Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Andrew Tam | -- | 3442 | 2023-11-16 20:18:25 | | | |
| 2 | Lindsay Dong | Meta information modification | 3442 | 2023-11-20 03:05:53 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Tam, A.; Mercier, B.D.; Thomas, R.M.; Tizpa, E.; Wong, I.G.; Shi, J.; Garg, R.; Hampel, H.; Gray, S.W.; Williams, T.; et al. Genomically-Guided Precision Radiation Treatment. Encyclopedia. Available online: https://encyclopedia.pub/entry/51708 (accessed on 03 July 2026).
Tam A, Mercier BD, Thomas RM, Tizpa E, Wong IG, Shi J, et al. Genomically-Guided Precision Radiation Treatment. Encyclopedia. Available at: https://encyclopedia.pub/entry/51708. Accessed July 03, 2026.
Tam, Andrew, Benjamin D. Mercier, Reeny M. Thomas, Eemon Tizpa, Irene G. Wong, Juncong Shi, Rishabh Garg, Heather Hampel, Stacy W. Gray, Terence Williams, et al. "Genomically-Guided Precision Radiation Treatment" Encyclopedia, https://encyclopedia.pub/entry/51708 (accessed July 03, 2026).
Tam, A., Mercier, B.D., Thomas, R.M., Tizpa, E., Wong, I.G., Shi, J., Garg, R., Hampel, H., Gray, S.W., Williams, T., Bazan, J.G., & Li, Y.R. (2023, November 16). Genomically-Guided Precision Radiation Treatment. In Encyclopedia. https://encyclopedia.pub/entry/51708
Tam, Andrew, et al. "Genomically-Guided Precision Radiation Treatment." Encyclopedia. Web. 16 November, 2023.
Copy Citation
Genetic information is seldom incorporated in formulating radiation treatment recommendations for patients with cancer, even though genetic information is now well established to be prognostic and predictive of cancer outcomes and response to systemic therapy. With the increasing accessibility to and use of genetic testing, tumor, and germline genetic data have the potential to inform clinical decisions by improving the efficacy of radiation treatment and ensuring the safety of treatment delivery.
radiation sensitivity
radiogenomics
genes
1. Introduction
In the last decade, the increased availability and rapid integration of genetic testing have revolutionized the landscape of cancer care, particularly in the management of patients with advanced and metastatic malignancies. It has paved the way for the development of personalized treatment recommendations by utilizing mutation profiles to target specific underlying molecular drivers of a tumor [1]. Despite the progress that has been made in our understanding of cancer genetics, the field of radiation oncology has lagged behind medical oncology in incorporating specific genetic information in formulating our decisions surrounding radiation treatment (RT). Even though the field has dramatically advanced in treatment delivery and planning techniques, RT is still largely prescribed today as it has been carried out for many decades, based on tumor size, disease type, histology, surgical margin status, disease stage, and proximity to normal anatomical structures [1][2].
It has long been observed that individuals with certain genetic disorders are predisposed to adverse side effects from RT. One of the most well-known examples is ataxia-telangiectasia (AT), which is a condition caused by homozygous or complete loss of function mutations in the ATM (ataxia telangiectasia, mutated) gene leading to impaired response to DNA (deoxyribonucleic acid) double-strand breaks [3]. Reports from as early as the 1960s have documented the radiation sensitivity of patients with AT developing more pronounced toxicities and increased risk of secondary malignancy from radiation than the general population [4][5]. Since then, many more mutations associated with radiation sensitivity have been identified and the list of potential “radio-sensitizing genes” continues to grow and is likely only to expand exponentially in the next decade.
Perhaps more importantly, radiation oncologists will be increasingly faced with clinical scenarios in which patients will present with existing genetic testing results. The radiation oncologist must then decide based on this information, whether specific mutations portend increased risks of treatment toxicity or resistance, whether radiation would be recommended, and whether adaptations in the treatment field or dose need to be made based on mutations in “radio-sensitizing genes”. Consensus on RT recommendations for patients with pathogenic variants, even those with mutations in genes shown in the laboratory to affect radiation response, such as ATM, is lacking. Clinical guidelines are needed to guide radiation oncologists on how to safely incorporate genetic information in treatment decision-making for patients with “radio-sensitizing” mutations.
2. Genetic Basis of Radiation Sensitivity
Radiation destroys cancer cells by damaging genomic DNA via direct DNA breakage or indirectly via the formation of free radicals and other reactive oxygen species (ROS) [6]. Radiation consequentially induces DNA base damage, single-strand breaks (SSBs), and double-strand breaks (DSBs) to the DNA [6]. Thus, “radiation sensitivity” (or “radio-sensitivity”) describes any toxic, cancerous, or aging effect resulting from radiation [7].
