Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Carla Fernandes | -- | 3156 | 2023-11-28 15:13:03 | | | |
| 2 | Wendy Huang | Meta information modification | 3156 | 2023-11-30 10:52:12 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Ribeiro, R.; Costa, L.; Pinto, E.; Sousa, E.; Fernandes, C. Therapeutic Potential of Marine-Derived Cyclic Peptides in Malaria. Encyclopedia. Available online: https://encyclopedia.pub/entry/52147 (accessed on 26 June 2026).
Ribeiro R, Costa L, Pinto E, Sousa E, Fernandes C. Therapeutic Potential of Marine-Derived Cyclic Peptides in Malaria. Encyclopedia. Available at: https://encyclopedia.pub/entry/52147. Accessed June 26, 2026.
Ribeiro, Ricardo, Lia Costa, Eugénia Pinto, Emília Sousa, Carla Fernandes. "Therapeutic Potential of Marine-Derived Cyclic Peptides in Malaria" Encyclopedia, https://encyclopedia.pub/entry/52147 (accessed June 26, 2026).
Ribeiro, R., Costa, L., Pinto, E., Sousa, E., & Fernandes, C. (2023, November 28). Therapeutic Potential of Marine-Derived Cyclic Peptides in Malaria. In Encyclopedia. https://encyclopedia.pub/entry/52147
Ribeiro, Ricardo, et al. "Therapeutic Potential of Marine-Derived Cyclic Peptides in Malaria." Encyclopedia. Web. 28 November, 2023.
Copy Citation
Malaria is a severe infectious disease which is endemic in tropical and subtropical regions. It can be caused by four species of the intracellular protozoan parasite from the Plasmodium family, with P. falciparum being the disease’s most dangerous form, and it is transmitted by mosquito bites. Parasitic diseases still compromise human health. Some of the available therapeutic drugs have limitations considering their adverse effects, questionable efficacy, and long treatment, which have encouraged drug resistance. There is an urgent need to find new, safe, effective, and affordable antiparasitic drugs. Marine-derived cyclic peptides have been increasingly screened as candidates for developing new drugs.
antiparasitic activity
cyclic peptides
malaria
parasitic diseases
antimalarial activity
1. Introduction
Parasitic diseases affect millions of people around the world, especially in developing countries where sanitary and hygiene conditions are very poor, resulting in a huge mortality rate [1][2]. Despite this, the therapeutic arsenal available to treat this type of disease has been the same for many years, having low effectiveness and many side effects [3]. In addition to all these problems, the resistances acquired by parasites over time are also a huge concern [4]. The therapeutic scenario still remains the same because there is low investment in the development of this class of drugs since these diseases have a higher incidence in poor countries, which are not seen as good markets for the pharmaceutical industry [5]. Fortunately, this reality is changing thanks to the investment of some individuals and organizations for the development of antiparasitic drugs and to the cooperation between industry and academic research groups [6].
The parasitic diseases can be subdivided into two categories: those caused by protozoan parasites, such as malaria (caused by Plasmodium), leishmaniasis (caused by Leishmania), sleeping sickness, Chagas disease (caused by Trypanosoma), and toxoplasmosis (caused by Toxoplasma gondii); and those caused by helminths, such as schistosomiasis (caused by Schistosoma mansoni) and taeniiasis (caused by Taenia solium) [7].
Malaria is a severe infectious disease which is endemic in tropical and subtropical regions [8][9]. It can be caused by four species of the intracellular protozoan parasite from the Plasmodium family, with P. falciparum being the disease’s most dangerous form, and it is transmitted by mosquito bites [10]. According to the World Malaria Report 2022, in 2021, there were 247 million cases and 619,000 deaths from malaria. Moreover, malaria cases have been on the rise since 2016 [11].
Antimalarial drug options are still very limited [12]. One of the oldest drugs is quinine, whose discovery is considered serendipitous. Quinine was isolated from a cinchona tree (Cinchona succirubra), in 1820, by two French chemists. This compound began to be used as an antimalarial and its mechanism of action is still unknown. Quinine has a low therapeutic index and causes many side effects [13]. Subsequently, quinine analogues such as chloroquine, amodiaquine, primaquine, and mefloquine have emerged and are still used in therapy today; we highlight chloroquine, a 4-aminoquinoline derivative of quinine, which was synthesized in 1934 [12]. In 1970, artemisinin was isolated from the plant Artemisia annua and was a very useful compound for the treatment of malaria. In addition, to improve its pharmacokinetic and pharmacodynamic properties, molecular modifications were performed in artemisinin which gave rise to derivatives such as artesunate, arteether, and artemether [14]. All these compounds are still used to treat severe cases of malaria [12]. It is important to note that quinine and artemisinin derivatives are the main drugs currently used to treat malaria [15]. Derivatives of tetracyclines are sometimes used, such as doxycycline, which is used for treatment and prophylaxis in combination with quinine or artesunate, when the treatment with artesunate is not effective [15]. There is also the option of antifolate drugs, which are dihydrofolate reductase inhibitors; however, they are susceptible to rapid development of resistance by the parasite. Therefore, these drugs are used in combination to overcome the resistance issue [12]. The resistance to the few antimalarials available is caused by spontaneous mutations that increase the parasite’s tolerance to the drug [16]. Thus, although there is an urgent need to devise new strategies for developing new drugs that act on already known targets, it is also important to discover new targets.
Currently, several strategies for developing new antimalarials are being explored [17]. For example, via synthesis, Wani et al. [18] obtained a methalocenic analogue of chloroquine, ferroquine, which is in clinical trials for the treatment of uncomplicated malaria [19]. Nature is also an immensely rich source of bioactive compounds, including antimalarials [20][21]. Natural products with antimalarial activity include various classes of compounds, such as alkaloids [22], terpenes [23], biflavonoids [24], lactones [25], coumarins [26], xanthones [27], quinones [28], and peptides [29][30]. Other strategies include the identification of new targets and the design of selective inhibitors for these targets [17] or the repositioning of drugs [31][32].
2. Antimalarial Activity of Marine-Derived Cyclic Peptides
One of the most described antiparasitic activities associated with marine-derived cyclic peptides is antimalarial activity [33]. Among the 25 marine-derived cyclic peptides, 21 peptides (1–21) exhibited promising results against P. falciparum (Figure 1). Some of them have also demonstrated activity against other parasites, or even displayed other biological activities.
