 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Stanislav A. Rybtsov | -- | 4678 | 2023-11-22 15:31:40 | | | |
| 2 | Lindsay Dong | Meta information modification | 4678 | 2023-11-23 09:22:44 | | |
Video Upload Options
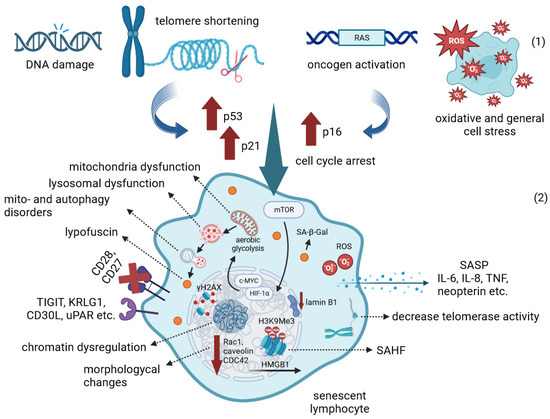
The accumulation of senescent cells increases age-related background inflammation, “Inflammaging”, causing lymphocyte exhaustion and cardiovascular, neurodegenerative, autoimmune and cancer diseases. Although modern research has not yet identified specific markers of aging lymphocytes, several sets of markers facilitate the separation of the aging population based on normal memory and exhausted cells for further genetic and functional analysis. The reasons for the higher predisposition of CD8+ T-lymphocytes to senescence compared to the CD4+ population are also discussed. The suppression of immune senescence is the most relevant area of research aimed at developing anti-aging and anti-cancer therapy for prolonging the lifespan of the global population.
1. Introduction
2. The Hallmarks of Senescent Immune Cells
2.1. Morphological Changes
2.2. Surface Markers
2.3. SASP
2.4. Decrease in Telomere Length and Telomerase Activity
2.5. Cell Cycle Arrest and Expression of p16, p21 and p53
2.6. Metabolic Changes/Disorders
2.6.1. Energy Metabolism Disruptions
2.6.2. Mitochondrial Dysfunction
2.6.3. Autophagy and Mitophagy Disorder
2.6.4. SA-β-Gal
2.7. Disorganization and Dysfunction of Chromatin
2.7.1. HMGB1
2.7.2. SAHF
2.7.3. Lamin B1
2.7.4. γH2AX
2.8. Multi-Omics Changes
2.8.1. Transcriptome
2.8.2. Epigenetic Changes
2.8.3. Chromatin Accessibility
3. Conclusions
References
- Budamagunta, V.; Foster, T.C.; Zhou, D. Cellular Senescence in Lymphoid Organs and Immunosenescence. Aging 2021, 13, 19920–19941.
- Josephson, A.M.; Bradaschia-Correa, V.; Lee, S.; Leclerc, K.; Patel, K.S.; Muinos Lopez, E.; Litwa, H.P.; Neibart, S.S.; Kadiyala, M.; Wong, M.Z.; et al. Age-Related Inflammation Triggers Skeletal Stem/Progenitor Cell Dysfunction. Proc. Natl. Acad. Sci. USA 2019, 116, 6995–7004.
- McElhaney, J.E.; Verschoor, C.P.; Andrew, M.K.; Haynes, L.; Kuchel, G.A.; Pawelec, G. The Immune Response to Influenza in Older Humans: Beyond Immune Senescence. Immun. Ageing 2020, 17, 10.
- Fulop, T.; Larbi, A.; Pawelec, G.; Cohen, A.A.; Provost, G.; Khalil, A.; Lacombe, G.; Rodrigues, S.; Desroches, M.; Hirokawa, K.; et al. Immunosenescence and Altered Vaccine Efficiency in Older Subjects: A Myth Difficult to Change. Vaccines 2022, 10, 607.
- Martínez-Zamudio, R.I.; Dewald, H.K.; Vasilopoulos, T.; Gittens-Williams, L.; Fitzgerald-Bocarsly, P.; Herbig, U. Senescence-associated Β-galactosidase Reveals the Abundance of Senescent CD8+ T Cells in Aging Humans. Aging Cell 2021, 20, e13344.
- Zhao, Y.; Shao, Q.; Peng, G. Exhaustion and Senescence: Two Crucial Dysfunctional States of T Cells in the Tumor Microenvironment. Cell Mol. Immunol. 2020, 17, 27–35.
- Yi, H.-S.; Kim, S.Y.; Kim, J.T.; Lee, Y.-S.; Moon, J.S.; Kim, M.; Kang, Y.E.; Joung, K.H.; Lee, J.H.; Kim, H.J.; et al. T-Cell Senescence Contributes to Abnormal Glucose Homeostasis in Humans and Mice. Cell Death Dis. 2019, 10, 249.
