The accumulation of senescent cells increases age-related background inflammation, “Inflammaging”, causing lymphocyte exhaustion and cardiovascular, neurodegenerative, autoimmune and cancer diseases. Although modern research has not yet identified specific markers of aging lymphocytes, several sets of markers facilitate the separation of the aging population based on normal memory and exhausted cells for further genetic and functional analysis. The reasons for the higher predisposition of CD8+ T-lymphocytes to senescence compared to the CD4+ population are also discussed. The suppression of immune senescence is the most relevant area of research aimed at developing anti-aging and anti-cancer therapy for prolonging the lifespan of the global population.
1. Introduction
To combat age-related diseases, morbidity and frailty, it is necessary to combine the efforts of society, science and new innovative approaches in anti-aging medicine and regenerative technologies to introduce anti-senescence therapy, the treatment of metabolic and inflammatory diseases. These efforts will definitely bridge the gap between lifespan and healthspan for future economic prosperity and equitable global wellbeing.
Aging also affects the immune system, reducing the effectiveness of immune surveillance and resulting in imbalanced immune responses, autoimmune disorders and anemia. Recently, it was documented that the accumulation of senescent cells in all immune populations is a hallmark of immune aging and an accelerator of inflammatory aging and immune dysfunction in older adults. Immunosenescent cells persist in a state of cell cycle arrest and cause a number of negative and destructive effects, such as a decrease in the ability to control malignancy and cleanse tissues of senescent cells, causing their accumulation in the entire body [
4]; stem cell senescence [
5]; the decreased efficiency of immune response to vaccination [
6,
7]; and increased susceptibility to various infectious diseases [
8].
The immunosenescent state impedes T-lymphocytes from successfully fighting tumor cells, which shows its pivotal role in cancer development [
9]. The role of immunosenescent cells has been shown in the development of diabetes [
10] and neurodegenerative [
11,
12], autoimmune [
13,
14,
15] and cardiovascular diseases [
16] in old age. Immunosenescence also likely contributes to an increased susceptibility to a severe form of coronavirus infection and increases morbidity and mortality from infectious diseases among elderly individuals [
17].
2. The Hallmarks of Senescent Immune Cells
2.1. Morphological Changes
Among the distinct features of senescent cells, the most noticeable external features are flattened morphology and increased size, which are associated, among other factors, with reduced expression of scaffold proteins such as caveolin-1, Rac1, and CDC42 [
22]. The appearance of enlarged and flattened cells was detected after prolonged exposure of the B-cell lymphoma line A20 to sodium arsenite [
23]. Along with decreased functionality, changes in the shape and size of immune cells during aging are probably one of the reasons for the age-related disruptions in the architectural organization and structure observed in tissues such as lymph nodes [
24] and the thymus [
25].
Significant changes affect many cellular organelles with senescence: the number and size of vacuoles, cytoplasmic filaments and nucleus and nucleolus enlargement (although an increase in the ratio of nucleus to cytoplasm also characterizes young cells such as naive lymphocytes and stem cells). Sometimes, senescent cells may be multinucleated and contain increased numbers of lysosomes and Golgi complexes [
26]. In addition, the mitochondria in lymphocytes of people over 60 are enlarged and irregularly spaced, and their cristae are replaced by a myelin-like structure (or other electron-dense substance) [
27].
The lysosomes of senescent cells accumulate lipofuscin, a yellow-brown “age pigment”, which is an undegradable aggregate of oxidized lipids, covalently cross-linked proteins, oligosaccharides and transition metals, formed by iron-catalyzed oxidation and polymerization of different cellular structures and macromolecules [
28,
29]. Lipofuscin granules and tubuloreticular inclusions (TRI) were found in the cytoplasm of lymphocytes from donors older than 60 years, and their content in cells increases with age [
27].