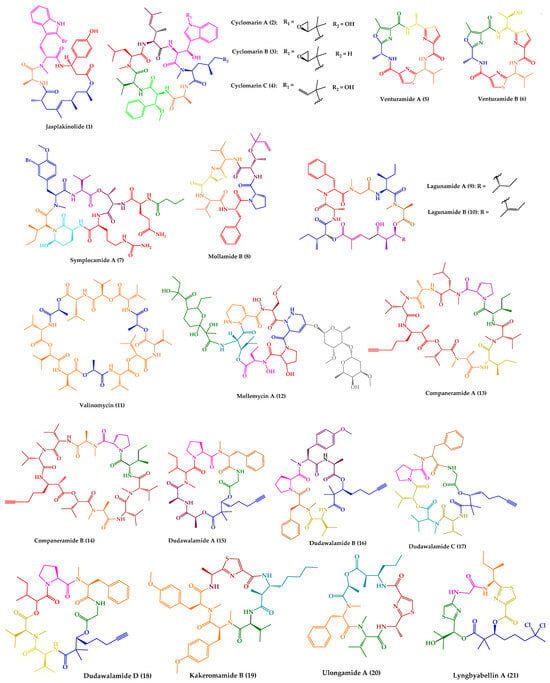
Figure 1. Structures of cyclic peptides with antimalarial activity (1–21).
Jasplakinolide (1), also named jaspamide, was isolated from the soft-bodied sponge species Jaspis collected off the shore of the island of Benga, Fiji [34]. This 19-membered macrocyclic depsipeptide, containing a macrolactam joined by a tripeptide unit and a non-peptide polypropionate sector (Figure 1), has been widely studied over the years. Diverse synthetic strategies for its total synthesis and analogs were explored, which were summarized in a recent review [35].
Regarding the biological activities, several studies can be found related to various activities of 1, such as antifungal [36][37], antitumor [38][39][40][41][42], neuroprotective [43], and antiparasitic [44][45][46] activities. Jasplakinolide (1) is a potent inducer of actin polymerization and filament-stabilizing drug [47][48], and this capability is relevant for its biological effects [49][50][51][52][53][54]. Compound 1 dramatically reduces the critical concentration of actin subunits necessary to drive polymerization and stabilizes filaments [47][54].
Regarding antiparasitic activity, it was found that 1 inhibits P. falciparum growth and impairs host cell invasion due to the stabilization of parasite actin filaments, in a time- and concentration-dependent manner. The decrease was remarkable at day 2 at concentrations of 0.3 µM and above, and parasites finally disappeared at day 4 [44]. P. falciparum and other parasites actively invade host cells, using a mechanism that depends on the interaction of the motor protein myosin and actin filaments which serve as tracks [55][56]. Thus, it has been proposed that the unstable nature of apicomplexan actin filaments is essential for parasite survival [55].
By electron cryomicroscopy, the near-atomic structure of jasplakinolide (1)-stabilized P. falciparum actin 1 filaments (PfAct1) was determined. Jasplakinolide (1) binds at regular intervals inside the filament to three adjacent actin subunits, reinforcing filament stability by hydrophobic interactions (Figure 2) [57].
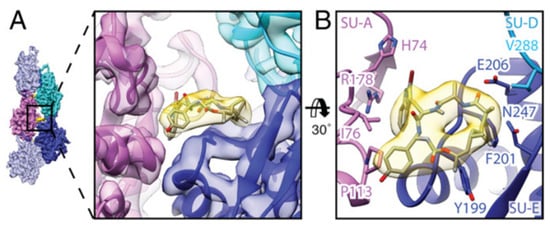
Figure 2. Interaction of jasplakinolide (1) with PfAct1. (A) 1 (yellow) binds noncovalently to three actin subunits (magenta, blue, and cyan). (B) Tilted top view of 1 and amino acids involved in the interaction [57].
In a previous study, jasplakinolide (1) also drastically decreased the T. gondii motility and host cell invasiveness [45]. Additionally, 1 induced an acrosomal process and allowed better visualization of the actin filaments, revealing the conoid and apical complex as major sites of actin polymerization [58].
Jasplakinolide (1) has also been reported to inhibit both the growth and encystation of Entamoeba histolytica and Entamoeba invadens, inducing the formation of F-actin aggregates [46][59]. Compound 1 inhibited the growth of E. histolytica in a dose-dependent manner, in which 0.1 and 0.3 mM of the drug had a similar inhibitory effect, whereas 0.5, 0.7, and 1 mM inhibited 62, 82, and 100% of control growth, respectively. The inhibition of E. invadens by 1 mM of 1 was 25% [59].
Cyclomarins A–C (2–4) were isolated from extracts of a cultured marine bacterium Streptomyces sp. CNB-982, collected in the vicinity of San Diego, CA. Cyclomarin A (2) is the major metabolite, while cyclomarins B and C (3–4) are only produced in lower percentages (2–3%). Cyclomarins A–C (2–4) are cyclic heptapeptides containing two proteinogenic amino acids, (S)-Ala and (S)-Val), an N-methylated (S)-Leu, in addition to four unusual (S)-amino acids (Figure 1). The planar structure of 2–4 was elucidated by 1D and 2D nuclear magnetic resonance (NMR) methods, and their stereochemistry was determined by X-ray crystallography of a diacetate derivative of cyclomarin A (2) [60].
Detailed studies at Novartis indicated a strong activity of compound 2 against P. falciparum strain NF54 in a nanomolar range, with a half-maximal inhibitory concentration (IC50) value of 40 nM [61]. By chemical proteomics, the biotarget was identified to be the diadenosine triphosphate hydrolase (PfAp3Aase) of the protozoan parasite, without affecting the human homolog hFHIT. Co-crystallization experiments revealed that one molecule of 2 binds a dimeric PfAp3Aase and prevents the formation of the enzyme–substrate complex (Figure 3) [61]. Cyclomarin C (4) has also been isolated from Streptomyces sp. BCC26924 and exhibited antimalarial activity against the multidrug-resistant P. falciparum K1 strain, with IC50 values of 0.24 µg/mL [62].
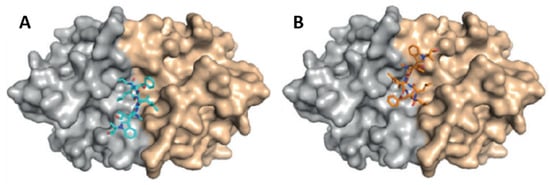
Figure 3. Crystal structure of PfAp3Aase in complex with cyclomarin A (2) (PDB ID: 5CS2). Surface representation of the PfAp3Aase homodimer with the two monomers colored in gold and grey with the fitted cyclomarin A molecules shown as a ball-and-stick model in cyan (A) and orange (B) for the two, mutually exclusive orientations [61].
In addition to antiparasitic activity, cyclomarin A (2) also displayed significant anti-inflammatory activity in both in vivo and in vitro assays [60] and anti-tuberculosis activity (minimum inhibitory concentration (MIC) of 0.1 μM) by targeting the ClpC1 subunit of the caseinolytic protease [63][64].
