Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Chiara Marabelli | -- | 1930 | 2023-11-28 09:10:23 | | | |
| 2 | Sirius Huang | -1 word(s) | 1929 | 2023-11-29 02:06:13 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Marabelli, C.; Santiago, D.J.; Priori, S.G. Calsequestrin Multimeric Organization. Encyclopedia. Available online: https://encyclopedia.pub/entry/52119 (accessed on 24 June 2026).
Marabelli C, Santiago DJ, Priori SG. Calsequestrin Multimeric Organization. Encyclopedia. Available at: https://encyclopedia.pub/entry/52119. Accessed June 24, 2026.
Marabelli, Chiara, Demetrio J. Santiago, Silvia G. Priori. "Calsequestrin Multimeric Organization" Encyclopedia, https://encyclopedia.pub/entry/52119 (accessed June 24, 2026).
Marabelli, C., Santiago, D.J., & Priori, S.G. (2023, November 28). Calsequestrin Multimeric Organization. In Encyclopedia. https://encyclopedia.pub/entry/52119
Marabelli, Chiara, et al. "Calsequestrin Multimeric Organization." Encyclopedia. Web. 28 November, 2023.
Copy Citation
Calsequestrin (CASQ) is a key intra-sarcoplasmic reticulum Ca2+-handling protein that plays a pivotal role in the contraction of cardiac and skeletal muscles. Its Ca2+-dependent polymerization dynamics shape the translation of electric excitation signals to the Ca2+-induced contraction of the actin-myosin architecture. Mutations in CASQ are linked to life-threatening pathological conditions, including tubular aggregate myopathy, malignant hyperthermia, and Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT).
calsequestrin
calcium
intracellular ion channels
ryanodine receptor
ion channel regulators
calcium-binding proteins
polymerization disorders
arrhythmias
tubular aggregate myopathy
malignant hyperthermia
1. An Introduction to CASQ
1.1. The Right Buffer at the Right Place
For muscle sarcomere contraction to occur, large quantities of Ca2+ must flow out of the sarcoplasmic reticulum (SR) lumen into the cytosol, driven by a very steep electrochemical gradient (SR/cytosol Nernst potential for Ca2+ is approx. 125 mV in a non-contracting cell). The maintenance of this on-demand Ca2+ supply partially relies on the highly acidic calsequestrin (CASQ) protein, and its interaction with each member of the Calcium Release Unit (Figure 1): Junctin (JNT), Triadin (TRDN), and RyR [1][2][3]. CASQ is relatively small (45 kDa), and yet is the most prominently expressed protein within the lumen of the “junctional” sarcoplasmic reticulum (jSR) (up to 100 mg/mL) [4][5]. Abundant quantities of Ca2+ cooperatively bind to CASQ with low affinity, allowing for a rapid back-and-forth (with each contraction–relaxation cycle) exchange of massive quantities of Ca2+ between CASQ and the matrix of the jSR. In mammals, two CASQ genes are differentially expressed in skeletal fast-twitch fibers (CASQ1) and cardiac muscle (CASQ2), whereas both isoforms are equally represented in slow-twitch fibers [6][7]. Measurements in CASQ2-KO mice indicate that CASQ2 stores about 50% of the cardiac SR Ca2+ content [8] while maintaining a free calcium concentration of approximately 1 mM [9]. Similar studies in CASQ1-KO fast-twitch skeletal muscles indicate that the Ca2+ stored in CSQ1 contributes 75% of the released calcium [10], while still maintaining a free Ca2+ concentration of approximately 1 mM in the resting fiber.