2.2. Surface Markers
Although no universal surface marker of senescence has been found for all cell types, markers specific to individual cell populations that correlate well with the senescence of these cells have been identified [
37]. Loss of the CD28 costimulatory molecule expression, which plays an important role in T-cell activation, was one of the first distinguishing features proposed to identify senescent cells [
38,
39]. These early studies found that during replicative senescence, the decrease in CD28 expression is more pronounced in a population of CD8+ T-lymphocytes (CD8+ CTLs) in comparison with CD4+ T-cells (CD4+ Ths) [
39]. Loss of CD27, another costimulatory molecule, occurs as a response to chronic antigenic load and is also considered a marker of senescent cells [
40]. Increased numbers of CD28- and CD27- T-cells are observed in the elderly [
41,
42]. As CD27 and CD28 are important co-stimulatory molecules essential for TCR signal transduction and T-cell priming, their loss by senescent T-cells is in agreement with the loss of their function and ability to activate. Naive CD28+ T-cells exist transiently and mature rapidly upon stress or antigen recognition. This could explain why no significant number of senescent cells were found among CD28+ T-cells. In contrast, long-term persistent memory cells are more likely to undergo replicative and metabolic stress or DNA damage, resulting in a switch to a senescent state. Indeed, memory cell markers such as CD45R0 have been found on senescent cells [
43].
Expression of the mature NK cell receptor CD57 correlates with the loss of cell proliferation ability and shorter telomere length [
44]. Upregulation of KLRG1 (killer-cell lectin like receptor G1) increases dramatically with age, mainly in the CD8+ CTL population and correlates with loss of T-cell proliferation capacity. Following this observation, KLRG1 has also been proposed as a marker of senescent cells and exhaustion of lymphocytes [
45,
46].
Senescent NK cells (natural killers) and T-lymphocytes also express TIGIT (immunoreceptor for T-cells with Ig and ITIM domains) on their surface. The number of TIGIT+ cells correlates with age, especially strongly in the CD8+ T-lymphocyte population [
47]. However, these surface markers do not accurately define senescent immune cells, since they also characterize the process of cell differentiation and overlap with the phenotype of memory cells [
48,
49]. Moreover, cells that have lost CD28 expression and/or exhibit various levels of expression of CD57 and other senescence markers described above retain the ability to proliferate, making it difficult to classify these cells as true senescent cells [
14,
50,
51].
2.3. SASP
In 2000, Claudio Franceschi et al. proposed the concept of “inflammaging”—a state of chronic mild low-grade aseptic inflammation that develops as a result of constant antigenic load and stress during life, contributing to the weakening of immune cell activity and the pathogenesis of age-related diseases, including cancer [
80,
81,
82]. Immune cells that have reached the senescence phase secrete various pro-inflammatory cytokines (TNF, IL-6, IL-1α/β, IFN-γ), chemokines (IL-8/CXCL8), MCP-1/CCL2, MIP-1α/CCL3, GROα/CXCL1), suppressive cytokines (TGF-β, IL-10), growth factors (GM-CSF, G-CSE, VEGF), matrix metalloproteinases (MMP-1, MMP-3, MMP-10), soluble receptors and ligands (ICAM-1/3, Fas, EGF-R), angiogenic factors, and other compounds that are now combined under the common term SASP (senescence-associated secretory phenotype) [
83,
84,
85]. In addition to increased concentrations of TNF, IL-1β, and IL-6, an increase in plasma levels of C-reactive protein (CRP) is typical of the “inflammaging” state [
86,
87,
88]. The accumulation of neopterin, secreted by macrophages in response to IFNγ stimulation, correlates with age regardless of gender and has been observed in physiologic aging and autoimmune diseases; therefore, it is also considered as another biomarker of age-associated chronic inflammation [
89]. Recently, a pronounced correlation with the age of urinary neopterin concentration has been shown for rhesus macaques [
90]. Thus, the senescent state is characterized by inadequate sterile inflammation with a simultaneous loss of the ability to activate in response to specific stimuli, such as pathogen invasion. The hypothesis of «trained innate memory» has been put forward as an explanation for the controversial production of proinflammatory factors by dysfunctional immune cells.
Selective inhibition of certain SASP components along with the use of senolytics is considered as one of the strategies to combat age-associated diseases and destructive effects of aging processes, including the immune system [
92]. Some drugs targeting different components of SASP, such as therapeutic monoclonal antibodies against IL-6 (Tocilizumab) and TNF (Infliximab), are already used in clinical practice [
93,
94,
95]. Various methods for determining the production of pro-inflammatory cytokines comprising SASP are widely used in the detection and characterization of senescent immune cells [
96].
2.4. Decrease in Telomere Length and Telomerase Activity
The “Hayflick” cell division limit is the number of times a normal cell divides before entering the senescent phase (usually 40–60). With each division, a decrease in the length of telomeres occurs until short telomeres cause DNA instability and the induction of a program of accelerated cellular senescence. Telomere length is a recognized marker of aging that correlates with chronological age [
97,
98].