Regarding the outstanding biological activities of cyclomarins, it is not surprising that synthetic studies have been performed to obtain these compounds in higher quantity as well as analogues for SAR studies [65][66]. The first report of stereoselective syntheses of four unusual amino acids constituents of cyclomarin A (2) was described by Yokokawa and co-workers [67], in 2002. A few years later, synthetic access to cyclomarin C (4) was achieved and optimized by Yao and co-workers [68][69]. Just over ten years later in 2016, Barbie and Kazmaier [70][71] accomplished the total synthesis of cyclomarin A (1) for the first time. They also reported the total synthesis of cyclomarin C (4), cyclomarin D, and other natural peptides [70]. Regarding the planning of derivatives, the first strategy was via semi-synthesis [64], and then by total synthesis [72]. Some addressed simplified structures, such as deoxycyclomarin C [73].
Venturamides A–B (5–6) were isolated from the crude organic extract of a Panamanian collection of Oscillatoria sp. from Buenaventura Bay, via an antimalarial bioassay-guided fractionation. Their isolation constitutes the first example of the identification of cyanobacterial peptides with selective antimalarial activity. Venturamides A–B (5–6) comprise 2,4-disubstituted thiazole units (Figure 1), and their complete structure elucidation was determined via 1D and 2D NMR analyses and Marfey’s method [74]. Venturamides A–B (5–6) were tested for their antimalarial activity against the W2 chloroquine-resistant strain of the malaria parasite. Compound 5 exhibited an IC50 value of 8.2 µM against P. falciparum, with only mild cytotoxicity to mammalian Vero cells (IC50 value of 86 µM). Compound 6 also displayed effective antimalarial activity against P. falciparum, with an IC50 value of 5.6 µM, and mild cytotoxicity to mammalian Vero cells, with an IC50 value of 56 µM. The positive control, chloroquine, showed IC50 values of 80–100 nM [74]. Liu et al. [75] described the total synthesis of both compounds via an effective one-pot procedure for enantiomerical synthesis of thiazole-containing amino acids.
Symplocamide A (7) (Figure 1) was isolated from the marine cyanobacterium Symploca sp., collected from Sunday Island in Papua New Guinea. The planar structure was elucidated by detailed NMR and MS analysis, and chiral high performance liquid chromatography (HPLC) was the method used for stereochemical elucidation of amino acid residues. Compound 7 was screened against three tropical parasites, specifically malaria, Chagas disease, and leishmaniasis, showing interesting results. An IC50 value of 0.95 µM was obtained regarding the inhibition of W2 P. falciparum, while for both T. cruzi and L. donovani, both IC50 values were higher than 9.5 µM [76]. Symplocamide A (7) is also a potent cancer cell cytotoxin to H-460 lung cancer cells, with an IC50 value of 40 nM, and neuro-2a neuroblastoma cells, with an IC50 value of 29 nM. Compound 7 also inhibits serine proteases with a 200-fold greater inhibition of chymotrypsin over trypsin [76]. The total synthesis of symplocamide A (7) using a solid-phase strategy was reported [77].
Mollamide B (8) (Figure 1) was isolated from the tunicate Didemnum molle, collected from Manado Bay, Indonesia. The planar structure was established using 1D and 2D NMR experiments. The absolute configurations of amino acid residues were assigned by Marfey’s method, while the relative configuration at the thiazoline moiety was determined using molecular modeling coupled with NMR-derived restraints. Mollamide B (8) showed inhibitory activity against P. falciparum, clones D6 and W2, with IC50 values of 2.0 and 21 µg/mL, respectively. Mollamide B (8) also exhibited slight activity against L. donovani, with an IC50 value of 18 µg/mL [78].
The cyclic pentadepsipeptides lagunamides A–B (9–10) (Figure 1) were isolated from the marine cyanobacteria Lyngbya majuscula obtained from Pulau Hantu Besar, Singapore. Extensive spectroscopic analysis, including 2D NMR experiments, in addition to Marfey’s method and 3JH-H coupling constant values, a modified method based on Mosher’s reagents, and analysis using liquid chromatography–mass spectrometry (LC–MS), allowed the total elucidation of these molecules. Compounds 9–10 displayed in vitro antimalarial properties, with IC50 values of 0.19 µM and 0.91 µM, respectively, against P. falciparum. Curiously, the only structural difference between these compounds is an additional olefinic group between C40 and C41 in 10. Consequently, this slight structural difference is responsible for an increase in antimalarial activity observed for 9 [79].
In addition to antimalarial activity, lagunamides A–B (9–10) displayed antiswarming activity when tested at 100 ppm against the Gram-negative bacterial strain Pseudomonas aeruginosa, which exerted 62% for 9 and 56% for 10, compared to control. Compounds 9–10 also exhibited potent cytotoxic activity against P388 murine leukemia cell lines, with IC50 values of 6.4 and 20.5 nM, respectively [79]. Further studies revealed that 9 exhibited a selective growth inhibitory activity against a panel of cancer cell lines, with IC50 values ranging from 1.6 nM to 6.4 nM [80]. Molecular mechanism studies suggested that the cytotoxic effect of these compounds might be via induction of mitochondrial mediated apoptosis [80][81].
The total synthesis and stereochemical revision of lagunamide A (9) was first described by Dai et al. [82]. Later, Lin and co-workers [83] reported the synthesis of 9 and five analogues. Although the total synthesis of lagunamide B (10) has not yet been described, a synthetic approach toward the total synthesis of a lagunamide B (10) analogue was reported [84].
A cyclic dodecadepsipeptide, valinomycin (11) (Figure 1), was identified from Streptomyces sp. strains isolated from Mediterranean sponges collected by SCUBA diving offshore of Rovinj, Croatia [85]. This was the first report of the isolation of 11 from a marine source [85]. Nevertheless, 11 had already been recovered from various soil-derived actinomycetes, being first reported in 1955 [86]. Over the years, this cyclic depsipeptide has been extensively studied and a wide range of issues were explored, including structural characterization, biogenesis, synthesis, and bioactivity. In a Scopus search (https://www.scopus.com, accessed on 22 October 2023, with the keyword “valinomycin” and searching within “article title”, 717 original articles were found. Recently, a review summarizing all the relevant features concerning this cyclic depsipeptide (11) was published [87]. Considering the subject of this review, it is important to highlight that 11 showed antiparasitic activity against P. falciparum, with an IC50 value of 5.3 ng/mL [88]. Moreover, 11 also exhibited inhibitory activity against both L. major, with an IC50 value lower than 0.11 μM, and T. brucei, with an IC50 of 3.2 nM [85]. Other relevant biological activities have been described for valinomycin [87], such as insecticidal [89], antiviral [90][91], antibacterial [91], antifungal [92], and antitumor [93] activities. Recent studies reported that 11, as a mitophagy activator, also played a positive role in the treatment of Parkinson’s and Alzheimer’s diseases [94][95].
