid Inhibits Growth and Metastasis of Esophageal Cancer Cells. Int. J. Mol. Sci. 2020, 21, 9409.
- Wang, J.; Jiang, Z.; Xiang, L.; Li, Y.; Ou, M.; Yang, X.; Shao, J.; Lu, Y.; Lin, L.; Chen, J.; et al. Synergism of ursolic acid derivative US597 with 2-deoxy-D-glucose to preferentially induce tumor cell death by dual-targeting of apoptosis and glycolysis. Sci. Rep. 2014, 4, 5006.
- Zheng, Q.-y.; Jin, F.-s.; Yao, C.; Zhang, T.; Zhang, G.-h.; Ai, X. Ursolic acid-induced AMP-activated protein kinase (AMPK) activation contributes to growth inhibition and apoptosis in human bladder cancer T24 cells. Biochem. Biophys. Res. Commun. 2012, 419, 741–747.
- Park, J.H.; Kwon, H.Y.; Sohn, E.J.; Kim, K.A.; Kim, B.; Jeong, S.J.; Song, J.H.; Koo, J.S.; Kim, S.H. Inhibition of Wnt/β-catenin signaling mediates ursolic acid-induced apoptosis in PC-3 prostate cancer cells. Pharmacol. Rep. PR 2013, 65, 1366–1374.
- Li, J.; Dai, C.; Shen, L. Ursolic Acid Inhibits Epithelial-Mesenchymal Transition through the Axl/NF-κB Pathway in Gastric Cancer Cells. Evid.-Based Complement. Altern. Med. 2019, 2019, 2474805.
- Wang, S.; Chang, X.; Zhang, J.; Li, J.; Wang, N.; Yang, B.; Pan, B.; Zheng, Y.; Wang, X.; Ou, H.; et al. Ursolic Acid Inhibits Breast Cancer Metastasis by Suppressing Glycolytic Metabolism via Activating SP1/Caveolin-1 Signaling. Front. Oncol. 2021, 11, 745584.
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453.
- Schmitt, C.A.; Wang, B.; Demaria, M. Senescence and cancer—Role and therapeutic opportunities. Nat. Rev. Clin. Oncol. 2022, 19, 619–636.
- Han, G.; Park, J.; Oh, K.; Lee, S.-J. Effect of Ursolic Acid on the Development of Mouse Embryonic Stem Cells under Hypoxia. J. Life Sci. 2013, 23, 1223–1229.
- Limami, Y.; Pinon, A.; Leger, D.Y.; Pinault, E.; Delage, C.; Beneytout, J.-L.; Simon, A.; Liagre, B. The P2Y2/Src/p38/COX-2 pathway is involved in the resistance to ursolic acid-induced apoptosis in colorectal and prostate cancer cells. Biochimie 2012, 94, 1754–1763.
- Zhou, X.; Ye, H.; Wang, X.; Sun, J.; Tu, J.; Lv, J. Ursolic acid inhibits human dermal fibroblasts hyperproliferation, migration, and collagen deposition induced by TGF-β via regulating the Smad2/3 pathway. Gene 2023, 867, 147367.
- Kim, H.; Ramirez, C.N.; Su, Z.Y.; Kong, A.N. Epigenetic modifications of triterpenoid ursolic acid in activating Nrf2 and blocking cellular transformation of mouse epidermal cells. J. Nutr. Biochem. 2016, 33, 54–62.
- Yie, Y.; Zhao, S.; Tang, Q.; Zheng, F.; Wu, J.; Yang, L.; Deng, S.; Hann, S.S. Ursolic acid inhibited growth of hepatocellular carcinoma HepG2 cells through AMPKα-mediated reduction of DNA methyltransferase 1. Mol. Cell. Biochem. 2015, 402, 63–74.
- Pal, D.; Raj, K.; Nandi, S.S.; Sinha, S.; Mishra, A.; Mondal, A.; Lagoa, R.; Burcher, J.T.; Bishayee, A. Potential of Synthetic and Natural Compounds as Novel Histone Deacetylase Inhibitors for the Treatment of Hematological Malignancies. Cancers 2023, 15, 2808.
More
Information
Subjects:
Cell Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
749
Revisions:
2 times
(View History)
Update Date:
05 Dec 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No