Chromatin is regarded as a heterogeneous and dynamic structure occupying a non-random position within the cell nucleus, where it plays a key role in regulating various functions of the genome. In addition to challenging early assumptions of chromatin being regular and static, high spatiotemporal resolution imaging made it possible to visualize and characterize different chromatin structures such as clutches, domains and compartments. More specifically, super-resolution microscopy facilitates the study of different cellular processes at a nucleosome scale, providing a multi-scale view of chromatin behavior within the nucleus in different environments.
- chromatin organization and dynamics
- high resolution imaging
- DNA repair
1. Introduction
2. Recent Fluorescence-Based Techniques to Study the Dynamic Organization of Chromatin at High Spatiotemporal Resolution
2.1. FRET-FLIM
2.2. FCS
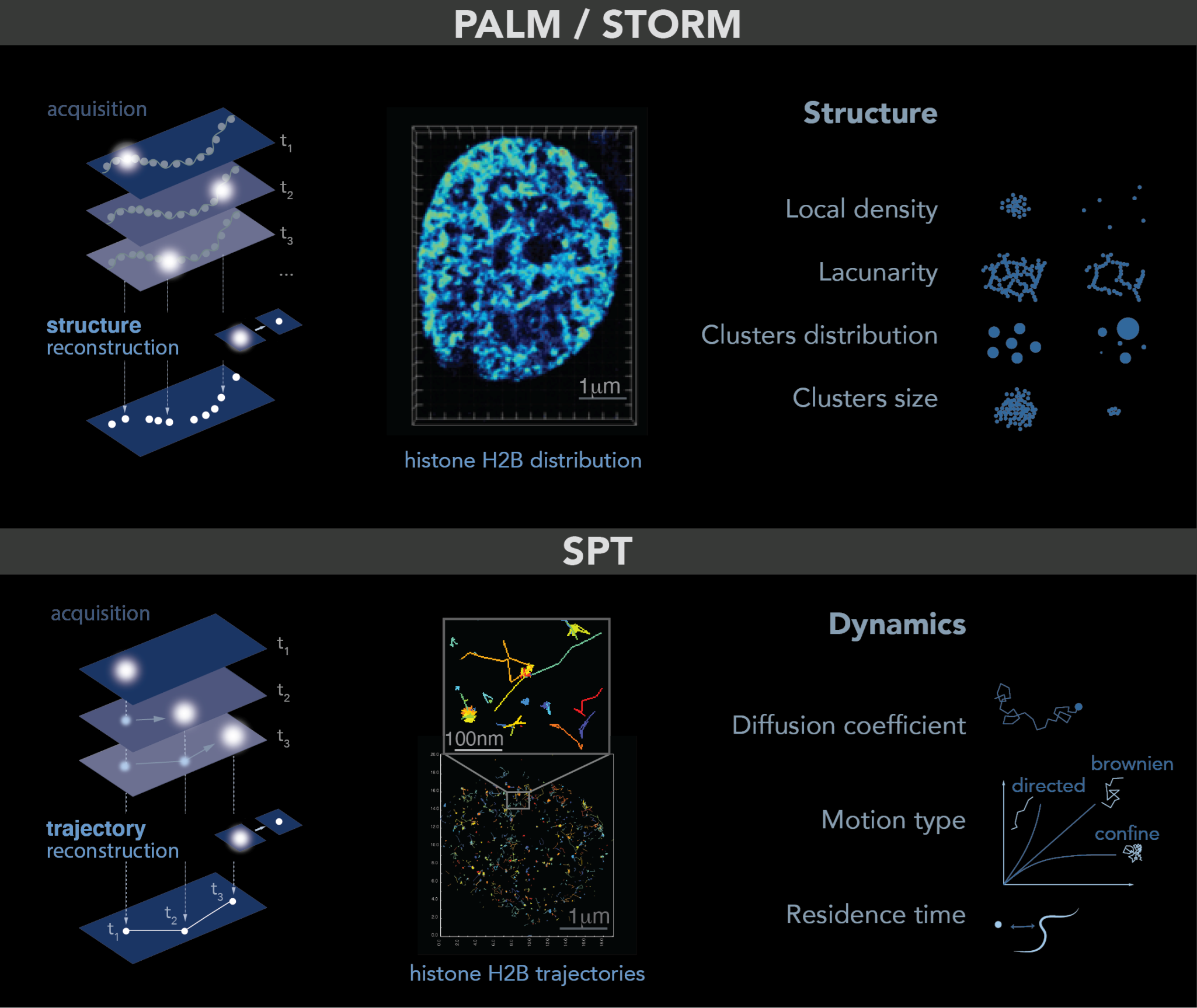
2.3. Single Molecule Localization Microscopy: PALM–STORM-SPT
2.4. Multiplexed FISH Combined with Super-Resolution Imaging
3. Chromatin Conformation as Proxy of Chromatin Accessibility
4. DNA Repair and Genome Stability
This entry is adapted from the peer-reviewed paper 10.3390/ijms242115975
References
- Dupont, C.; Chahar, D.; Trullo, A.; Gostan, T.; Surcis, C.; Grimaud, C.; Fisher, D.; Feil, R.; Llères, D. Evidence for Low Nanocompaction of Heterochromatin in Living Embryonic Stem Cells. EMBO J. 2023, 42, e110286.