The molecular mechanisms of a senescent phenotype emergence in response to telomere shortening remain poorly understood. However, it is known that chromosome fusion due to telomere dysfunction initiates DDR signaling and leads to the accumulation of chromatin fragments in the cytoplasm. These cytosolic DNA fragments containing DSBs are recognised by cyclic GMP-AMP synthase (cGAS) and activate the Stimulator of Interferon Genes (STING), leading to premature aging [
99].
Age-dependent telomere shortening occurs in all types of lymphocytes, including T-, B-, and NK cells [
101], and is associated with the risk of age-related and autoimmune diseases [
102,
103,
104], susceptibility and severity of infectious diseases [
105,
106], as well as mortality [
107,
108]. Telomerase activity and telomere length are higher and the rate of telomere shortening is lower in the T-cells of long-lived individuals and correlate with the health status of centenarians [
109]. However, the length of telomeres and the rate of telomeres shortening vary considerably among individuals, across different tissue types [
110] and even within the same organ [
111].
2.5. Cell Cycle Arrest and Expression of p16, p21 and p53
It is well known that the senescent state is characterized by cell cycle arrest. Inhibitors of cyclin-dependent kinases p16 and p21, as well as p53 protein, which are important elements of tumor suppressor pathways, are directly involved in its regulation and, consequently, in the control of cellular senescence [
117,
118]. Activation of the p53 pathway by phosphorylation has been shown to occur primarily in response to DNA damage (DNA damage response, DDR) and telomere dysfunction, whereas the p16 pathway is activated in response to mitogenic stress and reactive oxygen species (ROS) accumulation, exposure to oncogenes (e.g., RAS), and general cellular stress (including contact inhibition, cycling exhaustion and suboptimal culture conditions) [
119].
Increased expression of p16 and p21 is now considered a recognized marker of senescence and is widely used in various studies, including immune cells [
121]. It has been shown that physical activity decreases p16 and p21 expression in CD3+ T-lymphocytes [
122]. An important feature of p16 and p21 as markers of senescence is that their increased expression is not detectable in the exhaustion state, another dysfunctional cellular state distinct from classical senescence described in the literature [
9].
2.6. Metabolic Changes/Disorders
2.6.1. Energy Metabolism Disruptions
The energy supply of quiescent (non-proliferating) naïve lymphocytes is provided by oxidative phosphorylation (OXPHOS) [
125], whereas activation and differentiation of T-cells into effector cells is associated with the switch to the less energetically favorable process of glycolysis (aerobic glycolysis, known as the Warburg effect), which is probably necessary for optimal cytokine production [
126]. The transition of effector T-cells to a quiescent and memory state is characterized by resumption of OXPHOS, apparently mediated by IL-15 [
127]. In contrast to memory cells, senescent cells also carry out aerobic glycolysis through activation of the mTOR signaling pathway in response to DNA damage via induction of the two major transcription factors HIF1α and c-MYC. In addition, transcription of genes involved in the pentose phosphate pathway and de novo lipogenesis is enhanced [
128,
129]. It is interesting that EMRA effector CD8+ memory T-cells (CD45RA+CD27+/−), comprising the senescent population, show reduced proliferative potential, accumulate with age and are associated with several diseases, characterized by preferential energy acquisition through glycolysis in contrast to the EM (CD45RA+/−CD27−) population, which combines glycolysis and OXPHOS [
130]. Expression of the suppression and exhaustion marker PD-1 on the surface of activated T-lymphocytes inhibits glycolysis and induces the switch to lipolysis, allowing endogenous free fatty acids to be utilized in the β-oxidation process during the reduced ability of cells to absorb and utilize other classes of nutrients [
131]. Thus, the exhausted state of lymphocytes is characterized by a weakening of glycolysis, while the senescent state is characterized by its enhancement.
2.6.2. Mitochondrial Dysfunction
Decreased Ca
2+ uptake by mitochondria of T-lymphocytes during aging impairs Ca
2+ -dependent signaling and downstream induction of crucial pro-inflammatory transcription factors, such as NFAT1 and NF-κB [
135]. Naive CD4+ T-lymphocytes from old mice show reduced Ca
2+ ion uptake in response to stimulation by TCRs, consequently reducing T-cell proliferation and IL-2 production capacity [
136]. Memory T-cells derived from such old naive CD4+ T-lymphocytes are also dysfunctional, showing reduced IL-2 production and impaired response upon re-stimulation [
137]. However, senescence can also be caused by the excessive mitochondrial activity. For example, the accumulation of sphingolipid ceramides CerS6/C14 in the outer membrane due to increased expression of ceramide synthase over-activates mitophagy in T-cells, causing increased mitochondrial fission and senescence-like cellular dysfunction. The inhibition of ceramide metabolism prevents excessive mitophagy and restores the central memory phenotype in these cells [
138].