- Costantini, E.; D’Angelo, C.; Reale, M. The Role of Immunosenescence in Neurodegenerative Diseases. Mediat. Inflamm. 2018, 2018, 6039171.
- Fulop, T.; Larbi, A.; Khalil, A.; Plotka, A.; Laurent, B.; Ramassamy, C.; Bosco, N.; Hirokawa, K.; Frost, E.H.; Witkowski, J.M. Immunosenescence and Alzheimer’s Disease. In Healthy Longevity and Immune System; Bueno, V., Pawelec, G., Eds.; Healthy Ageing and Longevity; Springer International Publishing: Cham, Switzerland, 2022; Volume 16, pp. 177–199. ISBN 978-3-030-87531-2.
- Dema, M.; Eixarch, H.; Villar, L.M.; Montalban, X.; Espejo, C. Immunosenescence in Multiple Sclerosis: The Identification of New Therapeutic Targets. Autoimmun. Rev. 2021, 20, 102893.
- Gao, Y.; Cai, W.; Zhou, Y.; Li, Y.; Cheng, J.; Wei, F. Immunosenescence of T Cells: A Key Player in Rheumatoid Arthritis. Inflamm. Res. 2022, 71, 1449–1462.
- Lu, Y.; Ruan, Y.; Hong, P.; Rui, K.; Liu, Q.; Wang, S.; Cui, D. T-Cell Senescence: A Crucial Player in Autoimmune Diseases. Clin. Immunol. 2023, 248, 109202.
- Shirakawa, K.; Sano, M. T Cell Immunosenescence in Aging, Obesity, and Cardiovascular Disease. Cells 2021, 10, 2435.
- Cunha, L.L.; Perazzio, S.F.; Azzi, J.; Cravedi, P.; Riella, L.V. Remodeling of the Immune Response With Aging: Immunosenescence and Its Potential Impact on COVID-19 Immune Response. Front. Immunol. 2020, 11, 1748.
- Vellasamy, D.M.; Lee, S.-J.; Goh, K.W.; Goh, B.-H.; Tang, Y.-Q.; Ming, L.C.; Yap, W.H. Targeting Immune Senescence in Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 13059.
- Okamura, K.; Nohara, K. Long-Term Arsenite Exposure Induces Premature Senescence in B Cell Lymphoma A20 Cells. Arch. Toxicol. 2016, 90, 793–803.
- Cakala-Jakimowicz, M.; Kolodziej-Wojnar, P.; Puzianowska-Kuznicka, M. Aging-Related Cellular, Structural and Functional Changes in the Lymph Nodes: A Significant Component of Immunosenescence? An Overview. Cells 2021, 10, 3148.
- Rezzani, R.; Nardo, L.; Favero, G.; Peroni, M.; Rodella, L.F. Thymus and Aging: Morphological, Radiological, and Functional Overview. Age 2014, 36, 313–351.
- Augert, A.; Bernar, D. Immunosenescence and Senescence Immunosurveillance: One of the Possible Links Explaining the Cancer Incidence in Ageing Population. In Senescence and Senescence-Related Disorders; Zhiwei, W., Ed.; InTech: Middlesex, MA, USA, 2013; ISBN 978-953-51-0997-6.
- Beregi, E.; Regius, O.; Rajczy, K. Comparative Study of the Morphological Changes in Lymphocytes of Elderly Individuals and Centenarians. Age Ageing 1991, 20, 55–59.
- Salmonowicz, H.; Passos, J.F. Detecting Senescence: A New Method for an Old Pigment. Aging Cell 2017, 16, 432–434.
- Terman, A.; Brunk, U.T. Lipofuscin: Mechanisms of Formation and Increase with Age. APMIS 1998, 106, 265–276.
- Rybtsova, N.; Berezina, T.N.; Rybtsov, S. Molecular Markers of Blood Cell Populations Can Help Estimate Aging of the Immune System. Int. J. Mol. Sci. 2023, 24, 5708.
- Monteiro, J.; Batliwalla, F.; Ostrer, H.; Gregersen, P.K. Shortened Telomeres in Clonally Expanded CD28-CD8+ T Cells Imply a Replicative History that is Distinct from Their CD28+CD8+ Counterparts. J. Immunol. 1996, 156, 3587–3590.
- Vallejo, A.N.; Brandes, J.C.; Weyand, C.M.; Goronzy, J.J. Modulation of CD28 Expression: Distinct Regulatory Pathways During Activation and Replicative Senescence. J. Immunol. 1999, 162, 6572–6579.
- Hamann, D.; Kostense, S.; Wolthers, K.C.; Otto, S.A.; Baars, P.A.; Miedema, F.; Van Lier, R.A.W. Evidence That Human CD8+CD45RA+CD27– Cells are Induced by Antigen and Evolve through Extensive Rounds of Division. Int. Immunol. 1999, 11, 1027–1033.