- Llères, D.; James, J.; Swift, S.; Norman, D.G.; Lamond, A.I. Quantitative Analysis of Chromatin Compaction in Living Cells Using FLIM-FRET. J. Cell Biol. 2009, 187, 481–496.
- Audugé, N.; Padilla-Parra, S.; Tramier, M.; Borghi, N.; Coppey-Moisan, M. Chromatin Condensation Fluctuations Rather than Steady-State Predict Chromatin Accessibility. Nucleic Acids Res. 2019, 47, 6184–6194.
- Schwille, P.; Bieschke, J.; Oehlenschltiger, F. Kinetic Investigations by Fluorescence Correlation Spectroscopy: The Analytical and Diagnostic Potential of Diffusion Studies. Biophys. Chem. 1997, 66, 211–228.
- Magde, D.; Elson, E.L.; Webb, W.W. Fluorescence Correlation Spectroscopy. II. An Experimental Realization. Biopolymers 1974, 13, 29–61.
- Michelman-Ribeiro, A.; Mazza, D.; Rosales, T.; Stasevich, T.J.; Boukari, H.; Rishi, V.; Vinson, C.; Knutson, J.R.; McNally, J.G. Direct Measurement of Association and Dissociation Rates of DNA Binding in Live Cells by Fluorescence Correlation Spectroscopy. Biophys. J. 2009, 97, 337–346.
- D’Augustin, O.; Gaudon, V.; Siberchicot, C.; Smith, R.; Chapuis, C.; Depagne, J.; Veaute, X.; Busso, D.; Di Guilmi, A.M.; Castaing, B.; et al. Identification of Key Residues of the DNA Glycosylase OGG1 Controlling Efficient DNA Sampling and Recruitment to Oxidized Bases in Living Cells. Nucleic Acids Res. 2023, 51, 4942–4958.
- Yu, L.; Lei, Y.; Ma, Y.; Liu, M.; Zheng, J.; Dan, D.; Gao, P. A Comprehensive Review of Fluorescence Correlation Spectroscopy. Front. Phys. 2021, 9, 644450.
- Wachsmuth, M.; Knoch, T.A.; Rippe, K. Dynamic Properties of Independent Chromatin Domains Measured by Correlation Spectroscopy in Living Cells. Epigenetics Chromatin 2016, 9, 57.
- Bancaud, A.; Huet, S.; Daigle, N.; Mozziconacci, J.; Beaudouin, J.; Ellenberg, J. Molecular Crowding Affects Diffusion and Binding of Nuclear Proteins in Heterochromatin and Reveals the Fractal Organization of Chromatin. EMBO J. 2009, 28, 3785–3798.
- Betzig, E.; Patterson, G.; Sougrat, R.; Lindwasser, W.; Olenych, S.; Bonifacino, J.; Davidson, M.; Lippincott-Schwartz, J.; Hess, H. Imaging Intracellular Fluorescent Proteins at Nanometer Resolution. Science (1979) 2006, 313, 1638–1642.
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-Diffraction-Limit Imaging by Stochastic Optical Reconstruction Microscopy (STORM). Nat. Methods 2006, 3, 793–795.
- Miné-Hattab, J. Condensates: When fixation creates fiction. eLife 2023, 12, e85671.
- Andronov, L.; Orlov, I.; Lutz, Y.; Vonesch, J.-L.; Klaholz, B.P. ClusterViSu, a Method for Clustering of Protein Complexes by Voronoi Tessellation in Super-Resolution Microscopy. Sci. Rep. 2016, 6, 24084.
