Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Fabiola García Fernández | -- | 3287 | 2023-11-27 14:43:34 | | | |
| 2 | Peter Tang | Meta information modification | 3287 | 2023-11-28 01:54:35 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
García Fernández, F.; Huet, S.; Miné-Hattab, J. Multi-Scale Imaging of the Dynamic Organization of Chromatin. Encyclopedia. Available online: https://encyclopedia.pub/entry/52098 (accessed on 27 June 2026).
García Fernández F, Huet S, Miné-Hattab J. Multi-Scale Imaging of the Dynamic Organization of Chromatin. Encyclopedia. Available at: https://encyclopedia.pub/entry/52098. Accessed June 27, 2026.
García Fernández, Fabiola, Sébastien Huet, Judith Miné-Hattab. "Multi-Scale Imaging of the Dynamic Organization of Chromatin" Encyclopedia, https://encyclopedia.pub/entry/52098 (accessed June 27, 2026).
García Fernández, F., Huet, S., & Miné-Hattab, J. (2023, November 27). Multi-Scale Imaging of the Dynamic Organization of Chromatin. In Encyclopedia. https://encyclopedia.pub/entry/52098
García Fernández, Fabiola, et al. "Multi-Scale Imaging of the Dynamic Organization of Chromatin." Encyclopedia. Web. 27 November, 2023.
Copy Citation
Chromatin is regarded as a heterogeneous and dynamic structure occupying a non-random position within the cell nucleus, where it plays a key role in regulating various functions of the genome. In addition to challenging early assumptions of chromatin being regular and static, high spatiotemporal resolution imaging made it possible to visualize and characterize different chromatin structures such as clutches, domains and compartments. More specifically, super-resolution microscopy facilitates the study of different cellular processes at a nucleosome scale, providing a multi-scale view of chromatin behavior within the nucleus in different environments.
chromatin organization and dynamics
high resolution imaging
DNA repair
1. Introduction
Over the last two decades, extensive studies across different model systems have revealed that nuclear organization plays fundamental biological roles. Chromosomes, and the genes they host, are arranged within the three-dimensional space of the nucleus in a specific manner, occupying a preferred location. Far from being a polymer with a static organization, chromatin diffuses inside living cells with specific properties, and its dynamics are often altered following specific stresses or in cells from diseased tissue.
2. Recent Fluorescence-Based Techniques to Study the Dynamic Organization of Chromatin at High Spatiotemporal Resolution
2.1. FRET-FLIM
FRET-FLIM (Förster resonance energy transfer by fluorescence lifetime imaging) is the method of choice to measure the interactions between proteins in living cells. Fluorescence resonance energy transfer (FRET) involves the energy transfer through dipole–dipole coupling of a donor and acceptor chromophore. This transfer requires that the fluorescence emission spectra of the donor overlap with the absorption spectra of the acceptor molecule and that the distance between the two fluorophores is within a few nanometers. Since this ranges with the typical size of protein complexes, FRET is an ideal tool for the study of protein–protein interactions. Several methods have been developed to assess FRET efficiency, probably the most quantitative being the measurement of the fluorescence lifetime of the donor (FLIM), which has been used to measure the spatiotemporal dynamics of chromatin in living cells [1][2][3].
2.2. FCS
Fluorescence correlation spectroscopy (FCS) is based on the analysis of fluorescence fluctuations arising from fluorescently tagged proteins moving in and out of the constant laser beam of a confocal setup [4][5]. By calculating the autocorrelation of these fluctuations, it is possible to estimate the characteristic time spent by the tagged molecules within the focal volume as well as their average number within this volume. Further quantitative analysis of the autocorrelation curves can also reveal the mode of diffusion (Brownian motion, sub-diffusion, etc.) as well as the existence of sub-populations of molecules [6][7]. Applied to nuclear proteins, FCS provides information on the binding of proteins to chromatin and on their mobility within the chromosomal environment at the microsecond time scale [8]. For example, Wachsmuth et al. assessed the local movement of the chromatin fiber by analyzing fluorescence fluctuations arising from the linker histone variant H1.0 tagged with EGFP [9]. Similarly, Bancaud et al. showed how photoactivatable GFP dimers can easily diffuse throughout the nucleus within seconds, without dense structures such as heterochromatin not visibly slowing down their motion [10].
2.3. Single Molecule Localization Microscopy: PALM–STORM-SPT
Single molecule localization microscopy allows the imaging of single molecules in cells. The trick is to light only a small fraction of the fluorophores or dyes in the sample so that their relative distance is greater than 250 nm. In this case, each spot in the image corresponds to a single molecule that can be localized at high spatial resolution by determining the center of its point spread function (PSF)—a common tool to describe the response of an imaging system to a point source or object. To obtain a full image of the sample, a few molecules are lighted on, localized and photobleached: this process is repeated for each frame of a movie until the structure can be accurately reconstructed. This principle is named PALM [11] (photoactivated localization microscopy) or STORM (stochastic optical reconstruction microscopy) [12]. Both PALM and STORM need to be performed in fixed cells and allow the study of structures at up to 5 nm resolution, such as small organelles or local molecular clusters (Figure 1, top panel). Given the high spatial resolution accessible with these techniques and similar to what was reported for electron microscopy, it is important to keep in mind that some structures can be altered by the fixation process [13].