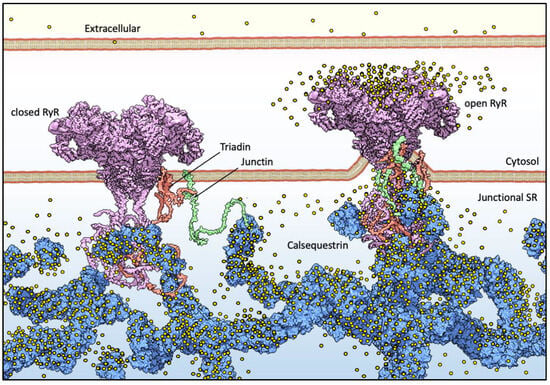
Figure 1. Basic components of Ca2+ release machinery in striated muscles. Excitation–contraction coupling (ECC) is the process by which an action potential at the sarcolemma leads to a massive release of intracellular Ca2+ which, in turn, activates cell-wide contraction. ECC occurs at subcellular structures that are periodically distributed inside muscle cells, called “junctions” (when referred to in structural terms) or Ca2+ Release Units (CRUs, when referred to in functional terms). Junctions are so termed because they are composed of specialized regions of the sarcolemma and of the “junctional” sarcoplasmic reticulum (jSR), both coalescing within nanometer distances. ECC occurs via different mechanisms in skeletal and cardiac muscles, so in the figure, the sarcolemmal portion of the junctions is not decorated with tissue-specific components. At the jSR membrane, CRUs are decorated by ordered arrays of ryanodine receptors (RyR, pink), which act as the SR Ca2+ release channels. In the lumen of the jSR, CASQ (blue) buffers Ca2+ ions (yellow spheres), whereas Junctin (green) and Triadin (red) are transmembrane proteins anchoring CASQ to the RyR and acting as signaling mediators between CASQ and RyR. The actual stoichiometry for the Jnt:Trd:RyR complex is unknown.
1.2. CASQ Macro-Architecture(s)
The first electron micrographs of the jSR revealed an electron-dense filling able to assume the most diverse conformations from wire-shaped structures immediately beneath and parallel to the junctional membrane to intricately branched filaments and spherical bodies or puncta [11][12][13][14][15][16][17]. Imaging in vivo and cross-linking experiments support the notion that this material is Ca2+-complexed CASQ [18][19][20]. In addition, these structures disassemble and disperse under luminal Ca2+ depletion conditions [15][21], which correlates with the fact that the recombinant protein can organize in vitro into multiple architectures of varying compactness, hierarchically ordered in response to rising Ca2+ concentrations from micromolar levels up to 20 mM [3][16][17][22] (Figure 2). As higher order polymeric structures are formed, the capacity and cooperativity of Ca2+-binding events show a parallel, stepwise increase, which has consequences for the modulation of intra-SR Ca2+ storage and release. But how are CASQ multimers organized?
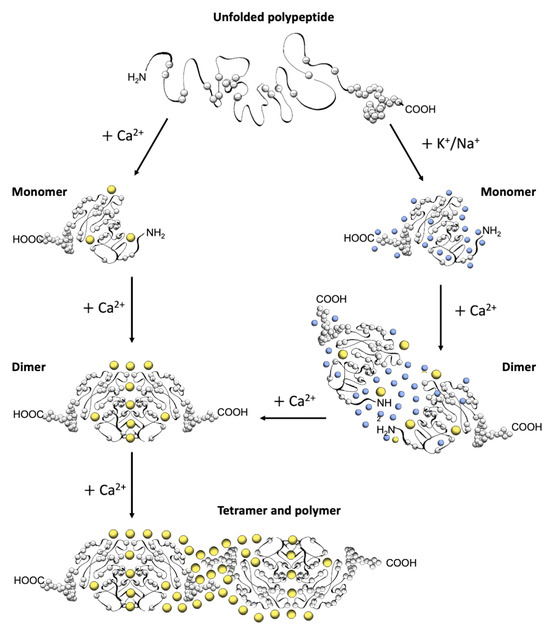
Figure 2. Schematic diagram of CASQ structural transitions guided by increasing amounts of calcium and/or monovalent cations. Negative charges along the polypeptide backbone are represented as white spheres. At close-to-neutral pH and low ionic strength, CASQ retains an unfolded conformation. Either monovalent ions such as Na+ or K+ (blue spheres) or divalent ions such as Ca2+ (yellow spheres) mask the negative charges of the abundant exposed glutamate and aspartate residues (103 and 102 acidic residues over 362 (CASQ1) and 380 (CASQ2) amino-acid-long polypeptides, respectively). In both cases, the ionic strength increase triggers formation of the same tertiary structure. To this end, the concentrations of Ca2+ required are about three orders of magnitude lower (about 0.1 mM) than that of other monovalent cations (100 mM). Further Ca2+ addition triggers dimerization through N-terminal tail swapping. A higher Ca2+ concentration is required for tight dimerization to occur when competing Na+ and K+ ions are present. The further increase in Ca2+ ions promotes “back-to-back” tetramerization, which concomitantly leads to an enormous increase in the calcium-binding capacity of each CASQ monomer. The protein’s intrinsically disordered C-termini, involved in stabilizing the Ca2+-dependent tetramer, are represented in a fixed conformation for clarity of representation.