- Levet, F.; Julien, G.; Galland, R.; Butler, C.; Beghin, A.; Chazeau, A.; Hoess, P.; Ries, J.; Giannone, G.; Sibarita, J.-B. A Tessellation-Based Colocalization Analysis Approach for Single-Molecule Localization Microscopy. Nat. Commun. 2019, 10, 2379.
- Heltberg, M.L.; Miné-Hattab, J.; Taddei, A.; Walczak, A.M.; Mora, T. Physical Observables to Determine the Nature of Membrane-Less Cellular Sub-Compartments. eLife 2021, 10, 69181.
- Lichter, P.; Cremer, T.; Borden, J.; Manuelidis, L.; Ward, D.C. Delineation of Individual Human Chromosomes in Metaphase and Interphase Cells by in Situ Suppression Hybridization Using Recombinant DNA Libraries. Hum. Genet. 1988, 80, 224–234.
- Beliveau, B.J.; Joyce, E.F.; Apostolopoulos, N.; Yilmaz, F.; Fonseka, C.Y.; McCole, R.B.; Chang, Y.; Li, J.B.; Senaratne, T.N.; Williams, B.R.; et al. Versatile Design and Synthesis Platform for Visualizing Genomes with Oligopaint FISH Probes. Proc. Natl. Acad. Sci. USA 2012, 109, 21301–21306.
- Boettiger, A.N.; Bintu, B.; Moffitt, J.R.; Wang, S.; Beliveau, B.J.; Fudenberg, G.; Imakaev, M.; Mirny, L.A.; Wu, C.T.; Zhuang, X. Super-Resolution Imaging Reveals Distinct Chromatin Folding for Different Epigenetic States. Nature 2016, 529, 418–422.
- Eng, C.H.L.; Lawson, M.; Zhu, Q.; Dries, R.; Koulena, N.; Takei, Y.; Yun, J.; Cronin, C.; Karp, C.; Yuan, G.C.; et al. Transcriptome-Scale Super-Resolved Imaging in Tissues by RNA SeqFISH+. Nature 2019, 568, 235–239.
- Shah, R.U.; Robinson, E.S.; Gu, P.; Robinson, A.L.; Apte, J.S.; Presto, A.A. High-Spatial-Resolution Mapping and Source Apportionment of Aerosol Composition in Oakland, California, Using Mobile Aerosol Mass Spectrometry. Atmos. Chem. Phys. 2018, 18, 16325–16344.
- Xia, C.; Fan, J.; Emanuel, G.; Hao, J.; Zhuang, X. Spatial Transcriptome Profiling by MERFISH Reveals Subcellular RNA Compartmentalization and Cell Cycle-Dependent Gene Expression. Proc. Natl. Acad. Sci. USA 2019, 116, 19490–19499.
- Takei, Y.; Shah, S.; Harvey, S.; Qi, L.S.; Cai, L. Multiplexed Dynamic Imaging of Genomic Loci by Combined CRISPR Imaging and DNA Sequential FISH. Biophys. J. 2017, 112, 1773–1776.
- Guan, J.; Liu, H.; Shi, X.; Feng, S.; Huang, B. Tracking Multiple Genomic Elements Using Correlative CRISPR Imaging and Sequential DNA FISH. Biophys. J. 2017, 112, 1077–1084.
- Ou, H.D.; Phan, S.; Deerinck, T.J.; Thor, A.; Ellisman, M.H.; O’Shea, C.C. ChromEMT: Visualizing 3D Chromatin Structure and Compaction in Interphase and Mitotic Cells. Science (1979) 2017, 357, eaag0025.
- Ricci, M.A.; Manzo, C.; García-Parajo, M.F.; Lakadamyali, M.; Cosma, M.P. Chromatin Fibers Are Formed by Heterogeneous Groups of Nucleosomes in Vivo. Cell 2015, 160, 1145–1158.
- Barth, R.; Bystricky, K.; Shaban, H.A. Coupling Chromatin Structure and Dynamics by Live Super-Resolution Imaging. Sci. Adv. 2020, 6, eaaz2196.
- Sedat, J.; McDonald, A.; Kasler, H.; Verdin, E.; Cang, H.; Arigovindan, M.; Murre, C.; Elbaum, M. A Proposed Unified Mitotic Chromosome Architecture. Proc. Natl. Acad. Sci. USA 2022, 119, e2119107119.