A glyco-hexadepsipeptide-polyketide, mollemycin A (12) (Figure 1), was isolated from a marine-derived Streptomyces sp. (CMB-M0244) collected off South Molle Island, Queensland. The structure of 12 was elucidated by detailed spectroscopic analysis, supported by chemical derivatization and degradation. C3 Marfey’s analysis was performed for stereochemical configuration assignments. Mollemycin A (12) exhibited exceptionally potent and selective growth inhibitory activity against drug-sensitive 3D7 and multidrug-resistant Dd2 clones of P. falciparum, with IC50 values of 7 and 9 nM, respectively. Remarkably, compound 12 exhibited greater activity when compared to the positive control, chloroquine, with IC50 values of 13 nM for 3D7 and 130 nM for Dd2. Moreover, lower cytotoxicity against a mammalian cell line (>20-fold) was observed [96].
Mollemycin A (12) also exhibited exceptionally potent and selective growth inhibitory activity against Gram-positive and Gram-negative bacteria (IC50 = 10–50 nM) [96].
Companeramides A–B (13–14) were isolated from a marine cyanobacterial assemblage comprising a small filament Leptolyngbya sp., from a reef pinnacle in Coiba National Park, Panama. Companeramides A–B (13–14) contain eight α-amino acid units, a 3-amino-2-methyl-7-octynoic acid, and hydroxy isovaleric acid (Figure 1). Their planar structures were elucidated by NMR spectroscopy and MS. The absolute configurations of the amino and hydroxy acid units in both compounds were determined using a combination of Marfey’s method and chiral HPLC. Companeramides A–B (13–14) were tested in vitro against three strains of the malaria parasite P. falciparum, D6, Dd2, and 7G8, showing high antiplasmodial activity, with IC50 values ranging from 0.22 to 0.70 µM for 13 and from 0.57 to 1.10 µM for 14. The positive control, chloroquine, showed IC50 values of 5 to 80 nM [97].
Dudawalamides A–D (15–18) were isolated from a Papua New Guinean field collection of the cyanobacterium Moorea producens, by a combination of bioassay-guided and spectroscopic approaches. Experiments using 1D and 2D NMR and MS analysis were performed for planar structure elucidation. Diverse techniques were used for the absolute configuration assignments, namely X-ray crystallography, modified Marfey’s analysis, chiral gas chromatography (GC)-MS, and chiral HPLC. Structurally, they consist of six amino acids and a 2,2-dimethyl-3-hydroxy-7-octynoic acid moiety (Figure 1). Dudawalamides A (15) and D (18) showed the most potent activities against P. falciparum, with IC50 values of 3.6 and 3.5 μM, respectively. Dudawalamides B (16) and C (17) were significantly less potent than 15 and 18 [98]. All exhibited minimal mammalian cell cytotoxicity. Regarding SAR features, it was found that slight changes in configuration and sequence of residues had a significant effect on the bioactivity of these marine cyclic peptides. For example, dudawalamides C (17) and D (18) only differ in one methyl group at one residue, specifically L-Hiva to Dallo-Hmpa; however, the single methyl group and stereochemical inversion resulted in a more than 3-fold difference in their P. falciparum inhibition [98]. It was found that dudawalamide D (18) was also relatively potent against L. donovani (IC50 = 2.6 μM) [98].
Recently, a new cyclic peptide, kakeromamide B (19) (Figure 1), was isolated from an extract of a marine cyanobacterium Moorea producens collected off the Northern Lau Islands of Fiji. The extract showed strong potency against P. falciparum and low toxicity to human liver cells [99]. The stereostructure of kakeromamide B (19) was assigned by different spectroscopic techniques, high-resolution electrospray ionization mass spectrometry (HRESIMS), and Marfey’s analysis. Kakeromamide B (19) exhibited activity against P. falciparum blood-stage and against P. berghei liver schizonts with effective concentration in 50% of population (EC50) values of 0.89 and 1.1 µM, respectively. By a threading-based computational method, FINDSITEcomb2.0, the binding of 19 to potentially druggable proteins of P. falciparum was predicted. Kakeromamide B (19) was predicted to bind to several Plasmodium actin-like proteins and a sortilin protein, suggesting possible interference with parasite invasion of host cells. In a mammalian actin polymerization assay, it was found that 19, in fact, stimulated actin polymerization in a dose-dependent manner [99].
The cyclic depsipeptides ulongamide A (20) and lyngbyabellin A (21) (Figure 1) were also identified from the same antimalarial extract of the Fijian marine cyanobacterium Moorea producens [99]. Ulongamide A (20) was previously isolated from Palauan collections of the marine cyanobacterium Lyngbya sp. [100] and lyngbyabellin A (21) from the marine cyanobacterium Lyngbya majuscula [101]. While 20 exhibited moderate activity against P. falciparum blood-stages with EC50 values of 0.99 µM, 21 was more potent with an EC50 value of 0.15 nM [99].
References
- Renslo, A.R.; McKerrow, J.H. Drug discovery and development for neglected parasitic diseases. Nat. Chem. Biol. 2006, 2, 701–710.
- Lee, S.-M.; Kim, M.-S.; Hayat, F.; Shin, D. Recent Advances in the Discovery of Novel Antiprotozoal Agents. Molecules 2019, 24, 3886.
- Pink, R.; Hudson, A.; Mouriès, M.-A.; Bendig, M. Opportunities and Challenges in Antiparasitic Drug Discovery. Nat. Rev. Drug Discov. 2005, 4, 727–740.
- Picot, S.; Beugnet, F.; Leboucher, G.; Bienvenu, A.-L. Drug resistant parasites and fungi from a one-health perspective: A global concern that needs transdisciplinary stewardship programs. One Health 2022, 14, 100368.
- McKerrow, J.H. Designing Drugs for Parasitic Diseases of the Developing World. PLoS Med. 2005, 2, e210.
- Goupil, L.S.; McKerrow, J.H. Introduction: Drug Discovery and Development for Neglected Diseases. Chem. Rev. 2014, 114, 11131–11137.
- Cummings, R.D.; Hokke, C.H.; Haslam, S.M. Parasitic Infections. In Essentials of Glycobiology, 4th ed.; Harbor, C.S., Ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2022.