2. CASQ Multimeric Organization
2.1. CASQ Secondary and Tertiary Folding Is Sustained by Cations
A high abundance of carboxylates, carried by aspartic and glutamic acids, characterizes all CASQ isoforms, with the human skeletal and cardiac proteins featuring an isoelectric point of 4.0 and 4.2, respectively (Figure 2). Due to repulsion between negative charges, the polypeptide retains an extended, random coil conformation at low ionic strength (i.e., lower than that provided by 100 mM KCl) [23]. Multiple, monovalent, or divalent cations can likewise guide the folding of three negatively charged, nearly identical, thioredoxin domains (Figure 3A,B) [24][25][26][27][28], where numerous hydrophobic interactions hold the interior of the domains [29]. The minimal ionic concentration sustaining secondary and tertiary protein folding varies with the coordination number and ionic radius of the cation [23][24][25][27][28][29][30], with Ca2+ featuring the lowest effective concentrations, plus distinctive binding cooperativity even at modest ionic strength [23][25]. Low concentrations of monovalent cations (i.e., 85–150 mM KCl) have a cooperative effect on Ca2+-binding events at the early stages of protein folding [23][29][31]. It appears, however, that similar or higher abundances of monovalent ions inhibit compaction of the critical, dimerization-competent monomer, which is ultimately stabilized by a minimal amount of Ca2+ [32].
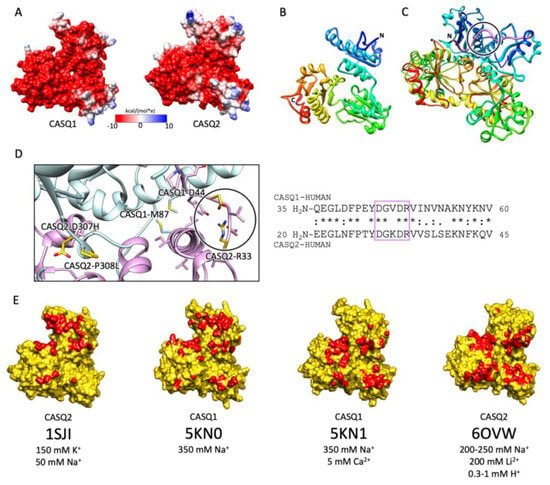
Figure 3. CASQ monomeric structure and salt-sensitive dimerization. (A) Representation of skeletal (left, PDB ID: 5KN0) and cardiac (right, PDB ID: 1SJI) CASQ surfaces. Colors mirror the coulombic potential values according to the color legend shown. The disordered C-terminus is not present in the experimental structures. (B) Ribbon representation of CASQ monomer. The PDB model is 1SJI for cardiac CASQ2. The polypeptide is conventionally rainbow-colored starting from its N-terminus (blue) to its C-terminus (red). (C) The corresponding cardiac CASQ dimer is shown (PDB ID: 1SJI). One of the two swapped N-terminal domains within the dimeric state is colored violet for ease of identification. The N-terminal portion within the circle corresponds to the dimerization switch. (D) Left: Zoomed view of the dimerization N-terminal switch (within the circle). The most relevant pathological missense mutation residues falling close or within the dimerization interface are evidenced in gold and represented as sticks. Right: The sequences of the N-termini for both skeletal and cardiac CASQ are compared. Identical or similar residues are evidenced by an asterisk or a colon, respectively. The dimerization switch is highlighted in the violet box. (E) Surface representation of single monomers, where the dimerization interface is, is evidenced in red. For each structure, the concentrations of the cations of crystallization conditions are indicated below the relative PDB ID.