Radiosensitivity can be subcategorized to refer to the responsiveness of tumor cells or the reaction of normal tissues to radiation. Radiosensitivity of tumor cells can enhance cell killing and thereby improve the efficacy of radiation, but conversely, radiosensitivity of healthy cells can lead to more prominent radiation-induced side effects. The molecular underpinning conferring radiosensitivity is complex and not fully understood. However, there is mounting evidence supporting the importance of genetic alterations in affecting clinical radiosensitivity. It has long been observed that patients with certain genetic syndromes develop adverse, sometimes, fatal toxicity following therapeutic radiation. In addition to the example of AT as mentioned above, cancer patients with Fanconi’s Anemia, a rare autosomal recessive DNA repair disorder, who received RT have been shown to exhibit hypersensitivity to radiation [8][9][10][11].
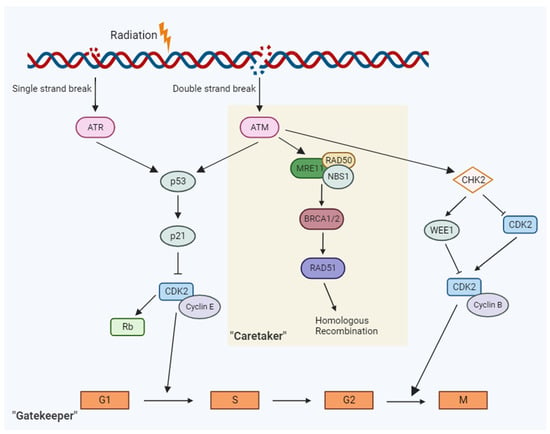
In brief, genomic characteristics that influence radiosensitivity are as follows [6][7][12] (Figure 1):
Figure 1. In response to DNA damage response from radiation, a cascade of events along the “gatekeep” and “caretaker” pathways are activated that ultimately leads to cell cycle arrest or DNA repair.
3. Gatekeeper and Caretaker Genes
3.1. Cell Cycle Checkpoint “Gatekeeper” Genes
Genes that regulate the cell cycle have critical roles in ensuring the DNA integrity of the cell prior to proceeding with cell division. Hence, they are often referred to as “gatekeeper” genes. The cell cycle is a series of cellular events that facilitate the replication of DNA and cell division to create two daughter cells [13]. The normal function of these checkpoint genes is to detect aberration in the DNA and control the progression of the cell cycle. Two examples of classical gatekeeper genes are p53 and ATM. Given their functions in mediating response to DNA damage, these genes have long been considered key players in radiosensitivity.
When DNA damage is detected, these gatekeeper genes either arrest the cell cycle to allow for DNA repair or induce cell death [13]. Checkpoints in the cell cycle include the Gap 1 (G1) and DNA Synthesis (S) phase junction (G1/S) to ensure to prevent replication of damaged DNA and the Gap 2 and mitosis (G2/M) for prevention of segregation of aberrant chromosomes into daughter cells [13].
Ataxia-telangiectasia, mutated (ATM gene) is the master controller for both “gatekeeper” and “caretaker” signaling pathways. In response to DNA damage, the ATM gene product, ATM, exhibits protein kinase activity and phosphorylates p53 at the G1-S checkpoint to induce cell cycle arrest and promote DNA repair [3][14]. Carriers of a germline mutation in ATM are notably at an increased risk of radiation sensitivity [3][14]. A study on female breast cancer patients revealed a significant correlation between the presence of ATM mutation and the development of Grade 3–4 toxicity, such as fibrosis (p = 0.001) following RT [15].
As mentioned above, p53 is one of the main downstream effectors of ATM. p53 is a gene product of the tumor suppressor gene TP53 and it signals multiple downstream targets in response to DNA damage that ultimately determines cell fate after signature radiation exposure [16]. Patients with germline TP53 mutations (for which single copy loss of function is the most common cause of Li-Fraumeni Syndrome) are predisposed to develop early-onset cancers of multiple tissue types [17]. However, the role of the TP53 mutations in radiosensitivity is not as fully understood as p53 has innumerable roles in normal cell physiology. O’Connor and colleagues studied the effect of ionizing gamma-radiation in 60 different cell lines, 39 of which contained a variant mutation of TP53 [18].
The Retinoblastoma tumor suppression gene (RB) is another cell cycle checkpoint gene heavily involved in the mediation of the G1/S checkpoint [19]. The products of this gene are integral to the antiproliferative process and have been characterized to be aberrant or even abrogate in approximately 20–35% of breast cancers as well as occupying a very prominent role in the carcinogenesis of many human lung adenocarcinomas [19][20]. RB is also known to act as an effector in the MAPK signal amplification pathway as well as possess an integral role in the formation of the DREAM complex alongside LIN37 [20][21]. The DREAM-p53 pathway is vital for the function of the G1/S checkpoint [21].