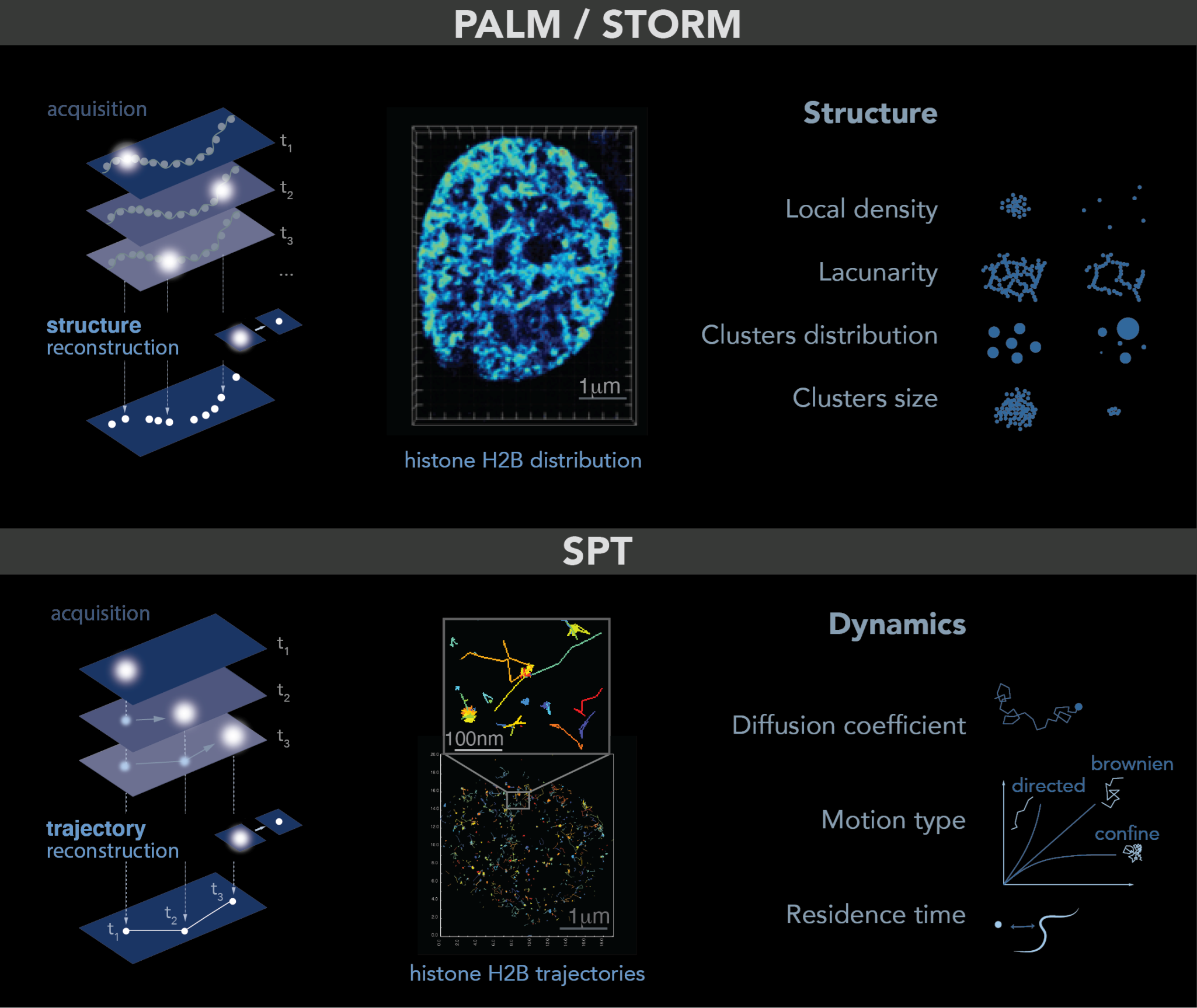
Figure 1. PALM and SPT: Principle, example and observables. Top: PALM/STORM principles. Left: a sparse subset of fluorescent probes is activated to produce single-particle images (represented by white circles) that do not overlap (left). After acquisition of images at a given time interval (t), a super-resolution image is reconstructed by plotting the measured positions of the fluorescent probes. Middle: example of histone H2B-mEOS distribution within a cell nucleus. (Right): further analysis of the final reconstructed images provides several parameters of the structure formed by the observed protein. Bottom: principle of SPT. (Left): during image acquisition, images are taken with a given exposure time (t) for the duration of several minutes. In each image, only a sparse number of emitters (white dots) are detected. Middle: using tracking and localization methods, it is possible to reconstruct the super-resolved trajectories of single molecules. Each histone H2B trajectory is represented within a cell nucleus by a colored trace. Right: different dynamic parameters can be extracted from SPT data by using mathematical approaches. (Image by Olga Markova).
The difference between PALM and STORM is the nature of the fluorescent probes used to realize the “light on–localization–bleaching” sequence. PALM uses fluorescent probes directly fused to the protein of interest and emitting light stochastically at low frequency under an illumination at 405 nm. PALM is also the best technique to estimate the number of tagged molecules inside cells or in sub-compartments. In contrast, STORM uses inorganic dyes indirectly linked to the protein of interest (through a Halo, Snap GFP-antiGFP nanobody system, etc.). These dyes emit light stochastically almost indefinitely, limiting the estimation of the number of molecules.
PALM/STORM microscopy gives access to several observables that are not easily accessible without such a single molecule approach. From the cloud of points obtained by PALM/STORM, the simplest observable is the local density of proteins, defined as the number of neighbors in a certain radius around each detection. To further quantify the distribution of points, a common method consists in calculating the Voronoi diagram of a cloud of points. This diagram is tiling of the plane into regions from a discrete set of points called seeds: Each region encloses a single germ and forms the set of points in the plane closer to this germ than any other. The Delaunay triangulation, also used for PALM/STORM analysis, is dual to the Voronoi diagram set. This computation geometry analysis allows then for quantifying clusters, to access the amount of space unoccupied by the protein of interest also called the lacunarity, and to characterize these wholes (number, distribution of sizes surfaces, volumes and shapes) [14][15].
The dynamic motion at a single molecular level can be tracked in living cells using single particle tracking (SPT): in that case, the same type of fluorescent probes can be used allowing the imaging of individual molecules one after another. However, instead of bleaching them immediately after lighting them, the aim is to follow their motion in time over multiple timeframes. To track the molecule correctly, the density of visible molecules needs to be low to avoid overlapping point emitters, i.e., mis-linking. This approach allows us to directly access the dynamics of individual molecules at unprecedented resolution, giving access to their types of motion, diffusion coefficient, residence time on a substrate, etc. (Figure 1, bottom panel). Using more advanced analysis, it is also possible to test the existence of an energy potential attracting or repelling molecules within distances smaller than the diffraction limit or to distinguish between different types of macromolecular condensates [16].
2.4. Multiplexed FISH Combined with Super-Resolution Imaging
Despite a very high spatial resolution, the main limitation of the imaging methods described above is their inability to identify specific genomic regions. In contrast, fluorescent in situ hybridization (FISH) and in situ sequencing-based approaches have the advantage of obtaining both the spatial and genomic information of the signal in single cells [17]. A catalyst of these approaches was the development of massively parallel oligonucleotide synthesis methods to generate customized complex oligonucleotide libraries such as Oligopaint [18], which greatly facilitate the detection of multiple non-repetitive nucleic acid species. However, these classical FISH techniques did not have the genomic and spatial resolution necessary to characterize the organization of chromatin at the sub-microscopic scale.
More recently, Beliveau et al. combined FISH and STORM to access the fine structure of chromosomes at known chromosomal loci [19]. They fluorescently marked 46 epigenetically defined genomic domains in Drosophila Kc167 cells and measured the physical volume occupied by each domain. Chromosomes exhibit fascinating behaviors on length scales of ~100 nm to 1 micron. Thus, adapting FISH and STORM techniques to image a whole chromosome will considerably widen our possibilities in the field of high spatial resolution imaging of chromosomes [20][21][22]. Another approach is to combine live cell chromatin imaging and multiplexed FISH labelling, which allows first to track the movement of chromosomal loci and then to resolve their identities [23][24].