2.2. The Ca2+-Specific Dimerization Switch Is Salt-Sensitive
The first and obligatory step of CASQ supra-molecular assembly is the Ca2+-driven “front-to-front” dimerization through N-terminal domain swapping (Figure 3C,D) [29][32][33][34]. Upon Ca2+ binding, a few alternatively charged residues of the N-terminus flip and establish a hydrogen bonding network. In turn, this secures the extended conformation of the N-terminal tail over the surface of the opposite monomer (Figure 3C,D). Multiple intercalated Ca2+ ions bridge in between the abundant carboxylate groups exposed from each monomer’s surface, as revealed by equilibrium dialysis [4][32]. The comparison between different crystal structures of the CASQ dimers reveals that the inter-monomer space could also be filled by other cations than Ca2+. Nonetheless, only divalent ions engage the two monomers in a tight architecture, allowing more hydrophobic residues to line the inter-monomer interface (Figure 3E) [31][32][35][36].
2.3. Poorly Understood Ca2+-Dependent Mechanisms Drive CASQ Polymerization
The bottleneck of CASQ polymerization is proper tetramer assembly [4][32]. The ordered “back-to-back” tetramerization necessitates the intrinsically disordered C-terminal tail [37][38]. Truncation mutants lacking the C-terminus indeed behave as constitutive dimers in the solution and fail to multimerize upon Ca2+ addition [4]. It has been hypothesized that the strong negatively charged tail repels the formation of improperly positioned dimers, yet the exact mechanism driving physiological polymerization has not been elucidated. Interestingly, the main difference between skeletal and cardiac isoforms lies precisely in the C-terminal segment (Figure 4). The longer and more negatively charged C-terminus of CASQ2 drives multimerization at higher Ca2+ concentrations (~2 mM) than CASQ1 (~0.7 mM) [37][38], and the swapping of this segment between isoforms causes the reciprocal exchange of their Ca2+-dependent polymerization kinetics [37]. As the coulombic properties of the C-terminal tail drive the specific conformational responsiveness to Ca2+, the other negative charges of the polypeptide shape the surface electric potential of the growing CASQ polymer, with CASQ1 polymers providing a more charged surface than CASQ2, onto which more Ca2+ ions can be adsorbed [4]. Since the dimer-to-tetramer transition, CASQ Ca2+ binding capacity and cooperativity increase; newly bound Ca2+ ions somehow stabilize further Ca2+-coordinating sites. The outcome is a multimeric species endowed with an enormous Ca2+-binding capacity; up to 60 Ca2+ ions are bound per molecule for human cardiac CASQ2, and up to 80 for the skeletal CASQ1 [4][29][32].
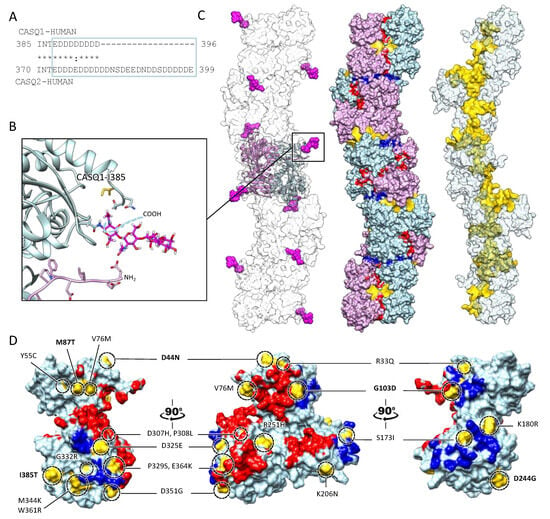
Figure 4. Multiple sites globally design the quaternary assembly of CASQ. (A) The C-terminal Intrinsically Disordered Region is highlighted in the blue box for skeletal and cardiac CASQ isoforms. (B) Inset from the glycosylated skeletal CASQ dimer structure (PDB ID: 3TRQ), showing the relative positions and orientations of the glycosyl moieties on Asn-316, with respect to the C-terminus of the glycosyl-carrying monomer and the N-terminus of the dimeric partner. The presence of a two-mannosyl chain is sufficient to fully stabilize the N-terminus, whereas the C-terminus retains a disordered conformation. The site of the known pathological missense mutation I385T in CASQ1 is close to the glycosylation site. (C) CASQ polymeric structure is represented for a portion corresponding to five dimers (PDB ID 6OWW). Left: The structure of native glycosylated skeletal CASQ1 (PDB ID: 3TRQ) is fitted within the 6OWW polymeric structure to visualize the position of the glycosyl moieties (magenta) with respect to the linear polymer surface (transparent). Centre: For each dimer, one monomer is colored light blue, and the second monomer is colored pink. A 90° rotation from one dimer to the adjacent one is evident along the polymer axis. The inter-monomer surfaces are colored red, whereas the inter-dimer interfaces are colored blue. The empty tunnel running within the structure is colored yellow. Entry/exit spaces in continuity with the internal tunnel are visible. Right: The internal tunnel is shown in yellow, and only one monomer for each dimer (corresponding to light blue monomers in the central polymer structure) is represented as a semi-transparent surface. (D) Surface missense pathological mutations are evidenced in gold on the surface of a CASQ2 monomer (PDB ID: 6OWW). The inter-monomer surfaces are colored red, whereas the inter-dimer interfaces are colored blue. CASQ1 mutations (D44N, M87Q, G103D, D244G, and I385T) are in bold for ease of identification among the numerous CASQ2 missense mutations.