- Effros, R.B.; Boucher, N.; Porter, V.; Zhu, X.; Spaulding, C.; Walford, R.L.; Kronenberg, M.; Cohen, D.; Schächter, F. Decline in CD28+ T Cells in Centenarians and in Long-Term T Cell Cultures: A Possible Cause for Both In Vivo and In Vitro Immunosenescence. Exp. Gerontol. 1994, 29, 601–609.
- Nijhuis, E.W.P.; Remarque, E.J.; Hinloopen, B.; Van Der Pouw-Kraan, T.; Van Lier, R.A.W.; Ligthart, G.J.; Nagelkerken, L. Age-Related Increase in the Fraction of CD27−CD4+ T Cells and IL-4 Production as a Feature of CD4+ T Cell Differentiation In Vivo. Clin. Exp. Immunol. 2008, 96, 528–534.
- Dunne, P.J.; Faint, J.M.; Gudgeon, N.H.; Fletcher, J.M.; Plunkett, F.J.; Soares, M.V.D.; Hislop, A.D.; Annels, N.E.; Rickinson, A.B.; Salmon, M.; et al. Epstein-Barr Virus–Specific CD8+ T Cells That Re-Express CD45RA Are Apoptosis-Resistant Memory Cells That Retain Replicative Potential. Blood 2002, 100, 933–940.
- Brenchley, J.M.; Karandikar, N.J.; Betts, M.R.; Ambrozak, D.R.; Hill, B.J.; Crotty, L.E.; Casazza, J.P.; Kuruppu, J.; Migueles, S.A.; Connors, M.; et al. Expression of CD57 Defines Replicative Senescence and Antigen-Induced Apoptotic Death of CD8+ T Cells. Blood 2003, 101, 2711–2720.
- Voehringer, D.; Koschella, M.; Pircher, H. Lack of Proliferative Capacity of Human Effector and Memory T Cells Expressing Killer Cell Lectinlike Receptor G1 (KLRG1). Blood 2002, 100, 3698–3702.
- Ouyang, Q.; Wagner, W.M.; Voehringer, D.; Wikby, A.; Klatt, T.; Walter, S.; Müller, C.A.; Pircher, H.; Pawelec, G. Age-Associated Accumulation of CMV-Specific CD8+ T Cells Expressing the Inhibitory Killer Cell Lectin-like Receptor G1 (KLRG1). Exp. Gerontol. 2003, 38, 911–920.
- Song, Y.; Wang, B.; Song, R.; Hao, Y.; Wang, D.; Li, Y.; Jiang, Y.; Xu, L.; Ma, Y.; Zheng, H.; et al. T-Cell Immunoglobulin and ITIM Domain Contributes to CD8+ T-Cell Immunosenescence. Aging Cell 2018, 17, e12716.
- Romero, P.; Zippelius, A.; Kurth, I.; Pittet, M.J.; Touvrey, C.; Iancu, E.M.; Corthesy, P.; Devevre, E.; Speiser, D.E.; Rufer, N. Four Functionally Distinct Populations of Human Effector-Memory CD8+ T Lymphocytes. J. Immunol. 2007, 178, 4112–4119.
- Gründemann, C.; Schwartzkopff, S.; Koschella, M.; Schweier, O.; Peters, C.; Voehringer, D.; Pircher, H. The NK Receptor KLRG1 Is Dispensable for Virus-induced NK and CD8+ T-cell Differentiation and Function In Vivo. Eur. J. Immunol. 2010, 40, 1303–1314.
- Chong, L.K.; Aicheler, R.J.; Llewellyn-Lacey, S.; Tomasec, P.; Brennan, P.; Wang, E.C.Y. Proliferation and Interleukin 5 Production by CD8hiCD57+ T Cells. Eur. J. Immunol. 2008, 38, 995–1000.
- Chou, J.P.; Effros, R.B. T Cell Replicative Senescence in Human Aging. Curr. Pharm. Des. 2013, 19, 1680–1698.
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-Aging: An Evolutionary Perspective on Immunosenescence. Ann. N. Y. Acad. Sci. 2006, 908, 244–254.
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A New Immune–Metabolic Viewpoint for Age-Related Diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590.
- Watanabe, S.; Kawamoto, S.; Ohtani, N.; Hara, E. Impact of Senescence-Associated Secretory Phenotype and Its Potential as a Therapeutic Target for Senescence-Associated Diseases. Cancer Sci. 2017, 108, 563–569.
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118.
- Prata, L.G.P.L.; Ovsyannikova, I.G.; Tchkonia, T.; Kirkland, J.L. Senescent Cell Clearance by the Immune System: Emerging Therapeutic Opportunities. Semin. Immunol. 2018, 40, 101275.
- Barbé-Tuana, F.; Funchal, G.; Schmitz, C.R.R.; Maurmann, R.M.; Bauer, M.E. The Interplay between Immunosenescence and Age-Related Diseases. Semin. Immunopathol. 2020, 42, 545–557.