3. Chromatin Conformation as Proxy of Chromatin Accessibility
It is well established now that the chromatin packing state is uneven within the cell nucleus. Single-cell techniques such as ChromEMT and super-resolution microscopy have allowed the mapping of chromatin within a cell nucleus according to its density [25][26][27][28][29]. In these maps, two types of regions previously observed by electron microscopy and soft X-ray tomography can be differentiated: highly dense regions such as the nuclear periphery and nucleolus, known as heterochromatin (HC), and much less dense regions dispersed in the center of the nucleoplasm, known as euchromatin (EC) [30][31][32]. However, a recent study applying FLIM-FRET microscopy to analyze the spatial organization at the nanometer-range proximity between nucleosomes, termed “nanocompaction”, showed that, contrary to expectations, constitutive HC is much less compacted than bulk chromatin [1]. This suggests a new view of the distribution in nucleosomes in HC versus EC: more frequent nucleosome–nucleosome contact would occur in EC (closer than 10 nm) than in HC, but these clusters would be more spaced, resulting in a less dense distribution at a larger scale. Interestingly, this recent evidence was found strictly in living cells, suggesting a bias caused by fixation on precedent studies.
Given that chromatin compaction and chromatin accessibility are in general strongly correlated, it is not surprising that DNA processes such as transcription, replication and repair occur differently in HC than in EC. During transcription, for example, gene expression is regulated by DNA accessibility and binding of transcription factors. Thus, HC is often associated with a transcriptionally inactive or repressed state decorated by histone H3 methylation (H3K9me2/3, H3K27me3) and heterochromatin protein-1 (HP1) association. Interestingly, in vitro and in vivo studies shown that HP1 protein forms phase-separated condensates upon binding DNA, indicating that gene silencing may occur in part through sequestration of compacted chromatin in HP1 droplets [33][34]. However, the role of HP1 in the compaction, accessibility and size of HC condensates was challenged by evidence in mice arguing that the HC condensates lack a separated liquid HP1 pool, and its compaction can alternate between two digital states (compacted or uncompacted) depending on the presence of a strong transcriptional activator [35]. Likewise, a study using biophysical modeling has recently proposed that the specific affinity of HP1 for H3K9me2/3 loci promotes the formation of stable HC condensates at HP1 levels well below those observed in vitro, highlighting that the H3K9me2/3 landscape governs the HC droplet rather than HP1 itself [36].
On the other hand, EC is characterized by active gene expression, histone depletion around transcriptional start sites, histone H3 acetylated and methylated histones marks (H3K27ac, H3K4me3) [29]. Likewise, some evidence across species has shown how replication timing is controlled by chromatin architecture. Thus, the open-structured and frequently transcribed EC replicates more rapidly than HC [37][38][39][40][41]. Similarly, during cell differentiation, there is a progressive transition of chromatin compaction accompanied by repression of certain genes and HC-landmarks, a process termed heterochromatinization [42][43][44][45]. Genome-wide chromatin modification assays and super-resolution imaging have compared the profiles of undifferentiated embryonic stem cells (ESCs) with those of differentiated cells. Both mouse and human ESCs revealed widespread active chromatin domains, characterized by dimmer, sparser histone H2B domains and enrichment of H3K27ac, H3K4me3. In contrast, highly condensed histone H2B domains and HC marks such as H3K9m and H3K27 become more abundant in differentiated cells [26][46]. Importantly, alterations in this transition have critical consequences on proper differentiation during development [47].
Chromatin compaction was also closely correlated to constrained movement at different chromatin scales [43][46][48][49][50][51]. Heatmaps resulting from either single histone H2B tracking or tracking of H2B-bound chromatin domains showed a big heterogeneity in chromatin motion inside a nucleus, showing less movement within HC-rich regions than regions elsewhere in the genome [46][48][49][51]. Interestingly, despite evidence showing heterogeneity in chromatin dynamics due to compaction, a recent study has described a steady-state motion of interphase chromatin, independent of changes in chromatin compaction, cell cycle and DNA replication [52]. This motion profile was discovered by tracking individual nucleosomes but also chromatin loci on a second (1 s) time scale. Beyond this time scale, steady-state motion is lost, which explains the discrepancy with previous work using longer time scales. In terms of function, the steady-state behavior of chromatin may allow cells to cope with changes in the nuclear environment in order to maintain their routine cellular functions in similar nuclear environments [52].
Thus, the organizational rearrangement of chromatin during different DNA process is not haphazard but seems to have a specific function mainly regulating chromatin accessibility and motion.
4. DNA Repair and Genome Stability
In fact, DNA is continuously threatened by endogenous and exogenous factors that can result in different types of lesions, double strand breaks (DSBs) being the most detrimental. A network of cellular mechanisms, named together the DNA damage response (DDR), monitor DNA lesions, guarantee faithful repair and, therefore, chromosome stability. Two of the most well-known DSB repair mechanisms are homologous recombination (HR) and non-homologous end joining (NHEJ). While HR repairs DNA breaks by copying the missing information across the lesion from an undamaged template, as from the replicated sister chromatid, NHEJ does it by ligation of the broken ends after their juxtaposition (reviewed in [53][54][55][56]).
To decipher the mechanisms underlying the DDR, several methods to induce DNA damage have been developed over the years. Methods such as ionizing radiations, crosslinking agents, radiomimetic compounds and localized UV laser micro-irradiation induce damage randomly throughout the genome. In contrast, homing endonucleases and restriction enzymes allow the induction of damage at a targeted position on the genome, which is useful for understanding local chromatin changes around the break [57].
As seen in in vitro studies, chromatin constitutes a barrier to the DDR machinery [58] and, therefore, needs to be remodeled to allow DNA accessibility. Thus, the chromatin structure undergoes dynamic changes that are crucial for the DDR progression. Certainly, the original architecture of a damaged chromatin domain and its near and distant environment affect signaling and repair kinetics. Thus, it is not surprising that damaged HC regions required more extensive remodeling than damaged EC ones [59][60][61]. Despite massive heterochromatin unfolding upon UV irradiation, its specific histone marks and transcriptional silencing are maintained [62]. However, recent evidence showed a significant drop in HC histone markers under oxidative stress and, on the contrary, a raise in H3K9Ac levels, suggesting a positive gene expression epigenetic profile [63].
