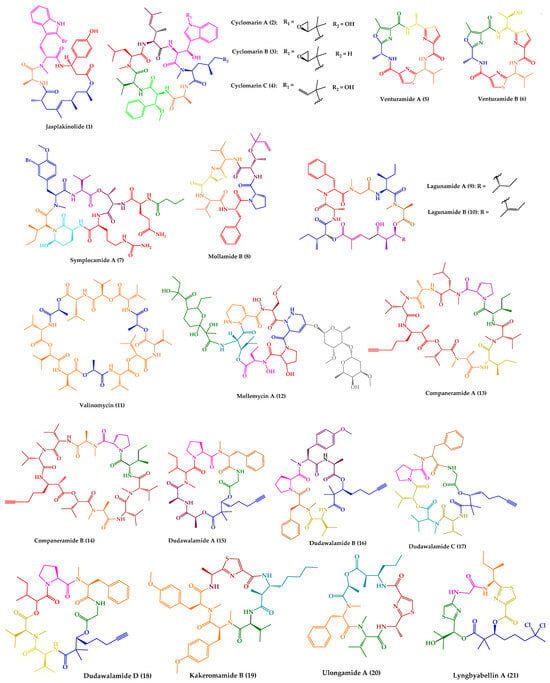
2. Antimalarial Activity of Marine-Derived Cyclic Peptides
One of the most described antiparasitic activities associated with marine-derived cyclic peptides is antimalarial activity
[33][153]. Among the 25 marine-derived cyclic peptides, 21 peptides (
1–
21) exhibited promising results against
P. falciparum (
Figure 1). Some of them have also demonstrated activity against other parasites, or even displayed other biological activities.
Figure 1.
Structures of cyclic peptides with antimalarial activity (
1
–
21
).
Jasplakinolide (
1), also named jaspamide, was isolated from the soft-bodied sponge species Jaspis collected off the shore of the island of Benga, Fiji
[34][127]. This 19-membered macrocyclic depsipeptide, containing a macrolactam joined by a tripeptide unit and a non-peptide polypropionate sector (
Figure 1), has been widely studied over the years. Diverse synthetic strategies for its total synthesis and analogs were explored, which were summarized in a recent review
[35][154].
Regarding the biological activities, several studies can be found related to various activities of
1, such as antifungal
[36][37][155,156], antitumor
[38][39][40][41][42][157,158,159,160,161], neuroprotective
[43][162], and antiparasitic
[44][45][46][128,129,130] activities. Jasplakinolide (
1) is a potent inducer of actin polymerization and filament-stabilizing drug
[47][48][163,164], and this capability is relevant for its biological effects
[49][50][51][52][53][54][165,166,167,168,169,170]. Compound
1 dramatically reduces the critical concentration of actin subunits necessary to drive polymerization and stabilizes filaments
[47][54][163,170].
Regarding antiparasitic activity, it was found that
1 inhibits
P. falciparum growth and impairs host cell invasion due to the stabilization of parasite actin filaments, in a time- and concentration-dependent manner. The decrease was remarkable at day 2 at concentrations of 0.3 µM and above, and parasites finally disappeared at day 4
[44][128].
P. falciparum and other parasites actively invade host cells, using a mechanism that depends on the interaction of the motor protein myosin and actin filaments which serve as tracks
[55][56][171,172]. Thus, it has been proposed that the unstable nature of apicomplexan actin filaments is essential for parasite survival
[55][171].
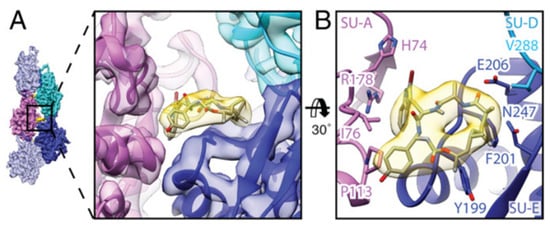
By electron cryomicroscopy, the near-atomic structure of jasplakinolide (
1)-stabilized
P. falciparum actin 1 filaments (PfAct1) was determined. Jasplakinolide (
1) binds at regular intervals inside the filament to three adjacent actin subunits, reinforcing filament stability by hydrophobic interactions (
Figure 2)
[57][173].