- Piber, D.; Olmstead, R.; Cho, J.H.-J.; Witarama, T.; Perez, C.; Dietz, N.; Seeman, T.E.; Breen, E.C.; Cole, S.W.; Irwin, M.R. Inflammaging: Age and Systemic, Cellular, and Nuclear Inflammatory Biology in Older Adults. J. Gerontol. Ser. A 2019, 74, 1716–1724.
- Furman, D.; Chang, J.; Lartigue, L.; Bolen, C.R.; Haddad, F.; Gaudilliere, B.; Ganio, E.A.; Fragiadakis, G.K.; Spitzer, M.H.; Douchet, I.; et al. Expression of Specific Inflammasome Gene Modules Stratifies Older Individuals into Two Extreme Clinical and Immunological States. Nat. Med. 2017, 23, 174–184.
- Mauer, J.; Chaurasia, B.; Goldau, J.; Vogt, M.C.; Ruud, J.; Nguyen, K.D.; Theurich, S.; Hausen, A.C.; Schmitz, J.; Brönneke, H.S.; et al. Signaling by IL-6 Promotes Alternative Activation of Macrophages to Limit Endotoxemia and Obesity-Associated Resistance to Insulin. Nat. Immunol. 2014, 15, 423–430.
- Spencer, M.E.; Jain, A.; Matteini, A.; Beamer, B.A.; Wang, N.-Y.; Leng, S.X.; Punjabi, N.M.; Walston, J.D.; Fedarko, N.S. Serum Levels of the Immune Activation Marker Neopterin Change With Age and Gender and Are Modified by Race, BMI, and Percentage of Body Fat. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2010, 65A, 858–865.
- Cooper, E.B.; Watowich, M.M.; Beeby, N.; Whalen, C.; Cayo Biobank Research Unit; Montague, M.J.; Brent, L.J.N.; Snyder-Mackler, N.; Higham, J.P. Concentrations of Urinary Neopterin, but Not suPAR, Positively Correlate with Age in Rhesus Macaques. Front. Ecol. Evol. 2022, 10, 1007052.
- Childs, B.G.; Durik, M.; Baker, D.J.; Van Deursen, J.M. Cellular Senescence in Aging and Age-Related Disease: From Mechanisms to Therapy. Nat. Med. 2015, 21, 1424–1435.
- Rivellese, F.; Surace, A.E.A.; Goldmann, K.; Sciacca, E.; Çubuk, C.; Giorli, G.; John, C.R.; Nerviani, A.; Fossati-Jimack, L.; Thorborn, G.; et al. Rituximab versus Tocilizumab in Rheumatoid Arthritis: Synovial Biopsy-Based Biomarker Analysis of the Phase 4 R4RA Randomized Trial. Nat. Med. 2022, 28, 1256–1268.
- Louis, E.; Resche-Rigon, M.; Laharie, D.; Satsangi, J.; Ding, N.; Siegmund, B.; D’Haens, G.; Picon, L.; Bossuyt, P.; Vuitton, L.; et al. Withdrawal of Infliximab or Concomitant Immunosuppressant Therapy in Patients with Crohn’s Disease on Combination Therapy (SPARE): A Multicentre, Open-Label, Randomised Controlled Trial. Lancet Gastroenterol. Hepatol. 2023, 8, 215–227.
- Onuora, S. Calprotectin Tracks Tocilizumab-Treated RA. Nat. Rev. Rheumatol. 2022, 18, 612.
- Zhou, D.; Borsa, M.; Simon, A.K. Hallmarks and Detection Techniques of Cellular Senescence and Cellular Ageing in Immune Cells. Aging Cell 2021, 20, e13316.
- Yadav, S.; Maurya, P.K. Correlation Between Telomere Length and Biomarkers of Oxidative Stress in Human Aging. Rejuvenation Res. 2022, 25, 25–29.
- Marioni, R.E.; Harris, S.E.; Shah, S.; McRae, A.F.; Von Zglinicki, T.; Martin-Ruiz, C.; Wray, N.R.; Visscher, P.M.; Deary, I.J. The Epigenetic Clock and Telomere Length Are Independently Associated with Chronological Age and Mortality. Int. J. Epidemiol. 2016, 45, 424–432.
- Abdisalaam, S.; Bhattacharya, S.; Mukherjee, S.; Sinha, D.; Srinivasan, K.; Zhu, M.; Akbay, E.A.; Sadek, H.A.; Shay, J.W.; Asaithamby, A. Dysfunctional Telomeres Trigger Cellular Senescence Mediated by Cyclic GMP-AMP Synthase. J. Biol. Chem. 2020, 295, 11144–11160.