Chromatin remodeling starts with a first step of chromatin relaxation within seconds after DNA damage [49][64][65]. Interestingly, single-molecule microscopy has shown that this relaxation effect is specific to damaged chromatin, with nuclear regions distal to the damage being more compact [64]. This first remodeling event is dependent on histone poly-ADP-ribosylation and crucial for the recruitment of downstream DDR factors such as the ATM kinase [66][67]. In a second step, ATM and other kinases mediate the phosphorylation of histone H2AX (ƴ-H2AX), which spreads over a megabase-size domain of chromatin surrounding DNA damage [68][69][70]. ATM signaling has been shown to lead to a more compacted fiber [71]. This compaction level is required for upstream signaling by facilitating the recruitment of some adaptor proteins such as 53BP1 [67]. Interestingly, chromatin compaction here was also involved in loop extrusion and TAD organization [72][73][74]. Indeed, super-resolution microscopy revealed that CTCF and cohesin, loop/TAD mediators, are juxtaposed to ƴ-H2AX foci, suggesting that the formation of these domains is governed by high-order structures [74]. The potential functions of loop extrusion around DNA damage are to amplify the DDR signaling by enhancing chromatin–protein interactions and to protect 3D genome integrity during DNA repair [75][76], although a reduction in TAD number and insulation was found under hyperosmotic stress [77]. Notably, whereas compacted chromatin boosts upstream DDR signaling, it impairs downstream repair and restoration [67]. Thus, a second step of chromatin relaxation is needed to complete repair. Although this last remodeling step has not been fully characterized, it was associated with histone SUMOylation and ubiquitination [78][79].
As expected, chromatin remodeling during the DDR is accompanied by changes in chromatin motion. Several studies have shown enhanced chromatin dynamics after DNA damage, which was shown to facilitate homology search during HR [61][80][81]. Rad51, the central protein of HR, has an essential role to promote increased mobility since in the absence of Rad51, no change is observed both at the damaged site but globally in the nucleus [80]. Miné-Hattab et al. proposed a model in which stiffening of the damaged ends by Rad51 polymerization along the single strand DNA tail, combined with globally increased stiffness, “act like a needle in a ball of yarn”, enhancing the ability of the break to traverse the chromatin meshwork. A global change in chromatin stiffening due to H2A phosphorylation has also been proposed to explain increased chromatin mobility upon DSB [82][83][84]. More recently, the direct visualization of Rad51 in living cells revealed that the dynamics of Rad51-ssDNA filaments constitute a robust search strategy, allowing DSB to rapidly explore the nuclear volume and thus enable efficient HR [85].
However, these notions come largely from studies in yeast [80][82][84][86][87][88] but remain controversial in mammals [89][90][91][92][93]. Notably, most of these studies have followed the dynamics of large regions of chromatin, while there is little evidence for the mobility of damaged chromatin at the nucleosomal level. A recent study performing histone PALM-SPT showed that motion heterogeneity changes also throughout the nucleus upon DNA damage. While heterogeneity decreases in regions around the break, the motion of regions elsewhere in the nucleus becomes homogenous [49]. This difference in the behavior of damaged and undamaged chromatin within the nucleus is proposed to be linked to the formation of repair foci or membrane-less condensates at the site of the DNA lesions. Indeed, many repair proteins relocalize from a diffuse nuclear distribution to highly concentrated nuclear foci in lesions (i.e., condensates). Consequently, these condensates change the structure of damaged chromatin, which may favor certain molecular interactions while preventing others [94]. The DDR protein 53BP1 forms one of the most studied condensates [95]. Live-cell microscopy revealed how 53BP1 condensates organize damaged chromatin into a larger repair compartment, while pushing undamaged chromatin regions away [96][97]. This is consistent with the notion of global compaction and homogenous motion in undamaged regions observed in [49][64]. Furthermore, a more recent study using STED and 3D SIM has shown that 53BP1 distribution stabilizes several neighboring loops at the break ends in an ordered circular arrangement [98]. Therefore, 53BP1 depletion disrupts this circular architecture, leading to persistent decompaction of damaged chromatin and aberrant spreading of DNA repair proteins. Indeed, inherited or acquired defects in nuclear chromatin organization during the DDR lead to genome instability. By optimizing STORM for imaging pathological tissues, a study revealed gradual chromatin decompaction and fragmentation throughout tumorigenesis. Importantly, this chromatin feature may improve diagnosis, risk stratification and cancer prevention [99].
References
- Dupont, C.; Chahar, D.; Trullo, A.; Gostan, T.; Surcis, C.; Grimaud, C.; Fisher, D.; Feil, R.; Llères, D. Evidence for Low Nanocompaction of Heterochromatin in Living Embryonic Stem Cells. EMBO J. 2023, 42, e110286.
- Llères, D.; James, J.; Swift, S.; Norman, D.G.; Lamond, A.I. Quantitative Analysis of Chromatin Compaction in Living Cells Using FLIM-FRET. J. Cell Biol. 2009, 187, 481–496.
- Audugé, N.; Padilla-Parra, S.; Tramier, M.; Borghi, N.; Coppey-Moisan, M. Chromatin Condensation Fluctuations Rather than Steady-State Predict Chromatin Accessibility. Nucleic Acids Res. 2019, 47, 6184–6194.
- Schwille, P.; Bieschke, J.; Oehlenschltiger, F. Kinetic Investigations by Fluorescence Correlation Spectroscopy: The Analytical and Diagnostic Potential of Diffusion Studies. Biophys. Chem. 1997, 66, 211–228.
- Magde, D.; Elson, E.L.; Webb, W.W. Fluorescence Correlation Spectroscopy. II. An Experimental Realization. Biopolymers 1974, 13, 29–61.
- Michelman-Ribeiro, A.; Mazza, D.; Rosales, T.; Stasevich, T.J.; Boukari, H.; Rishi, V.; Vinson, C.; Knutson, J.R.; McNally, J.G. Direct Measurement of Association and Dissociation Rates of DNA Binding in Live Cells by Fluorescence Correlation Spectroscopy. Biophys. J. 2009, 97, 337–346.
- D’Augustin, O.; Gaudon, V.; Siberchicot, C.; Smith, R.; Chapuis, C.; Depagne, J.; Veaute, X.; Busso, D.; Di Guilmi, A.M.; Castaing, B.; et al. Identification of Key Residues of the DNA Glycosylase OGG1 Controlling Efficient DNA Sampling and Recruitment to Oxidized Bases in Living Cells. Nucleic Acids Res. 2023, 51, 4942–4958.
- Yu, L.; Lei, Y.; Ma, Y.; Liu, M.; Zheng, J.; Dan, D.; Gao, P. A Comprehensive Review of Fluorescence Correlation Spectroscopy. Front. Phys. 2021, 9, 644450.
- Wachsmuth, M.; Knoch, T.A.; Rippe, K. Dynamic Properties of Independent Chromatin Domains Measured by Correlation Spectroscopy in Living Cells. Epigenetics Chromatin 2016, 9, 57.
- Bancaud, A.; Huet, S.; Daigle, N.; Mozziconacci, J.; Beaudouin, J.; Ellenberg, J. Molecular Crowding Affects Diffusion and Binding of Nuclear Proteins in Heterochromatin and Reveals the Fractal Organization of Chromatin. EMBO J. 2009, 28, 3785–3798.