- Xu, J.; Ma, H.; Jin, J.; Uttam, S.; Fu, R.; Huang, Y.; Liu, Y. Super-Resolution Imaging of Higher-Order Chromatin Structures at Different Epigenomic States in Single Mammalian Cells. Cell Rep. 2018, 24, 873–882.
- Harrison, C.J.; Allen’, T.D.; Britch, M.; Harris, R. High-resolution scanning electron microscopy of human metaphase chromosomes. J. Cell Sci. 1982, 56, 409–422.
- Sumner, A.T. Scanning Electron Microscopy of Mammalian Chromosomes from Prophase to Telophase. Chromosoma 1991, 100, 410–418.
- Le Gros, M.A.; Clowney, E.J.; Magklara, A.; Yen, A.; Markenscoff-Papadimitriou, E.; Colquitt, B.; Myllys, M.; Kellis, M.; Lomvardas, S.; Larabell, C.A. Soft X-Ray Tomography Reveals Gradual Chromatin Compaction and Reorganization during Neurogenesis In Vivo. Cell Rep. 2016, 17, 2125–2136.
- Larson, A.G.; Elnatan, D.; Keenen, M.M.; Trnka, M.J.; Johnston, J.B.; Burlingame, A.L.; Agard, D.A.; Redding, S.; Narlikar, G.J. Liquid Droplet Formation by HP1α Suggests a Role for Phase Separation in Heterochromatin. Nature 2017, 547, 236–240.
- Strom, A.R.; Emelyanov, A.V.; Mir, M.; Fyodorov, D.V.; Darzacq, X.; Karpen, G.H. Phase Separation Drives Heterochromatin Domain Formation. Nature 2017, 547, 241–245.
- Erdel, F.; Rademacher, A.; Vlijm, R.; Tünnermann, J.; Frank, L.; Weinmann, R.; Schweigert, E.; Yserentant, K.; Hummert, J.; Bauer, C.; et al. Mouse Heterochromatin Adopts Digital Compaction States without Showing Hallmarks of HP1-Driven Liquid-Liquid Phase Separation. Mol. Cell 2020, 78, 236–249.e7.
- Tortora, M.M.C.; Brennan, L.; Karpen, G.; Jost, D. Liquid-Liquid Phase Separation Recapitulates the Thermodynamics and Kinetics of Heterochromatin Formation. bioRxiv 2022.
- Kemp, M.G.; Ghosh, M.; Liu, G.; Leffak, M. The Histone Deacetylase Inhibitor Trichostatin A Alters the Pattern of DNA Replication Origin Activity in Human Cells. Nucleic Acids Res. 2005, 33, 325–336.
- Goren, A.; Tabib, A.; Hecht, M.; Cedar, H. DNA Replication Timing of the Human β-Globin Domain Is Controlled by Histone Modification at the Origin. Genes Dev. 2008, 22, 1319–1324.
- Sehwaiger, M.; Stadler, M.B.; Bell, O.; Kohler, H.; Oakeley, E.J.; Sehübeler, D. Chromatin State Marks Cell-Type- and Gender-Specific Replication of the Drosophila Genome. Genes Dev. 2009, 23, 589–601.
- Hansen, R.S.; Thomas, S.; Sandstrom, R.; Canfield, T.K.; Thurman, R.E.; Weaver, M.; Dorschner, M.O.; Gartler, S.M.; Stamatoyannopoulos, J.A. Sequencing Newly Replicated DNA Reveals Widespread Plasticity in Human Replication Timing. Proc. Natl. Acad. Sci. USA 2010, 107, 139–144.
- Lubelsky, Y.; Prinz, J.A.; DeNapoli, L.; Li, Y.; Belsky, J.A.; MacAlpine, D.M. DNA Replication and Transcription Programs Respond to the Same Chromatin Cues. Genome Res. 2014, 24, 1102–1114.
- Falk, M.; Lukášová, E.; Štefančíková, L.; Baranová, E.; Falková, I.; Ježková, L.; Davídková, M.; Bačíková, A.; Vachelová, J.; Michaelidesová, A.; et al. Heterochromatinization Associated with Cell Differentiation as a Model to Study DNA Double Strand Break Induction and Repair in the Context of Higher-Order Chromatin Structure. Appl. Radiat. Isot. 2014, 83, 177–185.