Figure 2. Interaction of jasplakinolide (
1) with PfAct1. (
A)
1 (yellow) binds noncovalently to three actin subunits (magenta, blue, and cyan). (
B) Tilted top view of
1 and amino acids involved in the interaction
[57][173].
In a previous study, jasplakinolide (
1) also drastically decreased the
T. gondii motility and host cell invasiveness
[45][129]. Additionally,
1 induced an acrosomal process and allowed better visualization of the actin filaments, revealing the conoid and apical complex as major sites of actin polymerization
[58][174].
Jasplakinolide (
1) has also been reported to inhibit both the growth and encystation of
Entamoeba histolytica and
Entamoeba invadens, inducing the formation of F-actin aggregates
[46][59][130,131]. Compound
1 inhibited the growth of
E. histolytica in a dose-dependent manner, in which 0.1 and 0.3 mM of the drug had a similar inhibitory effect, whereas 0.5, 0.7, and 1 mM inhibited 62, 82, and 100% of control growth, respectively. The inhibition of
E. invadens by 1 mM of
1 was 25%
[59][131].
Cyclomarins A–C (
2–
4) were isolated from extracts of a cultured marine bacterium
Streptomyces sp. CNB-982, collected in the vicinity of San Diego, CA. Cyclomarin A (
2) is the major metabolite, while cyclomarins B and C (
3–
4) are only produced in lower percentages (2–3%). Cyclomarins A–C (
2–
4) are cyclic heptapeptides containing two proteinogenic amino acids, (
S)-Ala and (
S)-Val), an
N-methylated (
S)-Leu, in addition to four unusual (
S)-amino acids (
Figure 1). The planar structure of
2–
4 was elucidated by 1D and 2D nuclear magnetic resonance (NMR) methods, and their stereochemistry was determined by X-ray crystallography of a diacetate derivative of cyclomarin A (
2)
[60][132].
Detailed studies at Novartis indicated a strong activity of compound
2 against
P. falciparum strain NF54 in a nanomolar range, with a half-maximal inhibitory concentration (IC
50) value of 40 nM
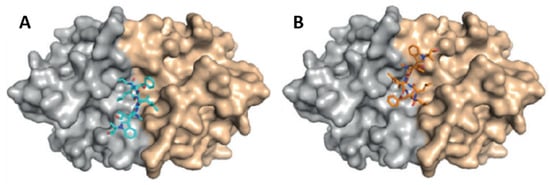
[61][133]. By chemical proteomics, the biotarget was identified to be the diadenosine triphosphate hydrolase (PfAp3Aase) of the protozoan parasite, without affecting the human homolog hFHIT. Co-crystallization experiments revealed that one molecule of
2 binds a dimeric PfAp3Aase and prevents the formation of the enzyme–substrate complex (
Figure 3)
[61][133]. Cyclomarin C (
4) has also been isolated from
Streptomyces sp. BCC26924 and exhibited antimalarial activity against the multidrug-resistant
P. falciparum K1 strain, with IC
50 values of 0.24 µg/mL
[62][134].
Figure 3. Crystal structure of PfAp3Aase in complex with cyclomarin A (
2) (PDB ID: 5CS2). Surface representation of the PfAp3Aase homodimer with the two monomers colored in gold and grey with the fitted cyclomarin A molecules shown as a ball-and-stick model in cyan (
A) and orange (
B) for the two, mutually exclusive orientations
[61][133].
In addition to antiparasitic activity, cyclomarin A (
2) also displayed significant anti-inflammatory activity in both in vivo and in vitro assays
[60][132] and anti-tuberculosis activity (minimum inhibitory concentration (MIC) of 0.1 μM) by targeting the ClpC1 subunit of the caseinolytic protease
[63][64][175,176].