- Ouyang, Q.; Baerlocher, G.; Vulto, I.; Lansdorp, P.M. Telomere Length in Human Natural Killer Cell Subsets. Ann. N. Y. Acad. Sci. 2007, 1106, 240–252.
- Weng, X.; Zhang, H.; Kan, M.; Ye, J.; Liu, F.; Wang, T.; Deng, J.; Tan, Y.; He, L.; Liu, Y. Leukocyte Telomere Length is Associated with Advanced Age-Related Macular Degeneration in the Han Chinese Population. Exp. Gerontol. 2015, 69, 36–40.
- Liao, Q.; He, J.; Tian, F.-F.; Bi, F.-F.; Huang, K. A Causal Relationship between Leukocyte Telomere Length and Multiple Sclerosis: A Mendelian Randomization Study. Front. Immunol. 2022, 13, 922922.
- Rossiello, F.; Jurk, D.; Passos, J.F.; d’Adda Di Fagagna, F. Telomere Dysfunction in Ageing and Age-Related Diseases. Nat. Cell Biol. 2022, 24, 135–147.
- Froidure, A.; Mahieu, M.; Hoton, D.; Laterre, P.-F.; Yombi, J.C.; Koenig, S.; Ghaye, B.; Defour, J.-P.; Decottignies, A. Short Telomeres Increase the Risk of Severe COVID-19. Aging 2020, 12, 19911–19922.
- Dos Santos, G.A.; Pimenta, R.; Viana, N.I.; Guimarães, V.R.; Romão, P.; Candido, P.; De Camargo, J.A.; Hatanaka, D.M.; Queiroz, P.G.; Teruya, A.; et al. Shorter Leukocyte Telomere Length is Associated with Severity of COVID-19 Infection. Biochem. Biophys. Rep. 2021, 27, 101056.
- Cawthon, R.M.; Smith, K.R.; O’Brien, E.; Sivatchenko, A.; Kerber, R.A. Association between Telomere Length in Blood and Mortality in People Aged 60 Years or Older. Lancet 2003, 361, 393–395.
- Barrett, E.L.B.; Burke, T.A.; Hammers, M.; Komdeur, J.; Richardson, D.S. Telomere Length and Dynamics Predict Mortality in a Wild Longitudinal Study. Mol. Ecol. 2013, 22, 249–259.
- Tedone, E.; Huang, E.; O’Hara, R.; Batten, K.; Ludlow, A.T.; Lai, T.-P.; Arosio, B.; Mari, D.; Wright, W.E.; Shay, J.W. Telomere Length and Telomerase Activity in T Cells Are Biomarkers of High-Performing Centenarians. Aging Cell 2019, 18, e12859.
- Demanelis, K.; Jasmine, F.; Chen, L.S.; Chernoff, M.; Tong, L.; Delgado, D.; Zhang, C.; Shinkle, J.; Sabarinathan, M.; Lin, H.; et al. Determinants of Telomere Length across Human Tissues. Science 2020, 369, eaaz6876.
- Dlouha, D.; Maluskova, J.; Kralova Lesna, I.; Lanska, V.; Hubacek, J.A. Comparison of the Relative Telomere Length Measured in Leukocytes and Eleven Different Human Tissues. Physiol. Res. 2014, 63, S343–S350.
- Campisi, J. Cellular Senescence as a Tumor-Suppressor Mechanism. Trends Cell Biol. 2001, 11, S27–S31.
- Campisi, J.; d’Adda Di Fagagna, F. Cellular Senescence: When Bad Things Happen to Good Cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740.
- Campisi, J. Senescent Cells, Tumor Suppression, and Organismal Aging: Good Citizens, Bad Neighbors. Cell 2005, 120, 513–522.
- Guan, L.; Crasta, K.C.; Maier, A.B. Assessment of Cell Cycle Regulators in Human Peripheral Blood Cells as Markers of Cellular Senescence. Ageing Res. Rev. 2022, 78, 101634.
- Englund, D.A.; Sakamoto, A.E.; Fritsche, C.M.; Heeren, A.A.; Zhang, X.; Kotajarvi, B.R.; Lecy, D.R.; Yousefzadeh, M.J.; Schafer, M.J.; White, T.A.; et al. Exercise Reduces Circulating Biomarkers of Cellular Senescence in Humans. Aging Cell 2021, 20, e13415.
- Verbist, K.C.; Wang, R.; Green, D.R. T Cell Metabolism and the Immune Response. Semin. Immunol. 2012, 24, 399–404.
- Chang, C.-H.; Curtis, J.D.; Maggi, L.B.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.-C.; van der Windt, G.J.W.; Blagih, J.; Qiu, J.; et al. Posttranscriptional Control of T Cell Effector Function by Aerobic Glycolysis. Cell 2013, 153, 1239–1251.