- Betzig, E.; Patterson, G.; Sougrat, R.; Lindwasser, W.; Olenych, S.; Bonifacino, J.; Davidson, M.; Lippincott-Schwartz, J.; Hess, H. Imaging Intracellular Fluorescent Proteins at Nanometer Resolution. Science (1979) 2006, 313, 1638–1642.
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-Diffraction-Limit Imaging by Stochastic Optical Reconstruction Microscopy (STORM). Nat. Methods 2006, 3, 793–795.
- Miné-Hattab, J. Condensates: When fixation creates fiction. eLife 2023, 12, e85671.
- Andronov, L.; Orlov, I.; Lutz, Y.; Vonesch, J.-L.; Klaholz, B.P. ClusterViSu, a Method for Clustering of Protein Complexes by Voronoi Tessellation in Super-Resolution Microscopy. Sci. Rep. 2016, 6, 24084.
- Levet, F.; Julien, G.; Galland, R.; Butler, C.; Beghin, A.; Chazeau, A.; Hoess, P.; Ries, J.; Giannone, G.; Sibarita, J.-B. A Tessellation-Based Colocalization Analysis Approach for Single-Molecule Localization Microscopy. Nat. Commun. 2019, 10, 2379.
- Heltberg, M.L.; Miné-Hattab, J.; Taddei, A.; Walczak, A.M.; Mora, T. Physical Observables to Determine the Nature of Membrane-Less Cellular Sub-Compartments. eLife 2021, 10, 69181.
- Lichter, P.; Cremer, T.; Borden, J.; Manuelidis, L.; Ward, D.C. Delineation of Individual Human Chromosomes in Metaphase and Interphase Cells by in Situ Suppression Hybridization Using Recombinant DNA Libraries. Hum. Genet. 1988, 80, 224–234.
- Beliveau, B.J.; Joyce, E.F.; Apostolopoulos, N.; Yilmaz, F.; Fonseka, C.Y.; McCole, R.B.; Chang, Y.; Li, J.B.; Senaratne, T.N.; Williams, B.R.; et al. Versatile Design and Synthesis Platform for Visualizing Genomes with Oligopaint FISH Probes. Proc. Natl. Acad. Sci. USA 2012, 109, 21301–21306.
- Boettiger, A.N.; Bintu, B.; Moffitt, J.R.; Wang, S.; Beliveau, B.J.; Fudenberg, G.; Imakaev, M.; Mirny, L.A.; Wu, C.T.; Zhuang, X. Super-Resolution Imaging Reveals Distinct Chromatin Folding for Different Epigenetic States. Nature 2016, 529, 418–422.
- Eng, C.H.L.; Lawson, M.; Zhu, Q.; Dries, R.; Koulena, N.; Takei, Y.; Yun, J.; Cronin, C.; Karp, C.; Yuan, G.C.; et al. Transcriptome-Scale Super-Resolved Imaging in Tissues by RNA SeqFISH+. Nature 2019, 568, 235–239.
- Shah, R.U.; Robinson, E.S.; Gu, P.; Robinson, A.L.; Apte, J.S.; Presto, A.A. High-Spatial-Resolution Mapping and Source Apportionment of Aerosol Composition in Oakland, California, Using Mobile Aerosol Mass Spectrometry. Atmos. Chem. Phys. 2018, 18, 16325–16344.
- Xia, C.; Fan, J.; Emanuel, G.; Hao, J.; Zhuang, X. Spatial Transcriptome Profiling by MERFISH Reveals Subcellular RNA Compartmentalization and Cell Cycle-Dependent Gene Expression. Proc. Natl. Acad. Sci. USA 2019, 116, 19490–19499.
- Takei, Y.; Shah, S.; Harvey, S.; Qi, L.S.; Cai, L. Multiplexed Dynamic Imaging of Genomic Loci by Combined CRISPR Imaging and DNA Sequential FISH. Biophys. J. 2017, 112, 1773–1776.
- Guan, J.; Liu, H.; Shi, X.; Feng, S.; Huang, B. Tracking Multiple Genomic Elements Using Correlative CRISPR Imaging and Sequential DNA FISH. Biophys. J. 2017, 112, 1077–1084.
- Ou, H.D.; Phan, S.; Deerinck, T.J.; Thor, A.; Ellisman, M.H.; O’Shea, C.C. ChromEMT: Visualizing 3D Chromatin Structure and Compaction in Interphase and Mitotic Cells. Science (1979) 2017, 357, eaag0025.
- Ricci, M.A.; Manzo, C.; García-Parajo, M.F.; Lakadamyali, M.; Cosma, M.P. Chromatin Fibers Are Formed by Heterogeneous Groups of Nucleosomes in Vivo. Cell 2015, 160, 1145–1158.
- Barth, R.; Bystricky, K.; Shaban, H.A. Coupling Chromatin Structure and Dynamics by Live Super-Resolution Imaging. Sci. Adv. 2020, 6, eaaz2196.
- Sedat, J.; McDonald, A.; Kasler, H.; Verdin, E.; Cang, H.; Arigovindan, M.; Murre, C.; Elbaum, M. A Proposed Unified Mitotic Chromosome Architecture. Proc. Natl. Acad. Sci. USA 2022, 119, e2119107119.
- Xu, J.; Ma, H.; Jin, J.; Uttam, S.; Fu, R.; Huang, Y.; Liu, Y. Super-Resolution Imaging of Higher-Order Chromatin Structures at Different Epigenomic States in Single Mammalian Cells. Cell Rep. 2018, 24, 873–882.
- Harrison, C.J.; Allen’, T.D.; Britch, M.; Harris, R. High-resolution scanning electron microscopy of human metaphase chromosomes. J. Cell Sci. 1982, 56, 409–422.
- Sumner, A.T. Scanning Electron Microscopy of Mammalian Chromosomes from Prophase to Telophase. Chromosoma 1991, 100, 410–418.
- Le Gros, M.A.; Clowney, E.J.; Magklara, A.; Yen, A.; Markenscoff-Papadimitriou, E.; Colquitt, B.; Myllys, M.; Kellis, M.; Lomvardas, S.; Larabell, C.A. Soft X-Ray Tomography Reveals Gradual Chromatin Compaction and Reorganization during Neurogenesis In Vivo. Cell Rep. 2016, 17, 2125–2136.
- Larson, A.G.; Elnatan, D.; Keenen, M.M.; Trnka, M.J.; Johnston, J.B.; Burlingame, A.L.; Agard, D.A.; Redding, S.; Narlikar, G.J. Liquid Droplet Formation by HP1α Suggests a Role for Phase Separation in Heterochromatin. Nature 2017, 547, 236–240.