- Lacerda, A.F.; Pelegrini, P.B.; de Oliveira, D.M.; Vasconcelos, É.A.R.; Grossi-de-Sá, M.F. Anti-parasitic Peptides from Arthropods and their Application in Drug Therapy. Front. Microbiol. 2016, 7, 91.
- Requena-Méndez, A.; Cattaneo, P.; Bogale, R.T.; Marti-Soler, H.; Wångdahl, A.; Buonfrate, D.; Bisoffi, Z.; Färnert, A.; Rodríguez-Cuadrado, A.; Monge-Maillo, B.; et al. Malaria parasite prevalence among migrants: A systematic review and meta-analysis. Clin. Microbiol. Infect. 2023.
- White, N.J.; Pukrittayakamee, S.; Hien, T.T.; Faiz, M.A.; Mokuolu, O.A.; Dondorp, A.M. Malaria. Lancet 2014, 383, 723–735.
- WHO World Malaria Report 2022. Available online: https://www.who.int/teams/global-malaria-programme/reports/world-malaria-report-2022 (accessed on 5 October 2023).
- Bloland, P.B. Drug Resistance in Malaria; WHO: Chamblee, GA, USA, 2011; Volume WHO/CDS/CSR/DRS/2001.4.
- Achan, J.; Talisuna, A.O.; Erhart, A.; Yeka, A.; Tibenderana, J.K.; Baliraine, F.N.; Rosenthal, P.J.; D’Alessandro, U. Quinine, an old anti-malarial drug in a modern world: Role in the treatment of malaria. Malar. J. 2011, 10, 144.
- Guo, Z. Artemisinin anti-malarial drugs in China. Acta Pharm. Sin. B 2016, 6, 115–124.
- Talapko, J.; Škrlec, I.; Alebić, T.; Jukić, M.; Včev, A. Malaria: The Past and the Present. Microorganisms 2019, 7, 179.
- Menard, D.; Dondorp, A. Antimalarial Drug Resistance: A Threat to Malaria Elimination. Cold Spring Harb. Perspect. Med. 2017, 7, a025619.
- Pandey, S.K.; Anand, U.; Siddiqui, W.A.; Tripathi, R. Drug Development Strategies for Malaria: With the Hope for New Antimalarial Drug Discovery-An Update. Adv. Med. 2023, 2023, 5060665.
- Wani, W.A.; Jameel, E.; Baig, U.; Mumtazuddin, S.; Hun, L.T. Ferroquine and its derivatives: New generation of antimalarial agents. Eur. J. Med. Chem. 2015, 101, 534–551.
- Adoke, Y.; Zoleko-Manego, R.; Ouoba, S.; Tiono, A.B.; Kaguthi, G.; Bonzela, J.E.; Duong, T.T.; Nahum, A.; Bouyou-Akotet, M.; Ogutu, B.; et al. A randomized, double-blind, phase 2b study to investigate the efficacy, safety, tolerability and pharmacokinetics of a single-dose regimen of ferroquine with artefenomel in adults and children with uncomplicated Plasmodium falciparum malaria. Malar. J. 2021, 20, 222.
- Tajuddeen, N.; Van Heerden, F.R. Antiplasmodial natural products: An update. Malar. J. 2019, 18, 404.
- Ribeiro, G.D.J.G.; Rei Yan, S.L.; Palmisano, G.; Wrenger, C. Plant Extracts as a Source of Natural Products with Potential Antimalarial Effects: An Update from 2018 to 2022. Pharmaceutics 2023, 15, 1638.
- Zahari, A.; Cheah, F.K.; Mohamad, J.; Sulaiman, S.N.; Litaudon, M.; Leong, K.H.; Awang, K. Antiplasmodial and antioxidant isoquinoline alkaloids from Dehaasia longipedicellata. Planta Med. 2014, 80, 599–603.
- Claudino, V.D.; da Silva, K.C.; Cechinel Filho, V.; Yunes, R.A.; Delle Monache, F.; Giménez, A.; Salamanca, E.; Gutierrez-Yapu, D.; Malheiros, A. Drimanes from Drimys brasiliensis with leishmanicidal and antimalarial activity. Mem. Inst. Oswaldo Cruz 2013, 108, 140–144.
- Konziase, B. Protective activity of biflavanones from Garcinia kola against Plasmodium infection. J. Ethnopharmacol. 2015, 172, 214–218.
- Du, Y.; Abedi, A.K.; Valenciano, A.L.; Fernández-Murga, M.L.; Cassera, M.B.; Rasamison, V.E.; Applequist, W.L.; Miller, J.S.; Kingston, D.G.I. Isolation of the New Antiplasmodial Butanolide, Malleastrumolide A, from Malleastrum sp. (Meliaceae) from Madagascar. Chem. Biodivers. 2017, 14, e1700331.
- Chung, I.M.; Ghimire, B.K.; Kang, E.Y.; Moon, H.I. Antiplasmodial and cytotoxic activity of khellactone derivatives from Angelica purpuraefolia Chung. Phytother. Res. 2010, 24, 469–471.
- Upegui, Y.; Robledo, S.M.; Gil Romero, J.F.; Quiñones, W.; Archbold, R.; Torres, F.; Escobar, G.; Nariño, B.; Echeverri, F. In vivo Antimalarial Activity of α-Mangostin and the New Xanthone δ-Mangostin. Phytother. Res. 2015, 29, 1195–1201.
- Dai, Y.; Harinantenaina, L.; Bowman, J.D.; Da Fonseca, I.O.; Brodie, P.J.; Goetz, M.; Cassera, M.B.; Kingston, D.G. Isolation of antiplasmodial anthraquinones from Kniphofia ensifolia, and synthesis and structure-activity relationships of related compounds. Bioorg. Med. Chem. 2014, 22, 269–276.
- Ibrahim, S.R.M.; Abdallah, H.M.; Elkhayat, E.S.; Al Musayeib, N.M.; Asfour, H.Z.; Zayed, M.F.; Mohamed, G.A. Fusaripeptide A: New antifungal and anti-malarial cyclodepsipeptide from the endophytic fungus Fusarium sp. J. Asian Nat. Prod. Res. 2018, 20, 75–85.
- Bracegirdle, J.; Casandra, D.; Rocca, J.R.; Adams, J.H.; Baker, B.J. Highly N-Methylated Peptides from the Antarctic Sponge Inflatella coelosphaeroides Are Active against Plasmodium falciparum. J. Nat. Prod. 2022, 85, 2454–2460.
- Parquet, V.; Henry, M.; Wurtz, N.; Dormoi, J.; Briolant, S.; Gil, M.; Baret, E.; Amalvict, R.; Rogier, C.; Pradines, B. Atorvastatin as a potential anti-malarial drug: In vitro synergy in combinational therapy with quinine against Plasmodium falciparum. Malar. J. 2010, 9, 139.