- Rosa, S.; Ntoukakis, V.; Ohmido, N.; Pendle, A.; Abranches, R.; Shaw, P. Cell Differentiation and Development in Arabidopsis Are Associated with Changes in Histone Dynamics at the Single-Cell Level. Plant Cell 2014, 26, 4821–4833.
- Dixon, J.R.; Jung, I.; Selvaraj, S.; Shen, Y.; Antosiewicz-Bourget, J.E.; Lee, A.Y.; Ye, Z.; Kim, A.; Rajagopal, N.; Xie, W.; et al. Chromatin Architecture Reorganization during Stem Cell Differentiation. Nature 2015, 518, 331–336.
- Fraser, J.; Ferrai, C.; Chiariello, A.M.; Schueler, M.; Rito, T.; Laudanno, G.; Barbieri, M.; Moore, B.L.; Kraemer, D.C.; Aitken, S.; et al. Hierarchical Folding and Reorganization of Chromosomes Are Linked to Transcriptional Changes in Cellular Differentiation. Mol. Syst. Biol. 2015, 11, 852.
- Nozaki, T.; Imai, R.; Tanbo, M.; Nagashima, R.; Tamura, S.; Tani, T.; Joti, Y.; Tomita, M.; Hibino, K.; Kanemaki, M.T.; et al. Dynamic Organization of Chromatin Domains Revealed by Super-Resolution Live-Cell Imaging. Mol. Cell 2017, 67, 282–293.e7.
- Chen, T.; Dent, S.Y.R. Chromatin Modifiers and Remodellers: Regulators of Cellular Differentiation. Nat. Rev. Genet. 2014, 15, 93–106.
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological Domains in Mammalian Genomes Identified by Analysis of Chromatin Interactions. Nature 2012, 485, 376–380.
- Locatelli, M.; Lawrimore, J.; Lin, H.; Sanaullah, S.; Seitz, C.; Segall, D.; Kefer, P.; Bloom, K.; Liu, J.; Bonin, K.; et al. DNA Damage Reduces Heterogeneity and Coherence of Chromatin Motions. Proc. Natl. Acad. Sci. USA 2022, 119, e2205166119.
- Chubb, J.R.; Boyle, S.; Perry, P.; Bickmore, W.A. Chromatin Motion Is Constrained by Association with Nuclear Compartments in Human Cells. Curr. Biol. 2002, 12, 439–445.
- Lerner, J.; Gomez-Garcia, P.A.; McCarthy, R.L.; Liu, Z.; Lakadamyali, M.; Zaret, K.S. Two-Parameter Mobility Assessments Discriminate Diverse Regulatory Factor Behaviors in Chromatin. Mol. Cell 2020, 79, 677–688.e6.
- Iida, S.; Shinkai, S.; Itoh, Y.; Tamura, S.; Kanemaki, M.T.; Onami, S.; Maeshima, K. Single-Nucleosome Imaging Reveals Steady-State Motion of Interphase Chromatin in Living Human Cells. Sci. Adv. 2022, 8, eabn5626.
- Symington, L.S.; Gautier, J. Double-Strand Break End Resection and Repair Pathway Choice. Annu. Rev. Genet. 2011, 45, 247–271.
- Mehta, A.; Haber, J.E. Sources of DNA Double-Strand Breaks and Models of Recombinational DNA Repair. Cold Spring Harb. Perspect. Biol. 2014, 6, a016428.
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64.
- García Fernandez, F.; Fabre, E. The Dynamic Behavior of Chromatin in Response to DNA Double-Strand Breaks. Genes 2022, 13, 215.
- Mladenov, E.; Magin, S.; Soni, A.; Iliakis, G. DNA Double-Strand-Break Repair in Higher Eukaryotes and Its Role in Genomic Instability and Cancer: Cell Cycle and Proliferation-Dependent Regulation. Semin. Cancer Biol. 2016, 37–38, 51–64.
- Adkins, N.L.; Niu, H.; Sung, P.; Peterson, C.L. Nucleosome Dynamics Regulates DNA Processing. Nat. Struct. Mol. Biol. 2013, 20, 836–842.
- Jakob, B.; Splinter, J.; Conrad, S.; Voss, K.O.; Zink, D.; Durante, M.; Löbrich, M.; Taucher-Scholz, G. DNA Double-Strand Breaks in Heterochromatin Elicit Fast Repair Protein Recruitment, Histone H2AX Phosphorylation and Relocation to Euchromatin. Nucleic Acids Res. 2011, 39, 6489–6499.