2.6.3. Autophagy and Mitophagy Disorder
A stable level of autophagy clears the cell of damaged membranes and misfolded proteins and keeps cells healthy. A decrease in the level of autophagy is characteristic of senescent cells. With age, cells also accumulate damaged and poorly functioning mitochondria. However, the process of mitophagy (digestion of damaged organelles) can both remove damaged mitochondria and stimulate mitochondrial renewal through division due to lack of energy supply. Healthy mitochondria neutralize reactive oxygen species and supply cells with enough high-energy molecules to power the cell’s biochemical machinery. Selective damage to mitochondria or inhibition of autophagy accelerate cellular senescence [
140,
141].
The disruption of autophagy correlates with increased intracellular ROS levels and inflammatory factor upregulation. The blockade of autophagy by Atg5 knockdown increases SA-β-Gal positive cell number, elevates IL-6 secretion, and induces senescence [
142]. Thus, disruption of autophagy itself causes cellular senescence. At the molecular level, the most important inhibitor of autophagy is mTOR [
144,
145].
2.6.4. SA-β-Gal
In healthy mammalian cells, pH 4.0 is optimal for lysosomal β-galactosidase activity. In 1995, Dimri et al. demonstrated that during replicative senescence of keratinocytes and fibroblasts, the optimum activity of this enzyme shifts to pH 6.0. In addition, this form of the enzyme is absent in pre-senescent and quiescent fibroblasts, as well as in terminally differentiated keratinocytes and proliferating cells. This β-galactosidase, which is active at pH 6.0, has been termed senescence-associated β-galactosidase (SA-β-Gal) [
148]. It was first hypothesized that SA-β-Gal might be an alternatively spliced form of lysosomal β-galactosidase, whose presence is attributed to the increased unspecific lysosomal activity found in senescent cells with aging [
148].
However, the view later developed that β-galactosidase activity at pH 6.0 is associated with the accumulation of specific glycoproteins and complex glycolipids in senescent cells [
149], such as lipofuscin granules (see above) and other lipoprotein complexes, and derived by disruption of cellular metabolism. It was shown that SA-β-Gal activity as a marker of senescence effectively reflects the completeness of cell immortalization and can be used to monitor their state at different stages of immortalization of cell lines. For instance, in cells derived from human ovarian surface epithelial cells (HOSE 6-3), a dramatic decrease in SA-β-Gal activity was observed after these cells acquired immortalized status. In addition, an inverse relationship between telomerase activity and SA-β-Gal was shown in immortalized cells [
150].
Despite the efficiency of SA-β-Gal as a senescent marker for most cell types, it is important to note that increased lysosomal beta-galactosidase activity is characteristic of some cells, such as active macrophages, Kupffer cells, and osteoclasts, in the normal state [
153,
154].
2.7. Disorganization and Dysfunction of Chromatin
2.7.1. HMGB1
Extremely conserved, high-mobility group protein B1 (HMGB1) is a non-histone protein with 99% identity among mammals. HMGB1 has two homologous DNA-binding domains and, when localized in the nucleus, binds to the small groove of B-type DNA, albeit with limited specificity, forming a 90° or more bend in the DNA double helix [
155,
156]. As a so-called chromatin architectural factor, HMGB1 directly interacts with a number of proteins such as transcription factors containing HOX or POU domains, p53, NF-kB and steroid hormone receptors, promoting their recruitment and facilitating interactions between these proteins and DNA. Furthermore, HMGB1, through its interaction with proteins that activate the RAG1/2 gene, is involved in enabling the V(D)J recombination process by enhancing specific recognition and facilitating DNA cleavage [
157,
158]. This may be of particular interest in the context of the well-known fact that the diversity of the T-cell receptor repertoire (TCRs) decreases with age [
159,
160].