- Pongratz, P.; Kurth, F.; Ngoma, G.M.; Basra, A.; Ramharter, M. In vitro activity of antifungal drugs against Plasmodium falciparum field isolates. Wien. Klin. Wochenschr. 2011, 123 (Suppl. 1), 26–30.
- Fotie, J. The potential of peptides and depsipeptides from terrestrial and marine organisms in the fight against human protozoan diseases. In Bioactive Natural Products: Chemistry and Biology; Wiley Blackwell: Hoboken, NJ, USA, 2015; pp. 279–320.
- Crews, P.; Manes, L.V.; Boehler, M. Jasplakinolide, a cyclodepsipeptide from the marine sponge, Jaspis sp. Tetrahedron Lett. 1986, 27, 2797–2800.
- Xu, Y.Y.; Liu, C.; Liu, Z.P. Advances in the total synthesis of cyclodepsipeptide (+)-jasplakinolide (jaspamide) and its analogs. Curr. Org. Synth. 2013, 10, 67–89.
- Scott, V.R.; Boehme, R.; Matthews, T.R. New class of antifungal agents: Jasplakinolide, a cyclodepsipeptide from the marine sponge, Jaspis species. Antimicrob. Agents Chemother. 1988, 32, 1154–1157.
- Zabriskie, T.M.; Ireland, C.M.; Klocke, J.A.; Marcus, A.H.; Molinski, T.F.; Faulkner, D.J.; Xu, C.; Clardy, J.C. Jaspamide, a Modified Peptide from a Jaspis Sponge, with Insecticidal and Antifungal Activity. J. Am. Chem. Soc. 1986, 108, 3123–3124.
- Yang, K.; Luo, M.; Li, H.; Abdulrehman, G.; Kang, L. Effects of jasplakinolide on cytotoxicity, cytoskeleton and apoptosis in two different colon cancer cell lines treated with m-THPC-PDT. Photodiagn. Photodyn. Ther. 2021, 35, 102425.
- Ali, R.; Mir, H.A.; Hamid, R.; Shah, R.A.; Khanday, F.A.; Bhat, S.S. Jasplakinolide Attenuates Cell Migration by Impeding Alpha-1-syntrophin Protein Phosphorylation in Breast Cancer Cells. Protein J. 2021, 40, 234–244.
- Odaka, C.; Sanders, M.L.; Crews, P. Jasplakinolide induces apoptosis in various transformed cell lines by a caspase-3-like protease-dependent pathway. Clin. Diagn. Lab. Immunol. 2000, 7, 947–952.
- Takeuchi, H.; Ara, G.; Sausville, E.A.; Teicher, B. Jasplakinolide: Interaction with radiation and hyperthermia in human prostate carcinoma and Lewis lung carcinoma. Cancer Chemother. Pharmacol. 1998, 42, 491–496.
- Senderowicz, A.M.J.; Kaur, G.; Sainz, E.; Laing, C.; Inman, W.D.; Rodriguez, J.; Crews, P.; Malspeis, L.; Grever, M.R.; Sausville, E.A.; et al. Jasplakinolide’s inhibition of the growth of prostate carcinoma cells in vitro with disruption of the actin cytoskeleton. J. Natl. Cancer Inst. 1995, 87, 46–51.
- Alvarinõ, R.; Alonso, E.; Tabudravu, J.N.; Pérez-Fuentes, N.; Alfonso, A.; Botana, L.M. Tavarua Deoxyriboside A and Jasplakinolide as Potential Neuroprotective Agents: Effects on Cellular Models of Oxidative Stress and Neuroinflammation. ACS Chem. Neurosci. 2021, 12, 150–162.
- Mizuno, Y.; Makioka, A.; Kawazu, S.I.; Kano, S.; Kawai, S.; Akaki, M.; Aikawa, M.; Ohtomo, H. Effect of jasplakinolide on the growth, invasion, and actin cytoskeleton of Plasmodium falciparum. Parasitol. Res. 2002, 88, 844–848.
- Poupel, O.; Tardieux, I. Toxoplasma gondii motility and host cell invasiveness are drastically impaired by jasplakinolide, a cyclic peptide stabilizing F-actin. Microbes Infect. 1999, 1, 653–662.
- Makioka, A.; Kumagai, M.; Ohtomo, H.; Kobayashi, S.; Takeuchi, T. Effect of jasplakinolide on the growth, encystation, and actin cytoskeleton of Entamoeba histolytica and Entamoeba invadens. J. Parasitol. 2001, 87, 399–405.
- Bubb, M.R.; Senderowicz, A.M.J.; Sausville, E.A.; Duncan, K.L.K.; Korn, E.D. Jasplakinolide, a cytotoxic natural product, induces actin polymerization and competitively inhibits the binding of phalloidin to F-actin. J. Biol. Chem. 1994, 269, 14869–14871.
- Holzinger, A. Jasplakinolide: An actin-specific reagent that promotes actin polymerization. Methods Mol. Biol. 2009, 586, 71–87.
- Pospich, S.; Merino, F.; Raunser, S. Structural Effects and Functional Implications of Phalloidin and Jasplakinolide Binding to Actin Filaments. Structure 2020, 28, 437–449.e5.
- Sun, Z.; Zhang, X.; Hu, L.; Wang, X.; Sun, Z.; Zhao, E.; Wang, J.; Wei, F. F-actin microfilament polymerized by jasplakinolide affects the expression of aquaporin-4 in astrocytic swelling after oxygen-glucose deprivation and reoxygenation. Mater. Express 2020, 10, 563–570.
- Zhou, C.; Wang, Y.; Pan, D.; Sun, Y.; Cao, J. The effect of Cytochalasin B and Jasplakinolide on depolymerization of actin filaments in goose muscles during postmortem conditioning. Food Res. Int. 2016, 90, 1–7.
- Zhang, X.; Cui, X.; Cheng, L.; Guan, X.; Li, H.; Li, X.; Cheng, M. Actin Stabilization by Jasplakinolide Affects the Function of Bone Marrow-Derived Late Endothelial Progenitor Cells. PLoS ONE 2012, 7, e50899.
- Jing, X.; Sun, J.L.; Zhang, X.Y.; Tang, K.X.; Yin, Q.L.; Wu, H.Y.; Wang, J.H.; Wang, J.W.; Cheng, M. Jasplakinolide affects the functions of HUVECs via actin stabilization. Chin. Pharmacol. Bull. 2013, 29, 1079–1083.
- Bubb, M.R.; Spector, I.; Beyer, B.B.; Fosen, K.M. Effects of jasplakinolide on the kinetics of actin polymerization. An explanation for certain in vivo observations. J. Biol. Chem. 2000, 275, 5163–5170.