- Zhang, Y.; Máté, G.; Müller, P.; Hillebrandt, S.; Krufczik, M.; Bach, M.; Kaufmann, R.; Hausmann, M.; Heermann, D.W. Radiation Induced Chromatin Conformation Changes Analysed by Fluorescent Localization Microscopy, Statistical Physics, and Graph Theory. PLoS ONE 2015, 10, e128555.
- Clouaire, T.; Rocher, V.; Lashgari, A.; Arnould, C.; Aguirrebengoa, M.; Biernacka, A.; Skrzypczak, M.; Aymard, F.; Fongang, B.; Dojer, N.; et al. Comprehensive Mapping of Histone Modifications at DNA Double-Strand Breaks Deciphers Repair Pathway Chromatin Signatures. Mol. Cell 2018, 72, 250–262.e6.
- Fortuny, A.; Chansard, A.; Caron, P.; Chevallier, O.; Leroy, O.; Renaud, O.; Polo, S.E. Imaging the Response to DNA Damage in Heterochromatin Domains Reveals Core Principles of Heterochromatin Maintenance. Nat. Commun. 2021, 12, 2428.
- Casali, C.; Siciliani, S.; Galgano, L.; Biggiogera, M. Oxidative Stress and Nuclear Reprogramming: A Pilot Study of the Effects of Reactive Oxygen Species on Architectural and Epigenetic Landscapes. Int. J. Mol. Sci. 2022, 24, 153.
- Hausmann, M.; Falk, M.; Neitzel, C.; Hofmann, A.; Biswas, A.; Gier, T.; Falkova, I.; Heermann, D.W. Elucidation of the Clustered Nano-Architecture of Radiation-Induced Dna Damage Sites and Surrounding Chromatin in Cancer Cells: A Single Molecule Localization Microscopy Approach. Int. J. Mol. Sci. 2021, 22, 3636.
- Smith, R.; Zentout, S.; Rother, M.; Bigot, N.; Chapuis, C.; Mihuț, A.; Zobel, F.F.; Ahel, I.; van Attikum, H.; Timinszky, G.; et al. HPF1-Dependent Histone ADP-Ribosylation Triggers Chromatin Relaxation to Promote the Recruitment of Repair Factors at Sites of DNA Damage. Nat. Struct. Mol. Biol. 2023, 30, 678–691.
- Sellou, H.; Lebeaupin, T.; Chapuis, C.; Smith, R.; Hegele, A.; Singh, H.R.; Kozlowski, M.; Bultmann, S.; Ladurner, A.G.; Timinszky, G.; et al. The Poly(ADP-Ribose)-Dependent Chromatin Remodeler Alc1 Induces Local Chromatin Relaxation upon DNA Damage. Mol. Biol. Cell 2016, 27, 3791–3799.
- Burgess, R.C.; Burman, B.; Kruhlak, M.J.; Misteli, T. Activation of DNA Damage Response Signaling by Condensed Chromatin. Cell Rep. 2014, 9, 1703–1717.
- Rogakou, E.P.; Boon, C.; Redon, C.; Bonner, W.M. Megabase Chromatin Domains Involved in DNA Double-Strand Breaks In Vivo. J. Cell Biol. 1999, 146, 905–915.
- Burma, S.; Chen, B.P.; Murphy, M.; Kurimasa, A.; Chen, D.J. ATM Phosphorylates Histone H2AX in Response to DNA Double-Strand Breaks. J. Biol. Chem. 2001, 276, 42462–42467.
- Shroff, R.; Arbel-Eden, A.; Pilch, D.; Ira, G.; Bonner, W.M.; Petrini, J.H.; Haber, J.E.; Lichten, M. Distribution and Dynamics of Chromatin Modification Induced by a Defined DNA Double-Strand Break. Curr. Biol. 2004, 14, 1703–1711.
- Khurana, S.; Kruhlak, M.J.; Kim, J.; Tran, A.D.; Liu, J.; Nyswaner, K.; Shi, L.; Jailwala, P.; Sung, M.H.; Hakim, O.; et al. A Macrohistone Variant Links Dynamic Chromatin Compaction to BRCA1-Dependent Genome Maintenance. Cell Rep. 2014, 8, 1049–1062.
