 +1 credit
+1 credit
Video Upload Options
Inflammasomes, a group of multiprotein complexes, are essential in regulating inflammation and immune responses. Several inflammasomes, including nucleotide-binding domain leucine-rich repeat-containing protein 1 (NLRP1), NLRP3, NLRP6, NLRP7, NLRP12, interferon-inducible protein 16 (IFI16), NOD-like receptor family CARD domain-containing protein 4 (NLRC4), absent in melanoma 2 (AIM2), and pyrin, have been studied in various inflammatory diseases. Activating inflammasomes leads to the processing and production of proinflammatory cytokines, such as interleukin (IL)-1β and IL-18. The NLRP3 inflammasome is the most extensively studied and well characterized. Consequently, targeting inflammasomes (particularly NLRP3) with several compounds, including small molecule inhibitors and natural compounds, has been studied as a potential therapeutic strategy.
1. Introduction
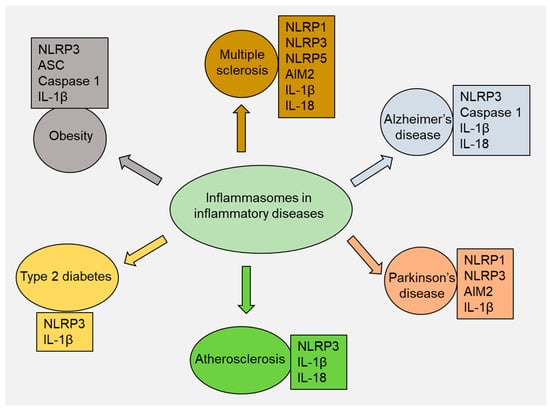
2. Multiple Sclerosis
3. Alzheimer’s Disease
4. Parkinson’s Disease
5. Atherosclerosis
6. Type 2 Diabetes
7. Obesity
8. Other Inflammatory Diseases
References
- Sutterwala, F.S.; Haasken, S.; Cassel, S.L. Mechanism of NLRP3 inflammasome activation. Ann. N. Y. Acad. Sci. 2014, 1319, 82–95.
- Ozaki, E.; Campbell, M.; Doyle, S.L. Targeting the NLRP3 inflammasome in chronic inflammatory diseases: Current perspectives. J. Inflamm. Res. 2015, 8, 15–27.
- Wang, Z.; Zhang, S.; Xiao, Y.; Zhang, W.; Wu, S.; Qin, T.; Yue, Y.; Qian, W.; Li, L. NLRP3 inflammasome and inflammatory diseases. Oxidative Med. Cell. Longev. 2020, 2020, 4063562.
- Gao, J.; Zhang, H.; Yang, Y.; Tao, J. Therapeutic potential of targeting the NLRP3 inflammasome in rheumatoid arthritis. Inflammation 2023, 46, 835–852.
- Gajofatto, A.; Benedetti, M.D. Treatment strategies for multiple sclerosis: When to start, when to change, when to stop? World J. Clin. Cases 2015, 3, 545.
- Cui, Y.; Yu, H.; Bu, Z.; Wen, L.; Yan, L.; Feng, J. Focus on the role of the NLRP3 inflammasome in multiple sclerosis: Pathogenesis, diagnosis, and therapeutics. Front. Mol. Neurosci. 2022, 15, 894298.
- Blevins, H.M.; Xu, Y.; Biby, S.; Zhang, S. The NLRP3 inflammasome pathway: A review of mechanisms and inhibitors for the treatment of inflammatory diseases. Front. Aging Neurosci. 2022, 14, 879021.
- Palomino-Antolin, A.; Narros-Fernández, P.; Farré-Alins, V.; Sevilla-Montero, J.; Decouty-Pérez, C.; Lopez-Rodriguez, A.B.; Fernandez, N.; Monge, L.; Casas, A.I.; Calzada, M.J. Time-dependent dual effect of NLRP3 inflammasome in brain ischaemia. Br. J. Pharmacol. 2022, 179, 1395–1410.
- Coll, R.C.; Hill, J.R.; Day, C.J.; Zamoshnikova, A.; Boucher, D.; Massey, N.L.; Chitty, J.L.; Fraser, J.A.; Jennings, M.P.; Robertson, A.A. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat. Chem. Biol. 2019, 15, 556–559.