Another important function of HMGB1 is intercellular signaling and its role as an alarmin. It is known that passive release of HMGB1 occurs during necrotic cell death, whereas in apoptosis HMGB1 remains bound to chromatin until it is eliminated by macrophages or non-professional phagocytic cells—scavengers [
161]. Activated macrophages, monocytes, and dendritic cells also serve as a source of extracellular HMGB1. HMGB1 danger signaling to surrounding cells occurs through interaction with its receptors RAGE, TLRs 2, 4 and 9, syndecan and thrombomodulin, followed by activation of the NF-κB signaling pathway [
158,
162,
163]. It has been shown that secretion of HMGB1 into the extracellular medium is accomplished by its acetylation on lysine residues. Under the influence of lipopolysaccharide on monocytes and macrophages, or under the exposure to trichostatin A histone deacetylase inhibitor (HDAC) on quiescent macrophages, HMGB1 is hypersacitylated and translocated to the cytosol with subsequent accumulation in secretory lysosomes [
164]. Extracellular HMGB1, as a ligand of TLRs, has been shown to promote sterile inflammation through the induction of IL-6, a key component of SASP. An important role of this protein in the production of inflammatory cytokines by immune cells has been described. The transition to cellular senescence due to telomere shortening, genomic instability, or DNA damage leads to loss of nuclear localisation of HMGB1 and increases the protein level in the extracellular space [
165], which enhances TLR/NF-κB-dependent production of SASP components [
166].
Senescence induced by X-ray irradiation, replicative depletion, or overexpression of p16 and the RAS oncogene results in a significant decrease in HMGB1 nuclear localization and its migration to the cytoplasm. Consequently, loss of nuclear HMGB1 characterizes the senescent state of cells regardless of the senescence inducer, but p16 overexpression by itself is not an activator of HMGB1 migration to the cytoplasm. Both depletion and overexpression of HMGB1 stimulated p53 expression in human mammary epithelial cells and mouse embryonic fibroblasts to the levels found in cells subjected to irradiation-induced or replicative senescence. Blocking p53 by RNA interference ensured the preservation of HMGB1 nuclear localization during X-ray irradiation-induced fibroblast senescence. Thus, p53 activity is regulated, among other things, by the expression level of HMGB1, and, at the same time, HMGB1 re-localization is directly dependent on p53 activity (in contrast to the expression of SASP components). In addition, decreased nuclear and serum HMGB1 was observed in vivo in old but not young mice [
166].
2.7.2. SAHF
In 2003, Narita et al. showed that senescent human fibroblasts are characterized by the formation of foci of a previously undescribed form of facultative chromatin enriched in heterochromatin protein 1 (HP1) and heterochromatin histone H3 trimethylatation at lysine 9 (H3K9me3), which provides a binding site for HP1. The euchromatic markers H3K9 and H3K4me3 are absent in these foci. These senescent specific foci are collectively called SAHF—senescence-associated heterochromatic foci. The accumulation of SAHF-positive cells after oncogene-induced senescence by RAS overexpression correlates well with the kinetics of other senescence markers such as senescence-associated beta-galactosidase (SA-β-gal) activity, p16 expression, Rb hypophosphorylation, and cell cycle arrest. RAS-induced senescence, SAHF formation, and SA-β-gal activity depend significantly on the activation of the p16/Rb pathway, while the influence of p53 on this process is minor. The knockdown of p16 or Rb substantially suppresses SAHF formation in RAS-induced senescence, but these p16 or Rb-deficient cells accumulate and have senescent morphological features and SA-β-gal activity [
168]. However, SHAF formation is not a universal marker of cellular senescence, characterizing the senescent state independently of cell type and stress exposure.
2.7.3. Lamin B1
The anchoring of heterochromatin on the nuclear lamina is an important element in ensuring the spatial organization of chromatin structure and the functioning of eukaryotic genomes. Inner nuclear membrane proteins are able to recognize specific protein lamina-associated domains (LADs) and bind lamins A/C or B, respectively. For lamin B1, such a protein is its receptor LBR [
171].
Lamin B1 (LB1) expression is known to be decreased during replicative and oncogene-induced senescence in various cell types [
171,
172,
173]. The induction of senescence also results in decreased LBR expression [
174]. The decreased expression of LB1 does not occur directly in response to DNA damage (DDR), activation of mitogen-activated protein kinase p38 (p38-MAPK) and nuclear factor-κB (NF-κB) or reactive oxygen species (ROS), which are all hallmarks of many senescent cells, but is observed upon direct stimulation of the p53 or p16/pRB signaling pathways: an elevated level of p53 or p16 expression was found to be a sufficient condition for the loss of LB1 [
172].