- Strom, A.R.; Emelyanov, A.V.; Mir, M.; Fyodorov, D.V.; Darzacq, X.; Karpen, G.H. Phase Separation Drives Heterochromatin Domain Formation. Nature 2017, 547, 241–245.
- Erdel, F.; Rademacher, A.; Vlijm, R.; Tünnermann, J.; Frank, L.; Weinmann, R.; Schweigert, E.; Yserentant, K.; Hummert, J.; Bauer, C.; et al. Mouse Heterochromatin Adopts Digital Compaction States without Showing Hallmarks of HP1-Driven Liquid-Liquid Phase Separation. Mol. Cell 2020, 78, 236–249.e7.
- Tortora, M.M.C.; Brennan, L.; Karpen, G.; Jost, D. Liquid-Liquid Phase Separation Recapitulates the Thermodynamics and Kinetics of Heterochromatin Formation. bioRxiv 2022.
- Kemp, M.G.; Ghosh, M.; Liu, G.; Leffak, M. The Histone Deacetylase Inhibitor Trichostatin A Alters the Pattern of DNA Replication Origin Activity in Human Cells. Nucleic Acids Res. 2005, 33, 325–336.
- Goren, A.; Tabib, A.; Hecht, M.; Cedar, H. DNA Replication Timing of the Human β-Globin Domain Is Controlled by Histone Modification at the Origin. Genes Dev. 2008, 22, 1319–1324.
- Sehwaiger, M.; Stadler, M.B.; Bell, O.; Kohler, H.; Oakeley, E.J.; Sehübeler, D. Chromatin State Marks Cell-Type- and Gender-Specific Replication of the Drosophila Genome. Genes Dev. 2009, 23, 589–601.
- Hansen, R.S.; Thomas, S.; Sandstrom, R.; Canfield, T.K.; Thurman, R.E.; Weaver, M.; Dorschner, M.O.; Gartler, S.M.; Stamatoyannopoulos, J.A. Sequencing Newly Replicated DNA Reveals Widespread Plasticity in Human Replication Timing. Proc. Natl. Acad. Sci. USA 2010, 107, 139–144.
- Lubelsky, Y.; Prinz, J.A.; DeNapoli, L.; Li, Y.; Belsky, J.A.; MacAlpine, D.M. DNA Replication and Transcription Programs Respond to the Same Chromatin Cues. Genome Res. 2014, 24, 1102–1114.
- Falk, M.; Lukášová, E.; Štefančíková, L.; Baranová, E.; Falková, I.; Ježková, L.; Davídková, M.; Bačíková, A.; Vachelová, J.; Michaelidesová, A.; et al. Heterochromatinization Associated with Cell Differentiation as a Model to Study DNA Double Strand Break Induction and Repair in the Context of Higher-Order Chromatin Structure. Appl. Radiat. Isot. 2014, 83, 177–185.
- Rosa, S.; Ntoukakis, V.; Ohmido, N.; Pendle, A.; Abranches, R.; Shaw, P. Cell Differentiation and Development in Arabidopsis Are Associated with Changes in Histone Dynamics at the Single-Cell Level. Plant Cell 2014, 26, 4821–4833.
- Dixon, J.R.; Jung, I.; Selvaraj, S.; Shen, Y.; Antosiewicz-Bourget, J.E.; Lee, A.Y.; Ye, Z.; Kim, A.; Rajagopal, N.; Xie, W.; et al. Chromatin Architecture Reorganization during Stem Cell Differentiation. Nature 2015, 518, 331–336.
- Fraser, J.; Ferrai, C.; Chiariello, A.M.; Schueler, M.; Rito, T.; Laudanno, G.; Barbieri, M.; Moore, B.L.; Kraemer, D.C.; Aitken, S.; et al. Hierarchical Folding and Reorganization of Chromosomes Are Linked to Transcriptional Changes in Cellular Differentiation. Mol. Syst. Biol. 2015, 11, 852.
- Nozaki, T.; Imai, R.; Tanbo, M.; Nagashima, R.; Tamura, S.; Tani, T.; Joti, Y.; Tomita, M.; Hibino, K.; Kanemaki, M.T.; et al. Dynamic Organization of Chromatin Domains Revealed by Super-Resolution Live-Cell Imaging. Mol. Cell 2017, 67, 282–293.e7.
- Chen, T.; Dent, S.Y.R. Chromatin Modifiers and Remodellers: Regulators of Cellular Differentiation. Nat. Rev. Genet. 2014, 15, 93–106.
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological Domains in Mammalian Genomes Identified by Analysis of Chromatin Interactions. Nature 2012, 485, 376–380.
- Locatelli, M.; Lawrimore, J.; Lin, H.; Sanaullah, S.; Seitz, C.; Segall, D.; Kefer, P.; Bloom, K.; Liu, J.; Bonin, K.; et al. DNA Damage Reduces Heterogeneity and Coherence of Chromatin Motions. Proc. Natl. Acad. Sci. USA 2022, 119, e2205166119.
- Chubb, J.R.; Boyle, S.; Perry, P.; Bickmore, W.A. Chromatin Motion Is Constrained by Association with Nuclear Compartments in Human Cells. Curr. Biol. 2002, 12, 439–445.
- Lerner, J.; Gomez-Garcia, P.A.; McCarthy, R.L.; Liu, Z.; Lakadamyali, M.; Zaret, K.S. Two-Parameter Mobility Assessments Discriminate Diverse Regulatory Factor Behaviors in Chromatin. Mol. Cell 2020, 79, 677–688.e6.
- Iida, S.; Shinkai, S.; Itoh, Y.; Tamura, S.; Kanemaki, M.T.; Onami, S.; Maeshima, K. Single-Nucleosome Imaging Reveals Steady-State Motion of Interphase Chromatin in Living Human Cells. Sci. Adv. 2022, 8, eabn5626.
- Symington, L.S.; Gautier, J. Double-Strand Break End Resection and Repair Pathway Choice. Annu. Rev. Genet. 2011, 45, 247–271.
- Mehta, A.; Haber, J.E. Sources of DNA Double-Strand Breaks and Models of Recombinational DNA Repair. Cold Spring Harb. Perspect. Biol. 2014, 6, a016428.
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64.
- García Fernandez, F.; Fabre, E. The Dynamic Behavior of Chromatin in Response to DNA Double-Strand Breaks. Genes 2022, 13, 215.
- Mladenov, E.; Magin, S.; Soni, A.; Iliakis, G. DNA Double-Strand-Break Repair in Higher Eukaryotes and Its Role in Genomic Instability and Cancer: Cell Cycle and Proliferation-Dependent Regulation. Semin. Cancer Biol. 2016, 37–38, 51–64.
- Adkins, N.L.; Niu, H.; Sung, P.; Peterson, C.L. Nucleosome Dynamics Regulates DNA Processing. Nat. Struct. Mol. Biol. 2013, 20, 836–842.