3.2. DNA Repair “Caretaker” Genes
DNA repair (or caretaker) genes often operate in conjunction with gatekeeper genes, and at times with overlapping purposes. Many gatekeeper genes function in the regulation of cell proliferation and cell cycle regulation. DNA repair genes encode proteins directly involved in DNA repair processes in repair mechanisms of both SSBs and DSBs, as well as DNA mismatch repair (MMR). Such repair mechanisms include non-homologous end joining (NHEJ), homologous recombination (HR), base excision repair, and nucleotide excision repair. The resulting encoded proteins from these genes often directly interface with the DNA molecule [22]. However, the line between DNA repair and gatekeeper genes is not always distinct. Gene products from genes such as BRCA1/2, TP53, RAD9, and ATM possess features of both cell cycle checkpoint and DNA repair regulation as well as intrinsic DNA repair features [23]. Mutations in DNA repair genes that cause functional attenuation will likely lead to the accumulation of mutations in other genes, which can contribute to cancer risk [24].
Lynch syndrome, or hereditary nonpolyposis colorectal cancer, is a well-documented inherited disorder that resulting from a constellation of mutations in MMR genes MLH1, MSH2, MSH6, or PMS2 [25]. The MMR proteins encoded by these genes act to directly interface with the damaged DNA molecule via heterodimerization with one another [26]. While Lynch syndrome was initially associated with the occurrence of colorectal cancer, with up to 20% of all patients with colon cancer being afflicted by this genetic condition, these patients are also at increased risk of other solid tumors, including endometrial, ovarian, and gastric cancers [25][27]. However, there is limited evidence suggesting increased radiosensitivity among patients with Lynch syndrome.
RAD51 is a gene family encoding proteins directly associated with DNA repair, specifically in HR in the setting of DSBs [28]. During HR, RAD51 is involved in the homology search, strand invasion, and strand pairing to facilitate repair [28][29]. RAD51 and its paralogs are heavily associated with tumorigenesis, as RAD51 tends to be downregulated in many cancers, causing a notable decline in DNA repair capacity, as well as overexpressed in others [23]. Overexpression of RAD51 is known to contribute to anomalous recombination between both short repetitive sequences and homologous sequencing, resulting in a significantly increased likelihood of tumorigenesis [23].
BRCA1/2 are the most commonly affected genes in hereditary breast and ovarian cancer, though these mutations impact the risk for many other cancers including prostate and pancreatic cancers [30]. Both BRCA1 and BRCA2 are involved in the maintenance of HR, a DSB repair pathway, at sites of DNA damage. BRCA1 encodes the breast cancer type 1 susceptibility protein (BRCA1), a phosphoprotein that helps maintain genomic stability as a component of the multi-subunit protein complex BRCA1-associated genome surveillance complex (BASC) [31]. BASC interacts with RNA polymerase II and histone deacetylase, displaying a role in transcription, repair of DNA double-stranded breaks (DSBs) [31], and recombination [31]. BRCA1 colocalizes with BRCA2 and the recombinase RAD51 to activate RAD51-mediated HR of DSBs by helping RAD51 assemble on ssDNA to search for a homologous DNA repair template and initiate strand exchange [32]. Tumors harboring BRCA1/2 mutations have been shown to have increased sensitivity to radiation.
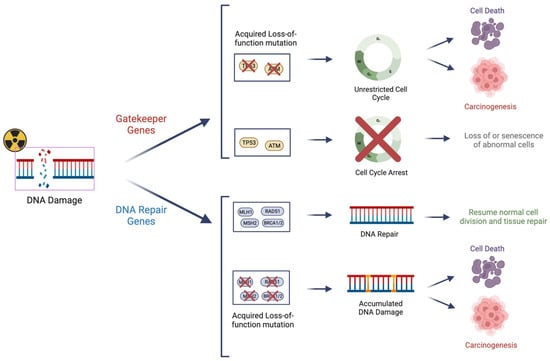
DNA repair genes and gatekeeper genes, while differing in terms of functionality, have similarly important roles in conferring the radiosensitivity of a given cell (Figure 2). With the recent advent of routine clinical cancer mutation profiling, a comprehensive assessment of gatekeeper and DNA repair genes can be employed to optimize cancer therapy.
Figure 2. Cell cycle checkpoint “gatekeeper” genes and DNA repair “caretaker” genes are important regulators in ensuring the integrity of the genome prior to cell division. Mutations of these genes can lead to their loss of function and dysfunction of the regulatory mechanism, as well as potential uncontrolled cell growth “carcinogenesis”. These genes also have important roles in conferring “radio-sensitivity” due to impaired surveillance and/or repair of radiation-induced DNA damage.
4. The Role of Molecular Testing in Identifying Radiosensitizing Genes
Despite the improved understanding of the molecular “gatekeeper” and “caretaker” pathways in recent years, establishing the role of a particular gene responsible for radiosensitivity remains a challenge, especially in light of potentially conflicting evidence. For example, patients with Fanconi anemia demonstrate radiosensitivity clinically, but with inconsistent findings on the extent of DNA repair in response to radiation from in vitro studies [33]. Another challenge is in identifying new potential genes that lead to radiosensitivity and ongoing efforts have aimed to tackle this by leveraging toxicity outcomes from large clinical datasets and also at the individual patient level.