2.7.4. γH2AX
Apart from the canonical histone H2A, the histone variant H2AX is widely represented in mammalian cells, accounting for about 2.5–25% of total H2A [
178]. The appearance of its phosphorylated form, called γH2AX, is one of the earliest events in response to DNA damage (DDR), such as various genotoxic stresses that induce double-strand breaks (DSBs). The occurrence of DSBs activates ATM, ATR, and DNA-PK kinases of the PI3K (phosphotidylinositol-3-kinase) family, which carry out phosphorylation of H2AX at the serine residue at position 139 (ser139). γH2AX foci can be detected in cell nuclei as early as 3 min after irradiation; then their number reaches a maximum within 30 min and remains unchanged for up to 60 min. The total number of γH2AX foci correlates with the total number of DSBs, and the size of γH2AX foci in nuclei at 3 min after irradiation is smaller than at 15 min. During DSBs repair, several phosphatases, such as PP2A, PP4, Wip1, and PP6, carry out the dephosphorylation of γH2AX [
179].
The formation and accumulation of γH2AX foci, which indicate the accumulation of persistent lesions and unrepairable double-stranded DNA breaks, respectively, were found to occur with increasing passage in various mouse and human cell lines, as well as resulting from chemical exposure (hydrogen peroxide). It has also been shown that γH2AX foci co-localize with repair factor proteins such as 53bp1, Mre11, Rad50 and Nbs1 [
180].
Along with other hallmarks of cellular senescence, γH2AX is widely used as a marker of senescent cells, including immune cells. Among the obvious advantages is the fact that γH2AX foci are formed within seconds after DSB formation; however, since they are initially rather small and difficult to visualize, for more reliable detection they are examined after 15–30 min in the case of senescence induced by irradiation.
2.8. Multi-Omics Changes
2.8.1. Transcriptome
The list of hallmarks of senescent cells would not be complete without a description of the transcriptional profile, which has been actively studied using single-cell RNAseq technologies that have recently become widespread. Casella G. et al. compared the transcriptomes of several types of human fibroblasts (WI-38, IMR-90, HAEC and HUVEC) in different types of senescence (replicative or induced by irradiation, doxorubicin and oncogenes) and found increased expression levels of 50 and decreased levels of 18 total transcripts. The most repressed gene was MCUB (Mitochondrial Calcium Uniporter Dominant Negative Subunit Beta) [
185], consistent with mitochondrial dysfunction and impaired regulation of mitochondrial calcium uptake during senescence, particularly as observed by Ron-Harel N. et al. in CD4+ T-lymphocytes [
186]. It has been established that suppression of the expression of nuclear fibrillarin methyltransferase (FBN) rRNA disrupts ribosome assembly and protein biosynthesis, which may be one of the reasons for the inability of senescent cells to perform their functions [
185]. FBN is an oncogene and its increased expression is characteristic of different types of tumors, which contributes to a significant increase in the rate of tumor proliferation. In addition, activation of the tumor suppressor p53 directly suppresses FBN expression [
187]. Thus, FBN suppression may be a key mechanism for cell cycle arrest in senescent cells.
Among the transcripts whose levels increased upon induction of senescence in a different type of fibroblasts, Casella G. et al. found SASP component TNF and its receptor (senescence is known to be associated with an inflammatory background) and the Rho family GTPase 3 (explaining the characteristic changes in the morphology of senescent cells) [
185]. Increased levels of Nicastrin, which acts as an oncogene through activation of the NOTCH and PI3K/Akt signaling pathways and inhibition of apoptosis, have also been found [
185,
189,
190].
Increased expression of these genes may reflect the cell’s attempt to escape from a state of cell cycle arrest by activating compensatory mechanisms. In addition, the activation of protoncogenes detected in cellular senescence can potentially induce mechanisms of oncogenesis, which, together with large-scale disorganization of chromatin and dysregulation of gene expression, confirms the connection between aging processes and malignant transformation of cells. Induction of fibroblast senescence increases the expression level of PURPL (lncRNA), which suppresses p53. PURPL is more actively produced in response to increased p53 levels.