- Caron, P.; Aymard, F.; Iacovoni, J.S.; Briois, S.; Canitrot, Y.; Bugler, B.; Massip, L.; Losada, A.; Legube, G. Cohesin Protects Genes against ΓH2AX Induced by DNA Double-Strand Breaks. PLoS Genet. 2012, 8, e1002460.
- Goloborodko, A.; Imakaev, M.V.; Marko, J.F.; Mirny, L. Compaction and Segregation of Sister Chromatids via Active Loop Extrusion. eLife 2016, 5, e14864.
- Natale, F.; Rapp, A.; Yu, W.; Maiser, A.; Harz, H.; Scholl, A.; Grulich, S.; Anton, T.; Hörl, D.; Chen, W.; et al. Identification of the Elementary Structural Units of the DNA Damage Response. Nat. Commun. 2017, 8, 15760.
- Sanders, J.T.; Freeman, T.F.; Xu, Y.; Golloshi, R.; Stallard, M.A.; Hill, A.M.; San Martin, R.; Balajee, A.S.; McCord, R.P. Radiation-Induced DNA Damage and Repair Effects on 3D Genome Organization. Nat. Commun. 2020, 11, 6178.
- Caron, P.; Choudjaye, J.; Clouaire, T.; Corte, F.; Daburon, V.; Aguirrebengoa, M.; Mangeat, T.; Iacovoni, J.S.; Alejandro, A. Non-Redundant Functions of ATM and DNA-PKcs in Response to DNA Double-Strand Breaks. Cell Rep. 2015, 13, 1598–1609.
- Amat, R.; Böttcher, R.; Le Dily, F.; Vidal, E.; Quilez, J.; Cuartero, Y.; Beato, M.; de Nadal, E.; Posas, F. Rapid Reversible Changes in Compartments and Local Chromatin Organization Revealed by Hyperosmotic Shock. Genome Res. 2019, 29, 18–28.
- Ayrapetov, M.K.; Gursoy-Yuzugullu, O.; Xu, C.; Xu, Y.; Price, B.D. DNA Double-Strand Breaks Promote Methylation of Histone H3 on Lysine 9 and Transient Formation of Repressive Chromatin. Proc. Natl. Acad. Sci. USA 2014, 111, 9169–9174.
- Goodarzi, A.A.; Jeggo, P.A. The Heterochromatic Barrier to DNA Double Strand Break Repair: How to Get the Entry Visa. Int. J. Mol. Sci. 2012, 13, 11844–11860.
- Miné-Hattab, J.; Rothstein, R.; Mine-Hattab, J.; Rothstein, R. Increased Chromosome Mobility Facilitates Homology Search during Recombination. Nat. Cell Biol. 2012, 14, 510–517.
- Clouaire, T.; Legube, G. A Snapshot on the Cis Chromatin Response to DNA Double-Strand Breaks. Trends Genet. 2019, 35, 330–345.
- García Fernández, F.; Almayrac, E.; Carré Simon, À.; Batrin, R.; Khalil, Y.; Boissac, M.; Fabre, E. Global Chromatin Mobility Induced by a DSB Is Dictated by Chromosomal Conformation and Defines the HR Outcome. eLife 2022, 11, e78015.
- García Fernández, F.; Lemos, B.; Khalil, Y.; Batrin, R.; Haber, J.E.; Fabre, E. Modified Chromosome Structure Caused by Phosphomimetic H2A Modulates the DNA Damage Response by Increasing Chromatin Mobility in Yeast. J. Cell Sci. 2021, 134, jcs258500.
- Herbert, S.; Brion, A.; Arbona, J.-M.; Lelek, M.; Veillet, A.; Lelandais, B.; Parmar, J.; Fernández, F.G.; Almayrac, E.; Khalil, Y.; et al. Chromatin Stiffening Underlies Enhanced Locus Mobility after DNA Damage in Budding Yeast. EMBO J. 2017, 36, 2595–2608.
- Liu, S.; Miné-Hattab, J.; Villemeur, M.; Guerois, R.; Pinholt, H.D.; Mirny, L.A.; Taddei, A. Publisher Correction: In Vivo Tracking of Functionally Tagged Rad51 Unveils a Robust Strategy of Homology Search. Nat. Struct. Mol. Biol. 2023, 30, 1607.
- Strecker, J.; Gupta, G.D.; Zhang, W.; Bashkurov, M.; Landry, M.-C.; Pelletier, L.; Durocher, D. DNA Damage Signalling Targets the Kinetochore to Promote Chromatin Mobility. Nat. Cell Biol. 2016, 18, 281–290.