As mentioned above, GWASs have revolutionized the search for candidate genes via “tagging” hundreds of thousands of SNPs through the use of microarray [34][35]. These studies utilize large patient data to identify potential genetic variants associated with a certain toxicity. The UK RAPPER study was one of the largest studies in identifying potential SNPs associated with chronic toxicities from adjuvant breast RT or definitive prostate RT [36][37].
While functional bioassays are routinely utilized to understand how specific DNA mutations can affect radiation response, the role of functional bioassays remains limited in clinical settings. For patient care, many other types of molecular tests are employed instead. At the genomic levels, whole exome, whole genome and targeted panel based sequencing employs next-generation sequencing (NGS) technology to interrogate mutations in the genome and subsequent filtering based on quality criteria such as by percentage of reads showing the variant, then by variants outside of coding regions and known variants to identify a potential gene of interest [38]. At the chromosomal level, fluorescence in situ hybridization (FISH) uses fluorescence tags to identify chromosomal aberration, such as translocations, amplifications, and deletions [39]. At the RNA level, RNA sequencing (RNAseq) applies the same principle as NGS to assess the functional levels of genetic variation [40].
5. Moving towards Genomically-Guided Radiation Treatment
5.1. Omission of Radiation Treatment
As described above, in some individuals with germline mutations in radio-sensitivity genes, radiation may lead to potentially significant clinical toxicities, including death [12]. The current NCCN guidelines advise avoidance of RT when possible among patients with Li-Fraumeni syndrome (i.e., most commonly a result of deleterious at least single copy loss of function mutation in TP53) due to concern for an increased risk of secondary malignancy [41][42]. However, there is no such recommendation for any other genetic syndrome or mutations on the basis of radiosensitivity.
With an increasing identification and understanding of the role of radio-sensitizing genes, targeted mutation profiling of these genomic regions can potentially provide valuable insights into the appropriateness of RT for patients who are known carriers. The utmost challenge is how to incorporate genetic information in weighing the risks versus benefits of omitting radiation when radiation is typically clinically indicated and/or is the best treatment option. In situations where patients are found to have predicted deleterious mutations in radio-sensitivity conveying genes and where there are good alternative treatment options that have equivalent patient outcomes, clinicians should carefully consider these instead of radiation.
5.2. Radiation Dose to Tumor
Another consideration is the use of genetic information in guiding radiation dose. The current practice of RT operates on the assumption that tumors of the same type respond similarly to radiation, regardless of the potential radiosensitivity of the targeted tumor or patients’ normal tissues. This one-size-fits-all approach leads to the same prescription dose across patients. However, this can pose challenges for patients with radiosensitivity, as it may exacerbate normal tissue damage and induce adverse side effects. A noteworthy study by Scott and colleagues addressed this concern by developing a clinical model to optimize RT dosages based on a retrospective cohort of non-small cell lung cancer patients [43]. Their findings revealed that a considerable number of patients were at risk of being overdosed, thereby increasing the likelihood of treatment-related toxicity [43].
The use of molecular data to guide radiation dosing is still in its infancy. A good recent example is the use of p16 overexpression to reduce treatment intensity in head and neck cancer, as such patients have been shown to have a more favorable disease prognosis and response rate compared to patients with smoking-associated malignancies. However, despite phase II trials demonstrating favorable outcomes with reducing the dose of radiation [44][45], the phase II multi-center randomized NRG-HN005 (NCT 03952585) did not reach a non-inferiority threshold and has been closed temporarily [46].
5.3. Volume Delineation
One of the most challenging and unfortunately largely subjective roles of a radiation oncologist is to define the target volumes for RT. RT is individualized to each patient with target volumes delineated based on patients’ anatomy, disease status, and other characteristics. Radiogenomic data can be utilized to further inform practitioners throughout these clinical decisions. One consideration is the dose to normal tissue, particularly to organs at risk (OARs). For patients harboring radio-sensitizing mutations, it could be prudent to have more conservative constraints for OARs to minimize adverse effects, such as the volume of small bowel receiving a high dose (50–55 Gy), or moderate dose (30 Gy). These more stringent constraints might also necessitate the need to adjust target volume contours (e.g., reduction of elective nodal volume coverage), and in some cases, avoidance of radiation completely as noted above. In oropharyngeal squamous cell carcinoma (SCC), recent efforts, such as the EVADER phase II clinical trial (NCT 03822897), aim to evaluate the efficacy of biomarkers expression as a guide for target volume coverage for RT [47].
5.4. Combining with Systemic Therapy
Genetic information can be leveraged to guide the selection of concurrent systemic therapies with RT to further enhance the effect of radiation. Synthetic lethality is a term used to describe therapeutic agents that kill target cells by taking advantage of existing cellular defects [48]. Such a combined effect of the somatic mutation and targeted therapy can also sensitize tumor cells to sublethal doses of DNA damage, a concept known as synthetic cytotoxicity [48]. Genetic information can guide the selection of a known radiosensitizers to optimize the therapeutic ratio in favor of tumor killing without worsening toxicity.