2.8.2. Epigenetic Changes
The aging process is accompanied by large-scale demethylation of genomic DNA [
195]. Recently, a large amount of data has been accumulated on specific CpG islands, the hyper- or hypomethylation of which significantly correlates with chronological age [
195,
196,
197,
198,
199,
200]. However, changes in methylation levels often do not correspond to differential gene expression.
Note that age-dependent changes in gene expression and DNA methylation landscape also affect the earliest populations of blood cell progenitors, including hematopoietic stem cells (HSCs), which often biases hematopoiesis toward preferential differentiation in one direction (e.g., myeloid).
2.8.3. Chromatin Accessibility
Developed about a decade ago, the latest technology for full genomic assessment of chromatin accessibility ATAC-seq (Assay for Transposase-Accessible Chromatin using sequencing) allowed additional characterization of chromatin loci of immune cells that are differentially open or closed depending on age [
204]. Chromatin closing with age largely affects T-lymphocyte activation genes (161 genes) and TCR signaling pathways (59 genes). CD4+ T-lymphocytes (both naive and memory cells) were found to exhibit significantly less chromatin remodeling during senescence compared to CD8+ T-cells.
Chromatin closure and associated reduction in expression of the homeostatic cytokine receptor IL-7 (IL-7R) gene were found in CD8+ T-lymphocytes, affecting both the naïve population and central and effector memory cells. This results in a decreased sensitivity of CD8+ CTLs to IL-7 with aging, which is not observed in the CD4+ lymphocyte population. Genes downstream of IL-7R signaling cascade, such as JAK1, JAK3, STAT5A, STAT5B, and PTK2B, also exhibit chromatin closure with age. Chromatin closure is also specific for reduction of signal transduction through TCRs and for IL-2, IL-9 signaling [
191,
204]. Importantly, chromatin closure was characterized by downregulation of histone genes (e.g., HIST1H3D, HIST1H3E, HIST4H4) as well as histone modifier genes (e.g., EZH1, SETD7) [
204].
3. Conclusions
The accumulation of senescent cells is observed during the aging process. Senescent immune cells undergo multiple abnormalities, which affect the overall function of the immune system, causing inflammation and immune balance disruption.
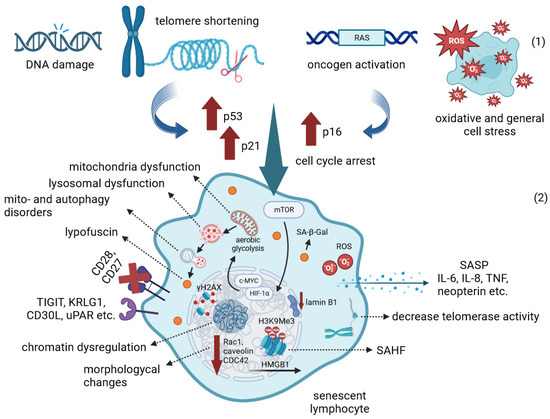
The accumulation of DNA damage and unrepaired DSBs (as indicated by γH2AX staining), telomere shortening, oncogene activation and oxidative stress activate DDR signaling in the cell, which leads to cellular senescence, cycle arrest and increased resistance to apoptosis. In addition to genomic instability, the epigenetic modifications of senescent immune cells lead to global dysregulation of gene expression and chromatin spatial structure disruption, including loss of HMGB1 nuclear localization and SAHF formation. Senescence is also accompanied by the widespread metabolic disorders. In response to stress, the mTOR signaling pathway becomes dominant and the transcription factors HIF1a and c-MYC are activated, which ultimately induces aerobic glycolysis. Global metabolic dysregulation impairs lysosomal and mitochondrial function and mito- and autophagy, resulting in the accumulation of lipofuscin granules. Cessation of intercellular interactions, morphological changes, cell cycle arrest, production of inflammatory cytokines and other SASP components are now recognized as common hallmarks of cellular senescence (Figure 1).
Figure 1. Cellular senescence: The first part (
1) illustrates the triggers of the senescent state. The second part (
2) shows multiple disregulations that cells undergo during senescence. The brown upward and down arrows show increased and decreased gene transcription, respectively. Dashed arrows show key characteristics of the senescent phenotype. Receptors recognized as surface markers of the senescent state are shown on the cell surface. The loss of surface CD27 and CD28 receptors observed in senescence is shown with a red cross. For more details, see the description in the text. Created with
BioRender.com (accessed on 5 October 2023).
This entry is adapted from the peer-reviewed paper 10.3390/ijms242115653