- Geginat, J.; Sallusto, F.; Lanzavecchia, A. Cytokine-Driven Proliferation and Differentiation of Human Naive, Central Memory, and Effector Memory CD4+ T Cells. J. Exp. Med. 2001, 194, 1711–1720.
- Düvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a Metabolic Gene Regulatory Network Downstream of mTOR Complex 1. Mol. Cell 2010, 39, 171–183.
- Hagiwara, A.; Cornu, M.; Cybulski, N.; Polak, P.; Betz, C.; Trapani, F.; Terracciano, L.; Heim, M.H.; Rüegg, M.A.; Hall, M.N. Hepatic mTORC2 Activates Glycolysis and Lipogenesis through Akt, Glucokinase, and SREBP1c. Cell Metab. 2012, 15, 725–738.
- Henson, S.M.; Lanna, A.; Riddell, N.E.; Franzese, O.; Macaulay, R.; Griffiths, S.J.; Puleston, D.J.; Watson, A.S.; Simon, A.K.; Tooze, S.A.; et al. P38 Signaling Inhibits mTORC1-Independent Autophagy in Senescent Human CD8+ T Cells. J. Clin. Investig. 2014, 124, 4004–4016.
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 Alters T-Cell Metabolic Reprogramming by Inhibiting Glycolysis and Promoting Lipolysis and Fatty Acid Oxidation. Nat. Commun. 2015, 6, 6692.
- Lewis, R.S. Calcium Oscillations in T-Cells: Mechanisms and Consequences for Gene Expression. Biochem. Soc. Trans. 2003, 31, 925–929.
- Haynes, L.; Linton, P.-J.; Eaton, S.M.; Tonkonogy, S.L.; Swain, S.L. Interleukin 2, but Not Other Common γ Chain–Binding Cytokines, Can Reverse the Defect in Generation of Cd4 Effector T Cells from Naive T Cells of Aged Mice. J. Exp. Med. 1999, 190, 1013–1024.
- Haynes, L.; Eaton, S.M.; Burns, E.M.; Randall, T.D.; Swain, S.L. CD4 T Cell Memory Derived from Young Naive Cells Functions Well into Old Age, but Memory Generated from Aged Naive Cells Functions Poorly. Proc. Natl. Acad. Sci. USA 2003, 100, 15053–15058.
- Vaena, S.; Chakraborty, P.; Lee, H.G.; Janneh, A.H.; Kassir, M.F.; Beeson, G.; Hedley, Z.; Yalcinkaya, A.; Sofi, M.H.; Li, H.; et al. Aging-Dependent Mitochondrial Dysfunction Mediated by Ceramide Signaling Inhibits Antitumor T Cell Response. Cell Rep. 2021, 35, 109076.
- Amin, S.; Liu, B.; Gan, L. Autophagy Prevents Microglial Senescence. Nat. Cell Biol. 2023, 25, 923–925.
- Miwa, S.; Kashyap, S.; Chini, E.; von Zglinicki, T. Mitochondrial Dysfunction in Cell Senescence and Aging. J. Clin. Investig. 2022, 132, e158447.
- Tai, H.; Wang, Z.; Gong, H.; Han, X.; Zhou, J.; Wang, X.; Wei, X.; Ding, Y.; Huang, N.; Qin, J.; et al. Autophagy Impairment with Lysosomal and Mitochondrial Dysfunction is an Important Characteristic of Oxidative Stress-Induced Senescence. Autophagy 2017, 13, 99–113.
- Nazio, F.; Strappazzon, F.; Antonioli, M.; Bielli, P.; Cianfanelli, V.; Bordi, M.; Gretzmeier, C.; Dengjel, J.; Piacentini, M.; Fimia, G.M.; et al. mTOR Inhibits Autophagy by Controlling ULK1 Ubiquitylation, Self-Association and Function through AMBRA1 and TRAF6. Nat. Cell Biol. 2013, 15, 406–416.
- Sung, J.Y.; Lee, K.Y.; Kim, J.-R.; Choi, H.C. Interaction between mTOR Pathway Inhibition and Autophagy Induction Attenuates Adriamycin-Induced Vascular Smooth Muscle Cell Senescence through Decreased Expressions of P53/P21/P16. Exp. Gerontol. 2018, 109, 51–58.
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A Biomarker that Identifies Senescent Human Cells in Culture and in Aging Skin In Vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367.
- Christov, K.T.; Shilkaitis, A.L.; Kim, E.S.; Steele, V.E.; Lubet, R.A. Chemopreventive Agents Induce a Senescence-like Phenotype in Rat Mammary Tumours. Eur. J. Cancer 2003, 39, 230–239.
- Litaker, J.R.; Pan, J.; Cheung, Y.; Zhang, D.K.; Liu, Y.; Wong, S.C.; Wan, T.S.; Tsao, S.W. Expression Profile of Senescence-Associated Beta-Galactosidase and Activation of Telomerase in Human Ovarian Surface Epithelial Cells Undergoing Immortalization. Int. J. Oncol. 1998, 13, 951–957.