In addition to targeted therapies, gene silencing can also be a potential strategy for potentiating the effect of RT. Genetic radiosensitizers work primarily by disrupting gene expressions through the use of short interfering RNAs (siRNAs) to further destabilize the genetic integrity of cancer cells to induce added DNA damage. An example of such a target is the Nijmegen breakage syndrome-1 (NBS1) gene which has an important role in HR repair of radiation-induced DSBs [49]. In non-small cell lung cancer cells, siRNA targeted for the NBS1 gene has been shown to enhance radiosensitivity [49].
6. Precision Medicine in Today’s Radiation Oncology Clinic—The Example of Genomically-Guided Radiation Treatment in Breast Cancer
Personalized medicine has a long and storied history in breast cancer medical oncology. Tamoxifen could be considered one of the field’s first truly personalized targeted therapies for patients with estrogen receptor-positive/human epidermal growth factor receptor-negative (ER+/HER2−) breast cancer. Tumor genomic assays, such as the 21-gene assay (commercially known as Oncotype DX®), currently play a significant role in adjuvant chemotherapy decisions for ER+/HER2− breast cancer [50]. For instance, breast cancer patients with germline BRCA1/2 mutations are candidates for a class of drugs called PARP inhibitors [51].
On the other hand, the translation of personalizing RT has been slow. Currently, radiation oncologists use standard doses of radiation to eradicate microscopic disease in the adjuvant setting or to treat gross disease with or without concurrent chemotherapy. However, data are accumulating for the use of genomic assays and/or other biomarkers to help identify patients with breast cancer who may not benefit from adjuvant RT.
Randomized trials are ongoing to determine whether the use of tumor genomic assays may help identify patients who are candidates for omission of adjuvant whole breast radiation. One such trial is the NRG Oncology BR007 (DEBRA) study, in which patients aged 50–69 years old with ER+/HER2−, stage I (≤2 cm and node-negative) breast cancers with Oncotype recurrence score (RS) ≤18 treated with lumpectomy are randomized to radiation versus omission of radiation (NCT04852887). The EXPERT trial is taking a similar approach for patients aged ≥50 years old with stage I (node-negative), grade 1–2, HR+/HER2− breast cancers and uses the Prosigna (PAM50) assay to identify luminal A patients with low risk of recurrence to randomize patients to omission of radiation versus adjuvant RT (NCTC02889874). The Oncotype RS is also currently being used for patients with node-positive breast cancer as a potential biomarker to help determine which patients may not benefi17t from regional nodal irradiation (RNI). The MA39 study randomizes patients aged ≥ 40 years old with ER+/HER2− breast cancer with 1–3+ nodes after lumpectomy or 1–2+ nodes after mastectomy with Oncotype RS ≤ 25 to whole breast irradiation +/− RNI (after lumpectomy) or postmastectomy radiation (PMRT) versus no PMRT after mastectomy (NCT03488693).
The assays described above were primarily developed to help guide adjuvant systemic therapy decisions in ER+/HER2− breast cancers. Several groups have also worked on developing radiation-specific genomic assays. These assays include the Radiation Sensitivity Index (RSI) [52], the Profile for the Omission of Local Adjuvant Radiation Treatment (POLAR) [53], and the Adjuvant Radiation Treatment Intensification Classifier (ARTIC) [54]. The ARTIC and POLAR assays are both prognostic and predictive, whereas the RSI is prognostic. For example, the POLAR assay is a 16-gene signature that was used to test and validate whether it could be used to identify patients with ER+/HER2−, node-negative breast cancers that may not benefit from adjuvant RT. Using data from two independent clinical trials, Sjostrom and colleagues demonstrated that patients defined as POLAR low-risk had low 10-year rates of local-regional recurrence (6–7%) without RT and that RT did not benefit these patients. However, patients identified as POLAR high-risk had higher rates of LRR and demonstrated a significant reduction in LRR risk with adjuvant RT from 19% to 8% (hazard ratio [HR] 0.43) in one of the trial cohort and from 22% to 8% (HR 0.25) in the second trial cohort [53].
References
- Burnet, N.G. Defining the tumour and target volumes for radiotherapy. Cancer Imaging 2004, 4, 153–161.
- Baskar, R.; Lee, K.A.; Yeo, R.; Yeoh, K.-W. Cancer and Radiation Therapy: Current Advances and Future Directions. Int. J. Med. Sci. 2012, 9, 193–199.
- Rothblum-Oviatt, C.; Wright, J.; Lefton-Greif, M.A.; McGrath-Morrow, S.A.; Crawford, T.O.; Lederman, H.M. Ataxia telangiectasia: A review. Orphanet J. Rare Dis. 2016, 11, 1–21.