Regarding the outstanding biological activities of cyclomarins, it is not surprising that synthetic studies have been performed to obtain these compounds in higher quantity as well as analogues for SAR studies
[65][66][177,178]. The first report of stereoselective syntheses of four unusual amino acids constituents of cyclomarin A (
2) was described by Yokokawa and co-workers
[67][179], in 2002. A few years later, synthetic access to cyclomarin C (
4) was achieved and optimized by Yao and co-workers
[68][69][180,181]. Just over ten years later in 2016, Barbie and Kazmaier
[70][71][182,183] accomplished the total synthesis of cyclomarin A (
1) for the first time. They also reported the total synthesis of cyclomarin C (
4), cyclomarin D, and other natural peptides
[70][182]. Regarding the planning of derivatives, the first strategy was via semi-synthesis
[64][176], and then by total synthesis
[72][184]. Some addressed simplified structures, such as deoxycyclomarin C
[73][185].
Venturamides A–B (
5–
6) were isolated from the crude organic extract of a Panamanian collection of
Oscillatoria sp. from Buenaventura Bay, via an antimalarial bioassay-guided fractionation. Their isolation constitutes the first example of the identification of cyanobacterial peptides with selective antimalarial activity. Venturamides A–B (
5–
6) comprise 2,4-disubstituted thiazole units (
Figure 1), and their complete structure elucidation was determined via 1D and 2D NMR analyses and Marfey’s method
[74][135]. Venturamides A–B (
5–
6) were tested for their antimalarial activity against the W2 chloroquine-resistant strain of the malaria parasite. Compound
5 exhibited an IC
50 value of 8.2 µM against
P. falciparum, with only mild cytotoxicity to mammalian Vero cells (IC
50 value of 86 µM). Compound
6 also displayed effective antimalarial activity against
P. falciparum, with an IC
50 value of 5.6 µM, and mild cytotoxicity to mammalian Vero cells, with an IC
50 value of 56 µM. The positive control, chloroquine, showed IC
50 values of 80–100 nM
[74][135]. Liu et al.
[75][186] described the total synthesis of both compounds via an effective one-pot procedure for enantiomerical synthesis of thiazole-containing amino acids.
Symplocamide A (
7) (
Figure 1) was isolated from the marine cyanobacterium
Symploca sp., collected from Sunday Island in Papua New Guinea. The planar structure was elucidated by detailed NMR and MS analysis, and chiral high performance liquid chromatography (HPLC) was the method used for stereochemical elucidation of amino acid residues. Compound
7 was screened against three tropical parasites, specifically malaria, Chagas disease, and leishmaniasis, showing interesting results. An IC
50 value of 0.95 µM was obtained regarding the inhibition of W2
P. falciparum, while for both
T. cruzi and
L. donovani, both IC
50 values were higher than 9.5 µM
[76][136]. Symplocamide A (
7) is also a potent cancer cell cytotoxin to H-460 lung cancer cells, with an IC
50 value of 40 nM, and neuro-2a neuroblastoma cells, with an IC
50 value of 29 nM. Compound
7 also inhibits serine proteases with a 200-fold greater inhibition of chymotrypsin over trypsin
[76][136]. The total synthesis of symplocamide A (
7) using a solid-phase strategy was reported
[77][187].
Mollamide B (
8) (
Figure 1) was isolated from the tunicate
Didemnum molle, collected from Manado Bay, Indonesia. The planar structure was established using 1D and 2D NMR experiments. The absolute configurations of amino acid residues were assigned by Marfey’s method, while the relative configuration at the thiazoline moiety was determined using molecular modeling coupled with NMR-derived restraints. Mollamide B (
8) showed inhibitory activity against
P. falciparum, clones D6 and W2, with IC
50 values of 2.0 and 21 µg/mL, respectively. Mollamide B (
8) also exhibited slight activity against L. donovani, with an IC
50 value of 18 µg/mL
[78][137].
The cyclic pentadepsipeptides lagunamides A–B (
9–
10) (
Figure 1) were isolated from the marine cyanobacteria
Lyngbya majuscula obtained from Pulau Hantu Besar, Singapore. Extensive spectroscopic analysis, including 2D NMR experiments, in addition to Marfey’s method and
3J
H-H coupling constant values, a modified method based on Mosher’s reagents, and analysis using liquid chromatography–mass spectrometry (LC–MS), allowed the total elucidation of these molecules. Compounds
9–
10 displayed in vitro antimalarial properties, with IC
50 values of 0.19 µM and 0.91 µM, respectively, against
P. falciparum. Curiously, the only structural difference between these compounds is an additional olefinic group between C
40 and C
41 in
10. Consequently, this slight structural difference is responsible for an increase in antimalarial activity observed for
9 [79][138].