- Skillman, K.M.; Diraviyam, K.; Khan, A.; Tang, K.; Sept, D.; Sibley, L.D. Evolutionarily divergent, unstable filamentous actin is essential for gliding motility in apicomplexan parasites. PLoS Pathog. 2011, 7, e1002280.
- Drewry, L.L.; Sibley, L.D. Toxoplasma actin is required for efficient host cell invasion. mBio 2015, 6, 00557-15.
- Pospich, S.; Kumpula, E.P.; Von Der Ecken, J.; Vahokoski, J.; Kursula, I.; Raunser, S. Near-atomic structure of jasplakinolide-stabilized malaria parasite F-actin reveals the structural basis of filament instability. Proc. Natl. Acad. Sci. USA 2017, 114, 10636–10641.
- Shaw, M.K.; Tilney, L.G. Induction of an acrosomal process in Toxoplasma gondii: Visualization of actin filaments in a protozoan parasite. Proc. Natl. Acad. Sci. USA 1999, 96, 9095–9099.
- Makioka, A.; Kumagai, M.; Ohtomo, H.; Kobayashi, S.; Takeuchi, T. Growth inhibition and actin aggregate formation of Entamoeba histolytica by jasplakinolide. Arch. Med. Res. 2000, 31, S145–S146.
- Renner, M.K.; Shen, Y.C.; Cheng, X.C.; Jensen, P.R.; Frankmoelle, W.; Kauffman, C.A.; Fenical, W.; Lobkovsky, E.; Clardy, J. Cyclomarins A-C, new antiinflammatory cyclic peptides produced by a marine bacterium (Streptomyces sp.). J. Am. Chem. Soc. 1999, 121, 11273–11276.
- Bürstner, N.; Roggo, S.; Ostermann, N.; Blank, J.; Delmas, C.; Freuler, F.; Gerhartz, B.; Hinniger, A.; Hoepfner, D.; Liechty, B.; et al. Gift from Nature: Cyclomarin A Kills Mycobacteria and Malaria Parasites by Distinct Modes of Action. ChemBioChem 2015, 16, 2433–2436.
- Intaraudom, C.; Rachtawee, P.; Suvannakad, R.; Pittayakhajonwut, P. Antimalarial and antituberculosis substances from Streptomyces sp. BCC26924. Tetrahedron 2011, 67, 7593–7597.
- Vasudevan, D.; Rao, S.P.S.; Noble, C.G. Structural basis of mycobacterial inhibition by Cyclomarin A. J. Biol. Chem. 2013, 288, 30883–30891.
- Schmitt, E.K.; Riwanto, M.; Sambandamurthy, V.; Roggo, S.; Miault, C.; Zwingelstein, C.; Krastel, P.; Noble, C.; Beer, D.; Rao, S.P.S.; et al. The natural product cyclomarin kills Mycobacterium tuberculosis by targeting the ClpC1 subunit of the caseinolytic protease. Angew. Chem. Int. Ed. 2011, 50, 5889–5891.
- Kazmaier, U.; Junk, L. Recent developments on the synthesis and bioactivity of ilamycins/rufomycins and cyclomarins, marine cyclopeptides that demonstrate anti-malaria and anti-tuberculosis activity. Mar. Drugs 2021, 19, 446.
- Kiefer, A.; Kazmaier, U. Syntheses of Cyclomarins—Interesting Marine Natural Products with Distinct Mode of Action towards Malaria and Tuberculosis. Synthesis 2019, 51, 107–121.
- Sugiyama, H.; Shioiri, T.; Yokokawa, F. Syntheses of four unusual amino acids, constituents of cyclomarin A. Tetrahedron Lett. 2002, 43, 3489–3492.
- Wen, S.J.; Hu, T.S.; Yao, Z.J. Macrocyclization studies and total synthesis of cyclomarin C, an anti-inflammatory marine cyclopeptide. Tetrahedron 2005, 61, 4931–4938.
- Wen, S.J.; Yao, Z.J. Total synthesis of cyclomarin C. Org. Lett. 2004, 6, 2721–2724.
- Barbie, P.; Kazmaier, U. Total synthesis of cyclomarins A, C and D, marine cyclic peptides with interesting anti-tuberculosis and anti-malaria activities. Org. Biomol. Chem. 2016, 14, 6036–6054.
- Barbie, P.; Kazmaier, U. Total Synthesis of Cyclomarin A, a Marine Cycloheptapeptide with Anti-Tuberculosis and Anti-Malaria Activity. Org. Lett. 2016, 18, 204–207.
- Kiefer, A.; Bader, C.D.; Held, J.; Esser, A.; Rybniker, J.; Empting, M.; Müller, R.; Kazmaier, U. Synthesis of New Cyclomarin Derivatives and Their Biological Evaluation towards Mycobacterium tuberculosis and Plasmodium falciparum. Chem. Eur. J. 2019, 25, 8894–8902.
- Barbie, P.; Kazmaier, U. Total synthesis of desoxycyclomarin C and the cyclomarazines A and B. Org. Biomol. Chem. 2016, 14, 6055–6064.
- Linington, R.G.; González, J.; Ureña, L.D.; Romero, L.I.; Ortega-Barría, E.; Gerwick, W.H. Venturamides A and B: Antimalarial constituents of the Panamanian marine cyanobacterium Oscillatoria sp. J. Nat. Prod. 2007, 70, 397–401.
- Liu, Y.; He, P.; Zhang, Y.; Zhang, X.; Liu, J.; Du, Y. One-pot enantiomeric synthesis of thiazole-containing amino acids: Total synthesis of venturamides A and B. J. Org. Chem. 2018, 83, 3897–3905.
- Linington, R.G.; Edwards, D.J.; Shuman, C.F.; McPhail, K.L.; Matainaho, T.; Gerwick, W.H. Symplocamide A, a potent cytotoxin and chymotrypsin inhibitor from the marine cyanobacterium Symploca sp. J. Nat. Prod. 2008, 71, 22–27.
- Stolze, S.C.; Meltzer, M.; Ehrmann, M.; Kaiser, M. Solid phase total synthesis of the 3-amino-6-hydroxy-2-piperidone (Ahp) cyclodepsipeptide and protease inhibitor Symplocamide A. Chem. Commun. 2010, 46, 8857–8859.
- Donia, M.S.; Wang, B.; Dunbar, D.C.; Desai, P.V.; Patny, A.; Avery, M.; Hamann, M.T. Mollamides B and C, Cyclic hexapeptides from the indonesian tunicate Didemnum molle. J. Nat. Prod. 2008, 71, 941–945.