References
- Boncompagni, S.; Thomas, M.; Lopez, J.R.; Allen, P.D.; Yuan, Q.; Kranias, E.G.; Franzini-Armstrong, C.; Perez, C.F. Triadin/Junctin double null mouse reveals a differential role for Triadin and Junctin in anchoring CASQ to the jSR and regulating Ca2+ homeostasis. PLoS ONE 2012, 7, e39962.
- Dulhunty, A.F.; Wei-LaPierre, L.; Casarotto, M.G.; Beard, N.A. Core skeletal muscle ryanodine receptor calcium release complex. Clin. Exp. Pharmacol. Physiol. 2016, 44, 3–12.
- Lee, K.W.; Maeng, J.-S.; Choi, J.Y.; Lee, Y.R.; Hwang, C.Y.; Park, S.S.; Park, H.K.; Chung, B.H.; Lee, S.-G.; Kim, Y.-S.; et al. Role of Junctin Protein Interactions in Cellular Dynamics of Calsequestrin Polymer upon Calcium Perturbation. J. Biol. Chem. 2012, 287, 1679–1687.
- Park, H.; Park, I.Y.; Kim, E.; Youn, B.; Fields, K.; Dunker, A.K.; Kang, C. Comparing skeletal and cardiac calsequestrin structures and their calcium binding: A proposed mechanism for coupled calcium binding and protein polymerization. J. Biol. Chem. 2004, 279, 18026–18033.
- MacLennan, D.H.; Wong, P.T.S. Isolation of a Calcium-Sequestering Protein from Sarcoplasmic Reticulum. Proc. Natl. Acad. Sci. USA 1971, 68, 1231–1235.
- Biral, D.; Volpe, P.; Damiani, E.; Margreth, A. Coexistence of two calsequestrin isoforms in rabbit slow-twitch skeletal muscle fibers. FEBS Lett. 1992, 299, 175–178.
- Paolini, C.; Quarta, M.; D’Onofrio, L.; Reggiani, C.; Protasi, F. Differential Effect of Calsequestrin Ablation on Structure and Function of Fast and Slow Skeletal Muscle Fibers. J. Biomed. Biotechnol. 2011, 2011, 634075.
- Knollmann, B.C.; Chopra, N.; Hlaing, T.; Akin, B.; Yang, T.; Ettensohn, K.; Knollmann, B.E.C.; Horton, K.D.; Weissman, N.J.; Holinstat, I.; et al. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J. Clin. Investig. 2006, 116, 2510–2520.
- Shannon, T.R.; Guo, T.; Bers, D.M. Ca2+ scraps: Local depletions of free in cardiac sarcoplasmic reticulum during contractions leave substantial Ca2+ reserve. Circ. Res. 2003, 93, 40–45.
- Manno, C.; Sztretye, M.; Figueroa, L.; Allen, P.D.; Ríos, E. Dynamic measurement of the calcium buffering properties of the sarcoplasmic reticulum in mouse skeletal muscle. J. Physiol. 2013, 591, 423–442.