- Morgan, J.L.; Holcomb, T.M.; Morrissey, R.W. Radiation Reaction in Ataxia Telangiectasia. Arch. Pediatr. Adolesc. Med. 1968, 116, 557–558.
- Swift, M.; Morrell, D.; Massey, R.B.; Chase, C.L. Incidence of Cancer in 161 Families Affected by Ataxia–Telangiectasia. N. Engl. J. Med. 1991, 325, 1831–1836.
- Hall, E.J.; Giaccia, A.J. Radiobiology for the Radiologist, 7th ed.; Wolters Kluwer Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2012.
- El-Nachef, L.; Al-Choboq, J.; Restier-Verlet, J.; Granzotto, A.; Berthel, E.; Sonzogni, L.; Ferlazzo, M.L.; Bouchet, A.; Leblond, P.; Combemale, P.; et al. Human Radiosensitivity and Radiosusceptibility: What Are the Differences? Int. J. Mol. Sci. 2021, 22, 7158.
- Alter, B.P. Radiosensitivity in Fanconi’s anemia patients. Radiother. Oncol. 2002, 62, 345–347.
- Bremer, M.; Klöpper, K.; Yamini, P.; Bendix-Waltes, R.; Dörk, T.; Karstens, J.H. Clinical radiosensitivity in breast cancer patients carrying pathogenic ATM gene mutations: No observation of increased radiation-induced acute or late effects. Radiother. Oncol. 2003, 69, 155–160.
- Gatti, R.A.; Boder, E.; Good, R.A. Immunodeficiency, radiosensitivity, and the XCIND syndrome. Immunol. Res. 2007, 38, 87–101.
- Pollard, J.M.; Gatti, R.A. Clinical Radiation Sensitivity With DNA Repair Disorders: An Overview. Int. J. Radiat. Oncol. 2009, 74, 1323–1331.
- Gonçalves, D.; Pires, A.S.; Marques, I.A.; Gomes, I.; Sousa, G.; Botelho, M.F.; Abrantes, A.M. An Overview on Radiation Sensitivity in Hereditary Breast and Ovarian Cancer Syndrome. Cancers 2022, 14, 3254.
- Pawlik, T.M.; Keyomarsi, K. Role of cell cycle in mediating sensitivity to radiotherapy. Int. J. Radiat. Oncol. 2004, 59, 928–942.
- Andreassen, C.N.; Rosenstein, B.S.; Kerns, S.L.; Ostrer, H.; De Ruysscher, D.; Cesaretti, J.A.; Barnett, G.C.; Dunning, A.M.; Dorling, L.; West, C.M.L.; et al. Individual patient data meta-analysis shows a significant association between the ATM rs1801516 SNP and toxicity after radiotherapy in 5456 breast and prostate cancer patients. Radiother. Oncol. 2016, 121, 431–439.
- Iannuzzi, C.M.; Atencio, D.P.; Green, S.; Stock, R.G.; Rosenstein, B.S. ATM mutations in female breast cancer patients predict for an increase in radiation-induced late effects. Int. J. Radiat. Oncol. 2002, 52, 606–613.
- Lee, C.-L.; Blum, J.M.; Kirsch, D.G. Role of p53 in regulating tissue response to radiation by mechanisms independent of apoptosis. Transl. Cancer Res. 2013, 2, 412–421.
- Guha, T.; Malkin, D. Inherited TP53 Mutations and the Li–Fraumeni Syndrome. Cold Spring Harb. Perspect. Med. 2017, 7, a026187.
- O’Connor, P.M.; Jackman, J.; Bae, I.; Myers, T.G.; Fan, S.; Mutoh, M.; A Scudiero, D.; Monks, A.; A Sausville, E.; Weinstein, J.N.; et al. Characterization of the p53 tumor suppressor pathway in cell lines of the National Cancer Institute anticancer drug screen and correlations with the growth-inhibitory potency of 123 anticancer agents. Cancer Res. 1997, 57, 4285–4300.
- Bosco, E.E.; Wang, Y.; Xu, H.; Zilfou, J.T.; Knudsen, K.E.; Aronow, B.J.; Lowe, S.W.; Knudsen, E.S. The retinoblastoma tumor suppressor modifies the therapeutic response of breast cancer. J. Clin. Investig. 2007, 117, 218–228.
- Walter, D.M.; Yates, T.J.; Ruiz-Torres, M.; Kim-Kiselak, C.; Gudiel, A.A.; Deshpande, C.; Wang, W.Z.; Cicchini, M.; Stokes, K.L.; Tobias, J.W.; et al. RB constrains lineage fidelity and multiple stages of tumour progression and metastasis. Nature 2019, 569, 423–427.
- Uxa, S.; Bernhart, S.H.; Mages, C.F.S.; Fischer, M.; Kohler, R.; Hoffmann, S.; Stadler, P.F.; Engeland, K.; A Müller, G. DREAM and RB cooperate to induce gene repression and cell-cycle arrest in response to p53 activation. Nucleic Acids Res. 2019, 47, 9087–9103.