- Tripathi, A.; Puddick, J.; Prinsep, M.R.; Rottmann, M.; Tan, L.T. Lagunamides A and B: Cytotoxic and antimalarial cyclodepsipeptides from the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2010, 73, 1810–1814.
- Tripathi, A.; Fang, W.; Leong, D.T.; Tan, L.T. Biochemical studies of the lagunamides, potent cytotoxic cyclic depsipeptides from the marine cyanobacterium Lyngbya majuscula. Mar. Drugs 2012, 10, 1126–1137.
- Huang, X.; Huang, W.; Li, L.; Sun, X.; Song, S.; Xu, Q.; Zhang, L.; Wei, B.G.; Deng, X. Structure Determinants of Lagunamide A for Anticancer Activity and Its Molecular Mechanism of Mitochondrial Apoptosis. Mol. Pharm. 2016, 13, 3756–3763.
- Dai, L.; Chen, B.; Lei, H.; Wang, Z.; Liu, Y.; Xu, Z.; Ye, T. Total synthesis and stereochemical revision of lagunamide A. Chem. Commun. 2012, 48, 8697–8699.
- Huang, W.; Ren, R.G.; Dong, H.Q.; Wei, B.G.; Lin, G.Q. Diverse synthesis of marine cyclic depsipeptide lagunamide A and its analogues. J. Org. Chem. 2013, 78, 10747–10762.
- Pal, S.; Chakraborty, T.K. Toward the total synthesis of a lagunamide B analogue. Tetrahedron Lett. 2014, 55, 3469–3472.
- Pimentel-Elardo, S.M.; Kozytska, S.; Bugni, T.S.; Ireland, C.M.; Moll, H.; Hentschel, U. Anti-parasitic compounds from Streptomyces sp. strains isolated from Mediterranean sponges. Mar. Drugs 2010, 8, 373–380.
- Brockmann, H.; Schmidt-Kastner, G. Valinomycin I, XXVII. Mitteil. über Antibiotica aus Actinomyceten. Chem. Berichte 1955, 88, 57–61.
- Huang, S.; Liu, Y.; Liu, W.; Neubauer, P.; Li, J. The nonribosomal peptide valinomycin: From discovery to bioactivity and biosynthesis. Microorganisms 2021, 9, 780.
- Gumila, C.; Ancelin, M.L.; Jeminet, G.; Delort, A.M.; Miquel, G.; Vial, H.J. Differential in vitro activities of ionophore compounds against Plasmodium falciparum and mammalian cells. Antimicrob. Agents Chemother. 1996, 40, 602–608.
- Heisey, R.M.; Mishra, S.K.; Keller, J.E.; Miller, J.R.; Putnam, A.R.; Huang, J.; D’Silva, T.D.J. Production of valinomycin, an insecticidal antibiotic, by Streptomyces griseus var. flexipertum var. nov. J. Agric. Food Chem. 1988, 36, 1283–1286.
- Zhang, D.; Ma, Z.; Chen, H.; Lu, Y.; Chen, X. Valinomycin as a potential antiviral agent against coronaviruses: A review. Biomed. J. 2020, 43, 414–423.
- Wibowo, J.T.; Kellermann, M.Y.; Köck, M.; Putra, M.Y.; Murniasih, T.; Mohr, K.I.; Wink, J.; Praditya, D.F.; Steinmann, E.; Schupp, P.J. Anti-Infective and Antiviral Activity of Valinomycin and Its Analogues from a Sea Cucumber-Associated Bacterium, Streptomyces sp. SV 21. Mar. Drugs 2021, 19, 81.
- Jeon, C.W.; Kim, D.R.; Kwak, Y.S. Valinomycin, produced by Streptomyces sp. S8, a key antifungal metabolite in large patch disease suppressiveness. World J. Microbiol. Biotechnol. 2019, 35, 128.
- Zhang, Q.W.; Baig, M.M.F.A.; Zhang, T.Q.; Zhai, T.T.; Qin, X.; Xia, X.H. Liposomal valinomycin mediated cellular K+ leak promoting apoptosis of liver cancer cells. J. Control. Release 2021, 337, 317–328.
- Rakovic, A.; Ziegler, J.; Mårtensson, C.U.; Prasuhn, J.; Shurkewitsch, K.; König, P.; Paulson, H.L.; Klein, C. PINK1-dependent mitophagy is driven by the UPS and can occur independently of LC3 conversion. Cell Death Differ. 2019, 26, 1428–1441.
- Xiong, X.; Li, S.; Han, T.L.; Zhou, F.; Zhang, X.; Tian, M.; Tang, L.; Li, Y. Study of mitophagy and ATP-related metabolomics based on β-amyloid levels in Alzheimer’s disease. Exp. Cell Res. 2020, 396, 112266.
- Raju, R.; Khalil, Z.G.; Piggott, A.M.; Blumenthal, A.; Gardiner, D.L.; Skinner-Adams, T.S.; Capon, R.J. Mollemycin A: An antimalarial and antibacterial glyco-hexadepsipeptide- polyketide from an Australian marine-derived Streptomyces sp. (CMB-M0244). Org. Lett. 2014, 16, 1716–1719.
- Vining, O.B.; Medina, R.A.; Mitchell, E.A.; Videau, P.; Li, D.; Serrill, J.D.; Kelly, J.X.; Gerwick, W.H.; Proteau, P.J.; Ishmael, J.E. Depsipeptide companeramides from a panamanian marine cyanobacterium associated with the coibamide producer. J. Nat. Prod. 2015, 78, 413–420.
- Almaliti, J.; Malloy, K.L.; Glukhov, E.; Spadafora, C.; Gutiérrez, M.; Gerwick, W.H. Dudawalamides A-D, Antiparasitic Cyclic Depsipeptides from the Marine Cyanobacterium Moorea producens. J. Nat. Prod. 2017, 80, 1827–1836.
- Sweeney-Jones, A.M.; Gagaring, K.; Antonova-Koch, J.; Zhou, H.; Mojib, N.; Soapi, K.; Skolnick, J.; McNamara, C.W.; Kubanek, J. Antimalarial peptide and polyketide natural products from the Fijian marine cyanobacterium Moorea producens. Mar. Drugs 2020, 18, 167.
- Luesch, H.; Williams, P.G.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. Ulongamides A-F, new β-amino acid-containing cyclodepsipeptides from Palauan collections of the marine cyanobacterium Lyngbya sp. J. Nat. Prod. 2002, 65, 996–1000.
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J.; Mooberry, S.L. Isolation, structure determination, and biological activity of lyngbyabellin A from the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2000, 63, 611–615.
More
Information
Subjects:
Infectious Diseases
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
432
Revisions:
2 times
(View History)
Update Date:
30 Nov 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No