- Jakob, B.; Splinter, J.; Conrad, S.; Voss, K.O.; Zink, D.; Durante, M.; Löbrich, M.; Taucher-Scholz, G. DNA Double-Strand Breaks in Heterochromatin Elicit Fast Repair Protein Recruitment, Histone H2AX Phosphorylation and Relocation to Euchromatin. Nucleic Acids Res. 2011, 39, 6489–6499.
- Zhang, Y.; Máté, G.; Müller, P.; Hillebrandt, S.; Krufczik, M.; Bach, M.; Kaufmann, R.; Hausmann, M.; Heermann, D.W. Radiation Induced Chromatin Conformation Changes Analysed by Fluorescent Localization Microscopy, Statistical Physics, and Graph Theory. PLoS ONE 2015, 10, e128555.
- Clouaire, T.; Rocher, V.; Lashgari, A.; Arnould, C.; Aguirrebengoa, M.; Biernacka, A.; Skrzypczak, M.; Aymard, F.; Fongang, B.; Dojer, N.; et al. Comprehensive Mapping of Histone Modifications at DNA Double-Strand Breaks Deciphers Repair Pathway Chromatin Signatures. Mol. Cell 2018, 72, 250–262.e6.
- Fortuny, A.; Chansard, A.; Caron, P.; Chevallier, O.; Leroy, O.; Renaud, O.; Polo, S.E. Imaging the Response to DNA Damage in Heterochromatin Domains Reveals Core Principles of Heterochromatin Maintenance. Nat. Commun. 2021, 12, 2428.
- Casali, C.; Siciliani, S.; Galgano, L.; Biggiogera, M. Oxidative Stress and Nuclear Reprogramming: A Pilot Study of the Effects of Reactive Oxygen Species on Architectural and Epigenetic Landscapes. Int. J. Mol. Sci. 2022, 24, 153.
- Hausmann, M.; Falk, M.; Neitzel, C.; Hofmann, A.; Biswas, A.; Gier, T.; Falkova, I.; Heermann, D.W. Elucidation of the Clustered Nano-Architecture of Radiation-Induced Dna Damage Sites and Surrounding Chromatin in Cancer Cells: A Single Molecule Localization Microscopy Approach. Int. J. Mol. Sci. 2021, 22, 3636.
- Smith, R.; Zentout, S.; Rother, M.; Bigot, N.; Chapuis, C.; Mihuț, A.; Zobel, F.F.; Ahel, I.; van Attikum, H.; Timinszky, G.; et al. HPF1-Dependent Histone ADP-Ribosylation Triggers Chromatin Relaxation to Promote the Recruitment of Repair Factors at Sites of DNA Damage. Nat. Struct. Mol. Biol. 2023, 30, 678–691.
- Sellou, H.; Lebeaupin, T.; Chapuis, C.; Smith, R.; Hegele, A.; Singh, H.R.; Kozlowski, M.; Bultmann, S.; Ladurner, A.G.; Timinszky, G.; et al. The Poly(ADP-Ribose)-Dependent Chromatin Remodeler Alc1 Induces Local Chromatin Relaxation upon DNA Damage. Mol. Biol. Cell 2016, 27, 3791–3799.
- Burgess, R.C.; Burman, B.; Kruhlak, M.J.; Misteli, T. Activation of DNA Damage Response Signaling by Condensed Chromatin. Cell Rep. 2014, 9, 1703–1717.
- Rogakou, E.P.; Boon, C.; Redon, C.; Bonner, W.M. Megabase Chromatin Domains Involved in DNA Double-Strand Breaks In Vivo. J. Cell Biol. 1999, 146, 905–915.
- Burma, S.; Chen, B.P.; Murphy, M.; Kurimasa, A.; Chen, D.J. ATM Phosphorylates Histone H2AX in Response to DNA Double-Strand Breaks. J. Biol. Chem. 2001, 276, 42462–42467.
- Shroff, R.; Arbel-Eden, A.; Pilch, D.; Ira, G.; Bonner, W.M.; Petrini, J.H.; Haber, J.E.; Lichten, M. Distribution and Dynamics of Chromatin Modification Induced by a Defined DNA Double-Strand Break. Curr. Biol. 2004, 14, 1703–1711.
- Khurana, S.; Kruhlak, M.J.; Kim, J.; Tran, A.D.; Liu, J.; Nyswaner, K.; Shi, L.; Jailwala, P.; Sung, M.H.; Hakim, O.; et al. A Macrohistone Variant Links Dynamic Chromatin Compaction to BRCA1-Dependent Genome Maintenance. Cell Rep. 2014, 8, 1049–1062.
- Caron, P.; Aymard, F.; Iacovoni, J.S.; Briois, S.; Canitrot, Y.; Bugler, B.; Massip, L.; Losada, A.; Legube, G. Cohesin Protects Genes against ΓH2AX Induced by DNA Double-Strand Breaks. PLoS Genet. 2012, 8, e1002460.
- Goloborodko, A.; Imakaev, M.V.; Marko, J.F.; Mirny, L. Compaction and Segregation of Sister Chromatids via Active Loop Extrusion. eLife 2016, 5, e14864.
- Natale, F.; Rapp, A.; Yu, W.; Maiser, A.; Harz, H.; Scholl, A.; Grulich, S.; Anton, T.; Hörl, D.; Chen, W.; et al. Identification of the Elementary Structural Units of the DNA Damage Response. Nat. Commun. 2017, 8, 15760.
- Sanders, J.T.; Freeman, T.F.; Xu, Y.; Golloshi, R.; Stallard, M.A.; Hill, A.M.; San Martin, R.; Balajee, A.S.; McCord, R.P. Radiation-Induced DNA Damage and Repair Effects on 3D Genome Organization. Nat. Commun. 2020, 11, 6178.
- Caron, P.; Choudjaye, J.; Clouaire, T.; Corte, F.; Daburon, V.; Aguirrebengoa, M.; Mangeat, T.; Iacovoni, J.S.; Alejandro, A. Non-Redundant Functions of ATM and DNA-PKcs in Response to DNA Double-Strand Breaks. Cell Rep. 2015, 13, 1598–1609.
- Amat, R.; Böttcher, R.; Le Dily, F.; Vidal, E.; Quilez, J.; Cuartero, Y.; Beato, M.; de Nadal, E.; Posas, F. Rapid Reversible Changes in Compartments and Local Chromatin Organization Revealed by Hyperosmotic Shock. Genome Res. 2019, 29, 18–28.
- Ayrapetov, M.K.; Gursoy-Yuzugullu, O.; Xu, C.; Xu, Y.; Price, B.D. DNA Double-Strand Breaks Promote Methylation of Histone H3 on Lysine 9 and Transient Formation of Repressive Chromatin. Proc. Natl. Acad. Sci. USA 2014, 111, 9169–9174.