In addition to antimalarial activity, lagunamides A–B (
9–
10) displayed antiswarming activity when tested at 100 ppm against the Gram-negative bacterial strain
Pseudomonas aeruginosa, which exerted 62% for
9 and 56% for
10, compared to control. Compounds
9–
10 also exhibited potent cytotoxic activity against P388 murine leukemia cell lines, with IC
50 values of 6.4 and 20.5 nM, respectively
[79][138]. Further studies revealed that
9 exhibited a selective growth inhibitory activity against a panel of cancer cell lines, with IC
50 values ranging from 1.6 nM to 6.4 nM
[80][188]. Molecular mechanism studies suggested that the cytotoxic effect of these compounds might be via induction of mitochondrial mediated apoptosis
[80][81][188,189].
The total synthesis and stereochemical revision of lagunamide A (
9) was first described by Dai et al.
[82][190]. Later, Lin and co-workers
[83][191] reported the synthesis of
9 and five analogues. Although the total synthesis of lagunamide B (
10) has not yet been described, a synthetic approach toward the total synthesis of a lagunamide B (
10) analogue was reported
[84][192].
A cyclic dodecadepsipeptide, valinomycin (
11) (
Figure 1), was identified from
Streptomyces sp. strains isolated from Mediterranean sponges collected by SCUBA diving offshore of Rovinj, Croatia
[85][140]. This was the first report of the isolation of
11 from a marine source
[85][140]. Nevertheless,
11 had already been recovered from various soil-derived actinomycetes, being first reported in 1955
[86][193]. Over the years, this cyclic depsipeptide has been extensively studied and a wide range of issues were explored, including structural characterization, biogenesis, synthesis, and bioactivity. In a Scopus search (
https://www.scopus.com, accessed on 22 October 2023, with the keyword “valinomycin” and searching within “article title”, 717 original articles were found. Recently, a review summarizing all the relevant features concerning this cyclic depsipeptide (
11) was published
[87][194]. Considering the subject of this review, it is important to highlight that
11 showed antiparasitic activity against
P. falciparum, with an IC
50 value of 5.3 ng/mL
[88][139]. Moreover,
11 also exhibited inhibitory activity against both
L. major, with an IC
50 value lower than 0.11 μM, and
T. brucei, with an IC
50 of 3.2 nM
[85][140]. Other relevant biological activities have been described for valinomycin
[87][194], such as insecticidal
[89][195], antiviral
[90][91][196,197], antibacterial
[91][197], antifungal
[92][198], and antitumor
[93][199] activities. Recent studies reported that
11, as a mitophagy activator, also played a positive role in the treatment of Parkinson’s and Alzheimer’s diseases
[94][95][200,201].
A glyco-hexadepsipeptide-polyketide, mollemycin A (
12) (
Figure 1), was isolated from a marine-derived
Streptomyces sp. (CMB-M0244) collected off South Molle Island, Queensland. The structure of
12 was elucidated by detailed spectroscopic analysis, supported by chemical derivatization and degradation. C3 Marfey’s analysis was performed for stereochemical configuration assignments. Mollemycin A (
12) exhibited exceptionally potent and selective growth inhibitory activity against drug-sensitive 3D7 and multidrug-resistant Dd2 clones of
P. falciparum, with IC
50 values of 7 and 9 nM, respectively. Remarkably, compound
12 exhibited greater activity when compared to the positive control, chloroquine, with IC
50 values of 13 nM for 3D7 and 130 nM for Dd2. Moreover, lower cytotoxicity against a mammalian cell line (>20-fold) was observed
[96][141].
Mollemycin A (
12) also exhibited exceptionally potent and selective growth inhibitory activity against Gram-positive and Gram-negative bacteria (IC
50 = 10–50 nM)
[96][141].