- Renken, C.; Hsieh, C.-E.; Marko, M.; Rath, B.; Leith, A.; Wagenknecht, T.; Frank, J.; Mannella, C.A. Structure of frozen–hydrated triad junctions: A case study in motif searching inside tomograms. J. Struct. Biol. 2009, 165, 53–63.
- Franzini-Armstrong, C.; Kenney, L.J.; Varriano-Marston, E. The structure of calsequestrin in triads of vertebrate skeletal muscle: A deep-etch study. J. Cell Biol. 1987, 105, 49–56.
- Perni, S.; Close, M.; Franzini-Armstrong, C. Novel Details of Calsequestrin Gel Conformation in Situ. J. Biol. Chem. 2013, 288, 31358–31362.
- Wagenknecht, T.; Hsieh, C.-E.; Rath, B.; Fleischer, S.; Marko, M. Electron Tomography of Frozen-Hydrated Isolated Triad Junctions. Biophys. J. 2002, 83, 2491–2501.
- Barone, V.; Del Re, V.; Gamberucci, A.; Polverino, V.; Galli, L.; Rossi, D.; Costanzi, E.; Toniolo, L.; Berti, G.; Malandrini, A.; et al. Identification and characterization of three novel mutations in the CASQ1 gene in four patients with tubular aggregate myopathy. Hum. Mutat. 2017, 38, 1761–1773.
- Manno, C.; Figueroa, L.C.; Gillespie, D.; Fitts, R.; Kang, C.; Franzini-Armstrong, C.; Rios, E. Calsequestrin depolymerizes when calcium is depleted in the sarcoplasmic reticulum of working muscle. Proc. Natl. Acad. Sci. USA 2017, 114, E638–E647.
- Chen, W.; Kudryashev, M. Structure of RyR1 in native membranes. EMBO Rep. 2020, 21, e49891.
- Maguire, P.B.; Briggs, F.N.; Lennon, N.J.; Ohlendieck, K. Oligomerization Is an Intrinsic Property of Calsequestrin in Normal and Transformed Skeletal Muscle. Biochem. Biophys. Res. Commun. 1997, 240, 721–727.
- Glover, L.; Culligan, K.; Cala, S.; Mulvey, C.; Ohlendieck, K. Calsequestrin binds to monomeric and complexed forms of key calcium-handling proteins in native sarcoplasmic reticulum membranes from rabbit skeletal muscle. Biochim. Biophys. Acta (BBA)-Biomembr. 2001, 1515, 120–132.
- O’Brian, J.J.; Ram, M.L.; Kiarash, A.; Cala, S.E. Mass Spectrometry of Cardiac Calsequestrin Characterizes Microheterogeneity Unique to Heart and Indicative of Complex Intracellular Transit. J. Biol. Chem. 2002, 277, 37154–37160.
- Mayfield, J.E.; Pollak, A.J.; Worby, C.A.; Xu, J.C.; Tandon, V.; Newton, A.C.; Dixon, J.E. Ca2+-dependent liquid-liquid phase separation underlies intracellular Ca2+ stores. bioRxiv 2021. bioRxiv:2021.07.06.451223. Available online: http://biorxiv.org/content/early/2021/07/06/2021.07.06.451223.abstract (accessed on 30 September 2023).
- Beard, N.; Laver, D.; Dulhunty, A. Calsequestrin and the calcium release channel of skeletal and cardiac muscle. Prog. Biophys. Mol. Biol. 2004, 85, 33–69.
- Aaron, B.M.; Oikawa, K.; A Reithmeier, R.; Sykes, B.D. Characterization of skeletal muscle calsequestrin by 1H NMR spectroscopy. J. Biol. Chem. 1984, 259, 11876–11881.
- Ikemoto, N.; Bhatnagar, G.M.; Nagy, B.; Gergely, J. Interaction of divalent cations with the 55,000-dalton protein component of the sarcoplasmic reticulum. Studies of fluorescence and circular dichroism. J. Biol. Chem. 1972, 247, 7835–7837.
- Hidalgo, C.; Donoso, P.; Rodriguez, P. Protons induce calsequestrin conformational changes. Biophys. J. 1996, 71, 2130–2137.