- Seeber, A.; Dion, V.; Gasser, S.M. Checkpoint Kinases and the INO80 Nucleosome Remodeling Complex Enhance Global Chromatin Mobility in Response to DNA Damage. Genes Dev. 2013, 27, 1999–2008.
- Dion, V.; Kalck, V.; Horigome, C.; Towbin, B.D.; Gasser, S.M. Increased Mobility of Double-Strand Breaks Requires Mec1, Rad9 and the Homologous Recombination Machinery. Nat. Cell Biol. 2012, 14, 502–509.
- Kruhlak, M.J.; Celeste, A.; Dellaire, G.; Fernandez-Capetillo, O.; Müller, W.G.; McNally, J.G.; Bazett-Jones, D.P.; Nussenzweig, A. Changes in Chromatin Structure and Mobility in Living Cells at Sites of DNA Double-Strand Breaks. J. Cell Biol. 2006, 172, 823–834.
- Soutoglou, E.; Dorn, J.F.; Sengupta, K.; Jasin, M.; Nussenzweig, A.; Ried, T.; Danuser, G.; Misteli, T. Positional Stability of Single Double-Strand Breaks in Mammalian Cells. Nat. Cell Biol. 2007, 9, 675–682.
- Jakob, B.; Splinter, J.; Durante, M.; Taucher-scholz, G. Live Cell Microscopy Analysis of Radiation-Induced DNA Double-Strand Break Motion. Proc. Natl. Acad. Sci. USA 2009, 106, 3172–3177.
- Roukos, V.; Voss, T.C.; Schmidt, C.K.; Lee, S.; Wangsa, D.; Misteli, T. Spatial Dynamics of Chromosome Translocations in Living Cells. Science (1979) 2013, 341, 660–664.
- Liu, J.; Vidi, P.A.; Lelièvre, S.A.; Irudayaraj, J.M.K. Nanoscale Histone Localization in Live Cells Reveals Reduced Chromatin Mobility in Response to DNA Damage. J. Cell Sci. 2015, 128, 599–604.
- Shin, Y.; Chang, Y.C.; Lee, D.S.W.; Berry, J.; Sanders, D.W.; Ronceray, P.; Wingreen, N.S.; Haataja, M.; Brangwynne, C.P. Liquid Nuclear Condensates Mechanically Sense and Restructure the Genome. Cell 2018, 175, 1481–1491.e13.
- Kilic, S.; Lezaja, A.; Gatti, M.; Bianco, E.; Michelena, J.; Imhof, R.; Altmeyer, M. Phase Separation of 53 BP 1 Determines Liquid-like Behavior of DNA Repair Compartments. EMBO J. 2019, 38, e101379.
- Altmeyer, M.; Neelsen, K.J.; Teloni, F.; Pozdnyakova, I.; Pellegrino, S.; Grøfte, M.; Rask, M.B.D.; Streicher, W.; Jungmichel, S.; Nielsen, M.L.; et al. Liquid Demixing of Intrinsically Disordered Proteins Is Seeded by Poly(ADP-Ribose). Nat. Commun. 2015, 6, 8088.
- Bobkova, E.; Depes, D.; Lee, J.H.; Jezkova, L.; Falkova, I.; Pagacova, E.; Kopecna, O.; Zadneprianetc, M.; Bacikova, A.; Kulikova, E.; et al. Recruitment of 53BP1 Proteins for DNA Repair and Persistence of Repair Clusters Differ for Cell Types as Detected by Single Molecule Localization Microscopy. Int. J. Mol. Sci. 2018, 19, 3713.
- Ochs, F.; Karemore, G.; Miron, E.; Brown, J.; Sedlackova, H.; Rask, M.B.; Lampe, M.; Buckle, V.; Schermelleh, L.; Lukas, J.; et al. Stabilization of Chromatin Topology Safeguards Genome Integrity. Nature 2019, 574, 571–574.
- Xu, J.; Ma, H.; Ma, H.; Jiang, W.; Mela, C.A.; Duan, M.; Zhao, S.; Gao, C.; Hahm, E.R.; Lardo, S.M.; et al. Super-Resolution Imaging Reveals the Evolution of Higher-Order Chromatin Folding in Early Carcinogenesis. Nat. Commun. 2020, 11, 1899.