Companeramides A–B (
13–
14) were isolated from a marine cyanobacterial assemblage comprising a small filament
Leptolyngbya sp., from a reef pinnacle in Coiba National Park, Panama. Companeramides A–B (
13–
14) contain eight α-amino acid units, a 3-amino-2-methyl-7-octynoic acid, and hydroxy isovaleric acid (
Figure 1). Their planar structures were elucidated by NMR spectroscopy and MS. The absolute configurations of the amino and hydroxy acid units in both compounds were determined using a combination of Marfey’s method and chiral HPLC. Companeramides A–B (
13–
14) were tested in vitro against three strains of the malaria parasite
P. falciparum, D6, Dd2, and 7G8, showing high antiplasmodial activity, with IC
50 values ranging from 0.22 to 0.70 µM for
13 and from 0.57 to 1.10 µM for
14. The positive control, chloroquine, showed IC
50 values of 5 to 80 nM
[97][142].
Dudawalamides A–D (
15–
18) were isolated from a Papua New Guinean field collection of the cyanobacterium
Moorea producens, by a combination of bioassay-guided and spectroscopic approaches. Experiments using 1D and 2D NMR and MS analysis were performed for planar structure elucidation. Diverse techniques were used for the absolute configuration assignments, namely X-ray crystallography, modified Marfey’s analysis, chiral gas chromatography (GC)-MS, and chiral HPLC. Structurally, they consist of six amino acids and a 2,2-dimethyl-3-hydroxy-7-octynoic acid moiety (
Figure 1). Dudawalamides A (
15) and D (
18) showed the most potent activities against
P. falciparum, with IC
50 values of 3.6 and 3.5 μM, respectively. Dudawalamides B (
16) and C (
17) were significantly less potent than
15 and
18 [98][143]. All exhibited minimal mammalian cell cytotoxicity. Regarding SAR features, it was found that slight changes in configuration and sequence of residues had a significant effect on the bioactivity of these marine cyclic peptides. For example, dudawalamides C (
17) and D (
18) only differ in one methyl group at one residue, specifically L-Hiva to Dallo-Hmpa; however, the single methyl group and stereochemical inversion resulted in a more than 3-fold difference in their
P. falciparum inhibition
[98][143]. It was found that dudawalamide D (
18) was also relatively potent against
L. donovani (IC
50 = 2.6 μM)
[98][143].
Recently, a new cyclic peptide, kakeromamide B (
19) (
Figure 1), was isolated from an extract of a marine cyanobacterium
Moorea producens collected off the Northern Lau Islands of Fiji. The extract showed strong potency against
P. falciparum and low toxicity to human liver cells
[99][144]. The stereostructure of kakeromamide B (
19) was assigned by different spectroscopic techniques, high-resolution electrospray ionization mass spectrometry (HRESIMS), and Marfey’s analysis. Kakeromamide B (
19) exhibited activity against
P. falciparum blood-stage and against
P. berghei liver schizonts with effective concentration in 50% of population (EC
50) values of 0.89 and 1.1 µM, respectively. By a threading-based computational method, FINDSITEcomb2.0, the binding of
19 to potentially druggable proteins of
P. falciparum was predicted. Kakeromamide B (
19) was predicted to bind to several
Plasmodium actin-like proteins and a sortilin protein, suggesting possible interference with parasite invasion of host cells. In a mammalian actin polymerization assay, it was found that
19, in fact, stimulated actin polymerization in a dose-dependent manner
[99][144].
The cyclic depsipeptides ulongamide A (
20) and lyngbyabellin A (
21) (
Figure 1) were also identified from the same antimalarial extract of the Fijian marine cyanobacterium
Moorea producens [99][144]. Ulongamide A (
20) was previously isolated from Palauan collections of the marine cyanobacterium
Lyngbya sp.
[100][202] and lyngbyabellin A (
21) from the marine cyanobacterium
Lyngbya majuscula [101][203]. While
20 exhibited moderate activity against
P. falciparum blood-stages with EC
50 values of 0.99 µM,
21 was more potent with an EC
50 value of 0.15 nM
[99][144].