- Hall, B.M.; Balan, V.; Gleiberman, A.S.; Strom, E.; Krasnov, P.; Virtuoso, L.P.; Rydkina, E.; Vujcic, S.; Balan, K.; Gitlin, I.I.; et al. P16(Ink4a) and Senescence-Associated β-Galactosidase Can be Induced in Macrophages as Part of a Reversible Response to Physiological Stimuli. Aging 2017, 9, 1867–1884.
- Kaufmann, S.; Cerny-Garcia, J. Senescent Cells. Kaufmann Protocol Publications; The Kaufmann Anti-Aging Institute: Malvern, VIC, Australia, 2019; pp. 1–30.
- Muller, S. New Embo Members’ Review: The Double Life of HMGB1 Chromatin Protein: Architectural Factor and Extracellular Signal. EMBO J. 2001, 20, 4337–4340.
- Muller, S.; Ronfani, L.; Bianchi, M.E. Regulated Expression and Subcellular Localization of HMGB1, a Chromatin Protein with a Cytokine Function. J. Intern. Med. 2004, 255, 332–343.
- Zayed, H. The DNA-Bending Protein HMGB1 Is a Cellular Cofactor of Sleeping Beauty Transposition. Nucleic Acids Res. 2003, 31, 2313–2322.
- Bianchi, M.E.; Manfredi, A.A. High-Mobility Group Box 1 (HMGB1) Protein at the Crossroads between Innate and Adaptive Immunity. Immunol. Rev. 2007, 220, 35–46.
- Vallejo, A.N. Age-Dependent Alterations of the T Cell Repertoire and Functional Diversity of T Cells of the Aged. Immunol. Res. 2006, 36, 221–228.
- Britanova, O.V.; Putintseva, E.V.; Shugay, M.; Merzlyak, E.M.; Turchaninova, M.A.; Staroverov, D.B.; Bolotin, D.A.; Lukyanov, S.; Bogdanova, E.A.; Mamedov, I.Z.; et al. Age-Related Decrease in TCR Repertoire Diversity Measured with Deep and Normalized Sequence Profiling. J. Immunol. 2014, 192, 2689–2698.
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of Chromatin Protein HMGB1 by Necrotic Cells Triggers Inflammation. Nature 2002, 418, 191–195.
- Hreggvidsdottir, H.S.; Östberg, T.; Wähämaa, H.; Schierbeck, H.; Aveberger, A.-C.; Klevenvall, L.; Palmblad, K.; Ottosson, L.; Andersson, U.; Harris, H.E. The Alarmin HMGB1 Acts in Synergy with Endogenous and Exogenous Danger Signals to Promote Inflammation. J. Leukoc. Biol. 2009, 86, 655–662.
- Raucci, A.; Palumbo, R.; Bianchi, M.E. HMGB1: A Signal of Necrosis: Review. Autoimmunity 2007, 40, 285–289.
- Bonaldi, T. Monocytic Cells Hyperacetylate Chromatin Protein HMGB1 to Redirect It towards Secretion. EMBO J. 2003, 22, 5551–5560.
- Polanská, E.; Dobšáková, Z.; Dvořáčková, M.; Fajkus, J.; Štros, M. HMGB1 Gene Knockout in Mouse Embryonic Fibroblasts Results in Reduced Telomerase Activity and Telomere Dysfunction. Chromosoma 2012, 121, 419–431.
- Davalos, A.R.; Kawahara, M.; Malhotra, G.K.; Schaum, N.; Huang, J.; Ved, U.; Beausejour, C.M.; Coppe, J.-P.; Rodier, F.; Campisi, J. P53-Dependent Release of Alarmin HMGB1 is a Central Mediator of Senescent Phenotypes. J. Cell Biol. 2013, 201, 613–629.
- Narita, M.; Nuñez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-Mediated Heterochromatin Formation and Silencing of E2F Target Genes during Cellular Senescence. Cell 2003, 113, 703–716.
- Lukášová, E.; Kovařík, A.; Kozubek, S. Consequences of Lamin B1 and Lamin B Receptor Downregulation in Senescence. Cells 2018, 7, 11.
- Freund, A.; Laberge, R.-M.; Demaria, M.; Campisi, J. Lamin B1 Loss is a Senescence-Associated Biomarker. Mol. Biol. Cell 2012, 23, 2066–2075.
- Shimi, T.; Butin-Israeli, V.; Adam, S.A.; Hamanaka, R.B.; Goldman, A.E.; Lucas, C.A.; Shumaker, D.K.; Kosak, S.T.; Chandel, N.S.; Goldman, R.D. The Role of Nuclear Lamin B1 in Cell Proliferation and Senescence. Genes Dev. 2011, 25, 2579–2593.