- Goodarzi, A.A.; Jeggo, P.A. The Heterochromatic Barrier to DNA Double Strand Break Repair: How to Get the Entry Visa. Int. J. Mol. Sci. 2012, 13, 11844–11860.
- Miné-Hattab, J.; Rothstein, R.; Mine-Hattab, J.; Rothstein, R. Increased Chromosome Mobility Facilitates Homology Search during Recombination. Nat. Cell Biol. 2012, 14, 510–517.
- Clouaire, T.; Legube, G. A Snapshot on the Cis Chromatin Response to DNA Double-Strand Breaks. Trends Genet. 2019, 35, 330–345.
- García Fernández, F.; Almayrac, E.; Carré Simon, À.; Batrin, R.; Khalil, Y.; Boissac, M.; Fabre, E. Global Chromatin Mobility Induced by a DSB Is Dictated by Chromosomal Conformation and Defines the HR Outcome. eLife 2022, 11, e78015.
- García Fernández, F.; Lemos, B.; Khalil, Y.; Batrin, R.; Haber, J.E.; Fabre, E. Modified Chromosome Structure Caused by Phosphomimetic H2A Modulates the DNA Damage Response by Increasing Chromatin Mobility in Yeast. J. Cell Sci. 2021, 134, jcs258500.
- Herbert, S.; Brion, A.; Arbona, J.-M.; Lelek, M.; Veillet, A.; Lelandais, B.; Parmar, J.; Fernández, F.G.; Almayrac, E.; Khalil, Y.; et al. Chromatin Stiffening Underlies Enhanced Locus Mobility after DNA Damage in Budding Yeast. EMBO J. 2017, 36, 2595–2608.
- Liu, S.; Miné-Hattab, J.; Villemeur, M.; Guerois, R.; Pinholt, H.D.; Mirny, L.A.; Taddei, A. Publisher Correction: In Vivo Tracking of Functionally Tagged Rad51 Unveils a Robust Strategy of Homology Search. Nat. Struct. Mol. Biol. 2023, 30, 1607.
- Strecker, J.; Gupta, G.D.; Zhang, W.; Bashkurov, M.; Landry, M.-C.; Pelletier, L.; Durocher, D. DNA Damage Signalling Targets the Kinetochore to Promote Chromatin Mobility. Nat. Cell Biol. 2016, 18, 281–290.
- Seeber, A.; Dion, V.; Gasser, S.M. Checkpoint Kinases and the INO80 Nucleosome Remodeling Complex Enhance Global Chromatin Mobility in Response to DNA Damage. Genes Dev. 2013, 27, 1999–2008.
- Dion, V.; Kalck, V.; Horigome, C.; Towbin, B.D.; Gasser, S.M. Increased Mobility of Double-Strand Breaks Requires Mec1, Rad9 and the Homologous Recombination Machinery. Nat. Cell Biol. 2012, 14, 502–509.
- Kruhlak, M.J.; Celeste, A.; Dellaire, G.; Fernandez-Capetillo, O.; Müller, W.G.; McNally, J.G.; Bazett-Jones, D.P.; Nussenzweig, A. Changes in Chromatin Structure and Mobility in Living Cells at Sites of DNA Double-Strand Breaks. J. Cell Biol. 2006, 172, 823–834.
- Soutoglou, E.; Dorn, J.F.; Sengupta, K.; Jasin, M.; Nussenzweig, A.; Ried, T.; Danuser, G.; Misteli, T. Positional Stability of Single Double-Strand Breaks in Mammalian Cells. Nat. Cell Biol. 2007, 9, 675–682.
- Jakob, B.; Splinter, J.; Durante, M.; Taucher-scholz, G. Live Cell Microscopy Analysis of Radiation-Induced DNA Double-Strand Break Motion. Proc. Natl. Acad. Sci. USA 2009, 106, 3172–3177.
- Roukos, V.; Voss, T.C.; Schmidt, C.K.; Lee, S.; Wangsa, D.; Misteli, T. Spatial Dynamics of Chromosome Translocations in Living Cells. Science (1979) 2013, 341, 660–664.
- Liu, J.; Vidi, P.A.; Lelièvre, S.A.; Irudayaraj, J.M.K. Nanoscale Histone Localization in Live Cells Reveals Reduced Chromatin Mobility in Response to DNA Damage. J. Cell Sci. 2015, 128, 599–604.
- Shin, Y.; Chang, Y.C.; Lee, D.S.W.; Berry, J.; Sanders, D.W.; Ronceray, P.; Wingreen, N.S.; Haataja, M.; Brangwynne, C.P. Liquid Nuclear Condensates Mechanically Sense and Restructure the Genome. Cell 2018, 175, 1481–1491.e13.
- Kilic, S.; Lezaja, A.; Gatti, M.; Bianco, E.; Michelena, J.; Imhof, R.; Altmeyer, M. Phase Separation of 53 BP 1 Determines Liquid-like Behavior of DNA Repair Compartments. EMBO J. 2019, 38, e101379.
- Altmeyer, M.; Neelsen, K.J.; Teloni, F.; Pozdnyakova, I.; Pellegrino, S.; Grøfte, M.; Rask, M.B.D.; Streicher, W.; Jungmichel, S.; Nielsen, M.L.; et al. Liquid Demixing of Intrinsically Disordered Proteins Is Seeded by Poly(ADP-Ribose). Nat. Commun. 2015, 6, 8088.
- Bobkova, E.; Depes, D.; Lee, J.H.; Jezkova, L.; Falkova, I.; Pagacova, E.; Kopecna, O.; Zadneprianetc, M.; Bacikova, A.; Kulikova, E.; et al. Recruitment of 53BP1 Proteins for DNA Repair and Persistence of Repair Clusters Differ for Cell Types as Detected by Single Molecule Localization Microscopy. Int. J. Mol. Sci. 2018, 19, 3713.
- Ochs, F.; Karemore, G.; Miron, E.; Brown, J.; Sedlackova, H.; Rask, M.B.; Lampe, M.; Buckle, V.; Schermelleh, L.; Lukas, J.; et al. Stabilization of Chromatin Topology Safeguards Genome Integrity. Nature 2019, 574, 571–574.
- Xu, J.; Ma, H.; Ma, H.; Jiang, W.; Mela, C.A.; Duan, M.; Zhao, S.; Gao, C.; Hahm, E.R.; Lardo, S.M.; et al. Super-Resolution Imaging Reveals the Evolution of Higher-Order Chromatin Folding in Early Carcinogenesis. Nat. Commun. 2020, 11, 1899.
More
Information
Subjects:
Cell Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
644
Revisions:
2 times
(View History)
Update Date:
28 Nov 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No