- Levitt, N.C.; Hickson, I.D. Caretaker tumour suppressor genes that defend genome integrity. Trends Mol. Med. 2002, 8, 179–186.
- Broustas, C.G.; Lieberman, H.B. DNA Damage Response Genes and the Development of Cancer Metastasis. Radiat. Res. 2014, 181, 111–130.
- Borrego-Soto, G.; Ortiz-López, R.; Rojas-Martínez, A. Ionizing radiation-induced DNA injury and damage detection in patients with breast cancer. Genet. Mol. Biol. 2015, 38, 420–432.
- Lynch, H.T.; De La Chapelle, A. Hereditary Colorectal Cancer. N. Engl. J. Med. 2003, 348, 919–932.
- Pal, T.; Permuth-Wey, J.; Sellers, T.A. A review of the clinical relevance of mismatch-repair deficiency in ovarian cancer. Cancer 2008, 113, 733–742.
- Kastrinos, F.; Mukherjee, B.; Tayob, N.; Wang, F.; Sparr, J.; Raymond, V.M.; Bandipalliam, P.; Stoffel, E.M.; Gruber, S.B.; Syngal, S. Risk of Pancreatic Cancer in Families With Lynch Syndrome. JAMA 2009, 302, 1790–1795.
- Bonilla, B.; Hengel, S.R.; Grundy, M.K.; Bernstein, K.A. RAD51 Gene Family Structure and Function. Annu. Rev. Genet. 2020, 54, 25–46.
- Symington, L.S.; Gautier, J. Double-Strand Break End Resection and Repair Pathway Choice. Annu. Rev. Genet. 2011, 45, 247–271.
- Petrucelli, N.; Daly, M.B.; Pal, T. BRCA1- and BRCA2-Associated Hereditary Breast and Ovarian Cancer. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993.
- De la Torre, C.; Pincheira, J.; López-Sáez, J.F. Human Syndromes with Genomic Instability and Multiprotein Machines That Repair DNA Double-Strand Breaks. Histol. Histopathol. 2003, 18, 225–243.
- Foo, T.K.; Xia, B. BRCA1-Dependent and Independent Recruitment of PALB2–BRCA2–RAD51 in the DNA Damage Response and Cancer. Cancer Res 2022, 82, 3191–3197.
- Zahnreich, S.; Weber, B.; Rösch, G.; Schindler, D.; Schmidberger, H. Compromised repair of radiation-induced DNA double-strand breaks in Fanconi anemia fibroblasts in G2. DNA Repair 2020, 96, 102992.
- Manolio, T.A. Genomewide Association Studies and Assessment of the Risk of Disease. N. Engl. J. Med. 2010, 363, 166–176.
- Forte, G.I.; Minafra, L.; Bravatà, V.; Cammarata, F.P.; Lamia, D.; Pisciotta, P.; Cirrone, G.A.P.; Cuttone, G.; Gilardi, M.C.; Russo, G. Radiogenomics: The utility in patient selection. Transl. Cancer Res. 2017, 6, S852–S874.
- Barnett, G.C.; E Coles, C.; Elliott, R.M.; Baynes, C.; Luccarini, C.; Conroy, D.; Wilkinson, J.S.; Tyrer, J.; Misra, V.; Platte, R.; et al. Independent validation of genes and polymorphisms reported to be associated with radiation toxicity: A prospective analysis study. Lancet Oncol. 2012, 13, 65–77.
- Barnett, G.C.; Thompson, D.; Fachal, L.; Kerns, S.; Talbot, C.; Elliott, R.M.; Dorling, L.; Coles, C.E.; Dearnaley, D.P.; Rosenstein, B.S.; et al. A genome wide association study (GWAS) providing evidence of an association between common genetic variants and late radiotherapy toxicity. Radiother. Oncol. 2014, 111, 178–185.
- Gilissen, C.; Hoischen, A.; Brunner, H.G.; A Veltman, J. Disease gene identification strategies for exome sequencing. Eur. J. Hum. Genet. 2012, 20, 490–497.
- Mighton, C.; Lerner-Ellis, J.P. Principles of molecular testing for hereditary cancer. Genes, Chromosom. Cancer 2022, 61, 356–381.
- Murdock, D.R. Enhancing Diagnosis Through RNA Sequencing. Clin. Lab. Med. 2020, 40, 113–119.
- Thariat, J.; Chevalier, F.; Orbach, D.; Ollivier, L.; Marcy, P.-Y.; Corradini, N.; Beddok, A.; Foray, N.; Bougeard, G. Avoidance or adaptation of radiotherapy in patients with cancer with Li-Fraumeni and heritable TP53-related cancer syndromes. Lancet Oncol. 2021, 22, e562–e574.
- NCCN. National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology. Genetic/Familial High-Risk Assessment. Version 3.2023. Available online: https://www.nccn.org/professionals/physician_gls/pdf/genetics_bop.pdf. (accessed on 21 June 2023).