- Lukášová, E.; Kovařík, A.; Bačíková, A.; Falk, M.; Kozubek, S. Loss of Lamin B Receptor is Necessary to Induce Cellular Senescence. Biochem. J. 2017, 474, 281–300.
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA Double-Stranded Breaks Induce Histone H2AX Phosphorylation on Serine 139. J. Biol. Chem. 1998, 273, 5858–5868.
- Firsanov, D.V.; Solovjeva, L.V.; Svetlova, M.P. H2AX Phosphorylation at the Sites of DNA Double-Strand Breaks in Cultivated Mammalian Cells and Tissues. Clin. Epigenet 2011, 2, 283–297.
- Sedelnikova, O.A.; Horikawa, I.; Zimonjic, D.B.; Popescu, N.C.; Bonner, W.M.; Barrett, J.C. Senescing Human Cells and Ageing Mice Accumulate DNA Lesions with Unrepairable Double-Strand Breaks. Nat. Cell Biol. 2004, 6, 168–170.
- Casella, G.; Munk, R.; Kim, K.M.; Piao, Y.; De, S.; Abdelmohsen, K.; Gorospe, M. Transcriptome Signature of Cellular Senescence. Nucleic Acids Res. 2019, 47, 7294–7305.
- Ron-Harel, N.; Sharpe, A.H.; Haigis, M.C. Mitochondrial Metabolism in T Cell Activation and Senescence: A Mini-Review. Gerontology 2015, 61, 131–138.
- Marcel, V.; Ghayad, S.E.; Belin, S.; Therizols, G.; Morel, A.-P.; Solano-Gonzàlez, E.; Vendrell, J.A.; Hacot, S.; Mertani, H.C.; Albaret, M.A.; et al. P53 Acts as a Safeguard of Translational Control by Regulating Fibrillarin and rRNA Methylation in Cancer. Cancer Cell 2013, 24, 318–330.
- Wang, X.; Wang, X.; Xu, Y.; Yan, M.; Li, W.; Chen, J.; Chen, T. Effect of Nicastrin on Hepatocellular Carcinoma Proliferation and Apoptosis through PI3K/AKT Signalling Pathway Modulation. Cancer Cell Int. 2020, 20, 91.
- Lombardo, Y.; Filipović, A.; Molyneux, G.; Periyasamy, M.; Giamas, G.; Hu, Y.; Trivedi, P.S.; Wang, J.; Yagüe, E.; Michel, L.; et al. Nicastrin Regulates Breast Cancer Stem Cell Properties and Tumor Growth In Vitro and In Vivo. Proc. Natl. Acad. Sci. USA 2012, 109, 16558–16563.
- Gentilini, D.; Mari, D.; Castaldi, D.; Remondini, D.; Ogliari, G.; Ostan, R.; Bucci, L.; Sirchia, S.M.; Tabano, S.; Cavagnini, F.; et al. Role of Epigenetics in Human Aging and Longevity: Genome-Wide DNA Methylation Profile in Centenarians and Centenarians’ Offspring. Age 2013, 35, 1961–1973.
- Jones, M.J.; Goodman, S.J.; Kobor, M.S. DNA Methylation and Healthy Human Aging. Aging Cell 2015, 14, 924–932.
- Bocklandt, S.; Lin, W.; Sehl, M.E.; Sánchez, F.J.; Sinsheimer, J.S.; Horvath, S.; Vilain, E. Epigenetic Predictor of Age. PLoS ONE 2011, 6, e14821.
- Horvath, S. DNA Methylation Age of Human Tissues and Cell Types. Genome Biol. 2013, 14, R115.
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.-B.; Gao, Y.; et al. Genome-Wide Methylation Profiles Reveal Quantitative Views of Human Aging Rates. Mol. Cell 2013, 49, 359–367.
- Unnikrishnan, A.; Freeman, W.M.; Jackson, J.; Wren, J.D.; Porter, H.; Richardson, A. The Role of DNA Methylation in Epigenetics of Aging. Pharmacol. Ther. 2019, 195, 172–185.
- Ucar, D.; Márquez, E.J.; Chung, C.-H.; Marches, R.; Rossi, R.J.; Uyar, A.; Wu, T.-C.; George, J.; Stitzel, M.L.; Palucka, A.K.; et al. The Chromatin Accessibility Signature of Human Immune Aging Stems from CD8+ T Cells. J. Exp. Med. 2017, 214, 3123–3144.
- Márquez, E.J.; Chung, C.; Marches, R.; Rossi, R.J.; Nehar-Belaid, D.; Eroglu, A.; Mellert, D.J.; Kuchel, G.A.; Banchereau, J.; Ucar, D. Sexual-Dimorphism in Human Immune System Aging. Nat. Commun. 2020, 11, 751.