- Krause, K.; Milos, M.; Luan-Rilliet, Y.; Lew, D.; Cox, J. Thermodynamics of cation binding to rabbit skeletal muscle calsequestrin. Evidence for distinct Ca2+- and Mg2+-binding sites. J. Biol. Chem. 1991, 266, 9453–9459.
- Ostwald, T.J.; MacLennan, D.H.; Dorrington, K.J. Effects of Cation Binding on the Conformation of Calsequestrin and the High Affinity Calcium-binding Protein of Sarcoplasmic Reticulum. J. Biol. Chem. 1974, 249, 5867–5871.
- Bal, N.C.; Jena, N.; Sopariwala, D.; Balaraju, T.; Shaikh, S.; Bal, C.; Sharon, A.; Gyorke, S.; Periasamy, M. Probing cationic selectivity of cardiac calsequestrin and its CPVT mutants. Biochem. J. 2011, 435, 391–399.
- Park, H.; Wu, S.; Dunker, A.; Kang, C. Polymerization of calsequestrin. Implications for Ca2+ regulation. J. Biol. Chem. 2003, 278, 42728.
- Donoso, P.; Beltrán, A.M.; Hidalgo, C. Luminal pH Regulates Calcium Release Kinetics in Sarcoplasmic Reticulum Vesicles. Biochemistry 1996, 35, 13419–13425.
- Titus, E.W.; Deiter, F.H.; Shi, C.; Wojciak, J.; Scheinman, M.; Jura, N.; Deo, R.C. The structure of a calsequestrin filament reveals mechanisms of familial arrhythmia. Nat. Struct. Mol. Biol. 2020, 27, 1142–1151.
- Sanchez, E.J.; Lewis, K.M.; Danna, B.R.; Kang, C. High-capacity Ca2+ Binding of Human Skeletal Calsequestrin. J. Biol. Chem. 2012, 287, 11592–11601.
- Kim, E.; Youn, B.; Kemper, L.; Campbell, C.; Milting, H.; Varsanyi, M.; Kang, C. Characterization of Human Cardiac Calsequestrin and its Deleterious Mutants. J. Mol. Biol. 2007, 373, 1047–1057.
- Bal, N.C.; Sharon, A.; Gupta, S.C.; Jena, N.; Shaikh, S.; Gyorke, S.; Periasamy, M. The Catecholaminergic Polymorphic Ventricular Tachycardia Mutation R33Q Disrupts the N-terminal Structural Motif That Regulates Reversible Calsequestrin Polymerization. J. Biol. Chem. 2010, 285, 17188–17196.
- Lewis, K.M.; Ronish, L.A.; Ríos, E.; Kang, C. Characterization of Two Human Skeletal Calsequestrin Mutants Implicated in Malignant Hyperthermia and Vacuolar Aggregate Myopathy. J. Biol. Chem. 2015, 290, 28665–28674.
- Ng, K.; Titus, E.W.; Lieve, K.V.; Roston, T.M.; Mazzanti, A.; Deiter, F.H.; Denjoy, I.; Ingles, J.; Till, J.; Robyns, T.; et al. An International Multicenter Evaluation of Inheritance Patterns, Arrhythmic Risks, and Underlying Mechanisms of CASQ2-Catecholaminergic Polymorphic Ventricular Tachycardia. Circulation 2020, 142, 932–947.
- Bal, N.C.; Jena, N.; Chakravarty, H.; Kumar, A.; Chi, M.; Balaraju, T.; Rawale, S.V.; Rawale, J.S.; Sharon, A.; Periasamy, M. The C-terminal calcium-sensitive disordered motifs regulate isoform-specific polymerization characteristics of calsequestrin. Biopolymers 2015, 103, 15–22.
- Newcombe, E.A.; Fernandes, C.B.; Lundsgaard, J.E.; Brakti, I.; Lindorff-Larsen, K.; Langkilde, A.E.; Skriver, K.; Kragelund, B.B. Insight into Calcium-Binding Motifs of Intrinsically Disordered Proteins. Biomolecules 2021, 11, 1173.
More
Information
Subjects:
Biophysics
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
502
Revisions:
2 times
(View History)
Update Date:
29 Nov 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No