- Scott, J.G.; Sedor, G.; Scarborough, J.A.; Kattan, M.W.; Peacock, J.; Grass, G.D.; Mellon, E.A.; Thapa, R.; Schell, M.; Waller, A.; et al. Personalizing Radiotherapy Prescription Dose Using Genomic Markers of Radiosensitivity and Normal Tissue Toxicity in NSCLC. J. Thorac. Oncol. 2020, 16, 428–438.
- Chera, B.S.; Amdur, R.J.; Tepper, J.; Qaqish, B.; Green, R.; Aumer, S.L.; Hayes, N.; Weiss, J.; Grilley-Olson, J.; Zanation, A.; et al. Phase 2 Trial of De-intensified Chemoradiation Therapy for Favorable-Risk Human Papillomavirus–Associated Oropharyngeal Squamous Cell Carcinoma. Int. J. Radiat. Oncol. 2015, 93, 976–985.
- Yom, S.S.; Torres-Saavedra, P.; Caudell, J.J.; Waldron, J.N.; Gillison, M.L.; Xia, P.; Truong, M.T.; Kong, C.; Jordan, R.; Subramaniam, R.M.; et al. Reduced-Dose Radiation Therapy for HPV-Associated Oropharyngeal Carcinoma (NRG Oncology HN002). J. Clin. Oncol. 2021, 39, 956–965.
- PROTOCOL NRG-HN005 – TEMPORARY CLOSURE. NRG Oncology. Available online: https://myemail.constantcontact.com/PROTOCOL-NRG-HN005---TEMPORARY-CLOSURE.html?soid=1139396743412&aid=YHEDCt657NI (accessed on 6 June 2023).
- Bratman, S.V.; Berthelet, E.; Butler, J.B.; de Almeida, J.R.; Karam, I.; Metser, U.; Olson, R.A.; Pochini, C.; Waldron, J.; Yu, E.; et al. CCTG HN.10: A phase II single-arm trial of elective volume adjusted de-escalation radiotherapy (EVADER) in patients with low-risk HPV-related oropharyngeal squamous cell carcinoma (NCT03822897). J. Clin. Oncol. 2020, 38, TPS6592.
- Li, X.; O’neil, N.J.; Moshgabadi, N.; Hieter, P. Synthetic Cytotoxicity: Digenic Interactions with TEL1/ATM Mutations Reveal Sensitivity to Low Doses of Camptothecin. Genetics 2014, 197, 611–623.
- Ohnishi, K.; Scuric, Z.; Schiestl, R.H.; Okamoto, N.; Takahashi, A.; Ohnishi, T. siRNA Targeting NBS1 or XIAP Increases Radiation Sensitivity of Human Cancer Cells Independent of TP53 Status. Radiation Res. 2006, 166, 454–462.
- Paik, S.; Shak, S.; Tang, G.; Kim, C.; Baker, J.; Cronin, M.; Baehner, F.L.; Walker, M.G.; Watson, D.; Park, T.; et al. A Multigene Assay to Predict Recurrence of Tamoxifen-Treated, Node-Negative Breast Cancer. N. Engl. J. Med. 2004, 351, 2817–2826.
- Geyer, C.; Gelber, R.; Yothers, G.; Taboada, M.; Ross, L.; Rastogi, P.; Cui, K.; Arahmani, A.; Aktan, G.; Arnedos, M.; et al. Overall survival in the OlympiA phase III trial of adjuvant olaparib in patients with germline pathogenic variants in BRCA1/2 and high-risk, early breast cancer. Ann. Oncol. 2022, 33, 1250–1268.
- Torres-Roca, J.F.; Fulp, W.J.; Caudell, J.J.; Servant, N.; Bollet, M.A.; van de Vijver, M.; Naghavi, A.O.; Harris, E.E.; Eschrich, S.A. Integration of a Radiosensitivity Molecular Signature Into the Assessment of Local Recurrence Risk in Breast Cancer. Int. J. Radiat. Oncol. 2015, 93, 631–638.
- Sjöström, M.; Fyles, A.; Liu, F.-F.; McCready, D.; Shi, W.; Rey-McIntyre, K.; Chang, S.L.; Feng, F.Y.; Speers, C.W.; Pierce, L.J.; et al. Development and Validation of a Genomic Profile for the Omission of Local Adjuvant Radiation in Breast Cancer. J. Clin. Oncol. 2023, 41, 1533–1540.
- Sjöström, M.; Chang, S.L.; Fishbane, N.; Davicioni, E.; Zhao, S.G.; Hartman, L.; Holmberg, E.; Feng, F.Y.; Speers, C.W.; Pierce, L.J.; et al. Clinicogenomic Radiotherapy Classifier Predicting the Need for Intensified Locoregional Treatment After Breast-Conserving Surgery for Early-Stage Breast Cancer. J. Clin. Oncol. 2019, 37, 3340–3349.
More
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
770
Revisions:
2 times
(View History)
Update Date:
20 Nov 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No