- Pinke, K.H.; Zorzella-Pezavento, S.F.G.; de Campos Fraga-Silva, T.F.; Mimura, L.A.N.; De Oliveira, L.R.C.; Ishikawa, L.L.W.; Fernandes, A.A.H.; Lara, V.S.; Sartori, A. Calming down mast cells with ketotifen: A potential strategy for multiple sclerosis therapy? Neurotherapeutics 2020, 17, 218–234.
- Desu, H.L.; Plastini, M.; Illiano, P.; Bramlett, H.M.; Dietrich, W.D.; de Rivero Vaccari, J.P.; Brambilla, R.; Keane, R.W. IC100: A novel anti-ASC monoclonal antibody improves functional outcomes in an animal model of multiple sclerosis. J. Neuroinflamm. 2020, 17, 143.
- Kumari, P.; Russo, A.J.; Shivcharan, S.; Rathinam, V.A. AIM2 in health and disease: Inflammasome and beyond. Immunol. Rev. 2020, 297, 83–95.
- Ma, C.; Li, S.; Hu, Y.; Ma, Y.; Wu, Y.; Wu, C.; Liu, X.; Wang, B.; Hu, G.; Zhou, J. AIM2 controls microglial inflammation to prevent experimental autoimmune encephalomyelitis. J. Exp. Med. 2021, 218, e20201796.
- Sharma, B.R.; Karki, R.; Kanneganti, T.D. Role of AIM2 inflammasome in inflammatory diseases, cancer and infection. Eur. J. Immunol. 2019, 49, 1998–2011.
- Govindarajan, V.; de Rivero Vaccari, J.P.; Keane, R.W. Role of inflammasomes in multiple sclerosis and their potential as therapeutic targets. J. Neuroinflammation 2020, 17, 260.
- Maver, A.; Lavtar, P.; Ristić, S.; Stopinšek, S.; Simčič, S.; Hočevar, K.; Sepčić, J.; Drulović, J.; Pekmezović, T.; Novaković, I. Identification of rare genetic variation of NLRP1 gene in familial multiple sclerosis. Sci. Rep. 2017, 7, 3715.
- Zhang, L.; Jiao, C.; Liu, L.; Wang, A.; Tang, L.; Ren, Y.; Huang, P.; Xu, J.; Mao, D.; Liu, L. NLRC5: A potential target for central nervous system disorders. Front. Immunol. 2021, 12, 704989.
- Beh, S.C.; Greenberg, B.M.; Frohman, T.; Frohman, E.M. Transverse myelitis. Neurol. Clin. 2013, 31, 79–138.
- Goverman, J. Autoimmune T cell responses in the central nervous system. Nat. Rev. Immunol. 2009, 9, 393–407.
- Kany, S.; Vollrath, J.T.; Relja, B. Cytokines in inflammatory disease. Int. J. Mol. Sci. 2019, 20, 6008.
- Kucuksezer, U.C.; Aktas Cetin, E.; Esen, F.; Tahrali, I.; Akdeniz, N.; Gelmez, M.Y.; Deniz, G. The role of natural killer cells in autoimmune diseases. Front. Immunol. 2021, 12, 622306.
- McLaughlin, K.A.; Wucherpfennig, K.W. B cells and autoantibodies in the pathogenesis of multiple sclerosis and related inflammatory demyelinating diseases. Adv. Immunol. 2008, 98, 121–149.
- Barclay, W.; Shinohara, M.L. Inflammasome activation in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE). Brain Pathol. 2017, 27, 213–219.
- Voet, S.; Srinivasan, S.; Lamkanfi, M.; van Loo, G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. 2019, 11, e10248.
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta 2014, 1843, 2563–2582.
- Inoue, M.; Shinohara, M.L. Nlrp3 inflammasome and MS/EAE. Autoimmune Dis. 2013, 2013, 859145.
- Barclay, W.E.; Aggarwal, N.; Deerhake, M.E.; Inoue, M.; Nonaka, T.; Nozaki, K.; Luzum, N.A.; Miao, E.A.; Shinohara, M.L. The AIM2 inflammasome is activated in astrocytes during the late phase of EAE. JCI Insight 2022, 7, e155563.
- Madsen, P.M.; Desu, H.L.; de Rivero Vaccari, J.P.; Florimon, Y.; Ellman, D.G.; Keane, R.W.; Clausen, B.H.; Lambertsen, K.L.; Brambilla, R. Oligodendrocytes modulate the immune-inflammatory response in EAE via TNFR2 signaling. Brain Behav. Immun. 2020, 84, 132–146.
- Jha, S.; Srivastava, S.Y.; Brickey, W.J.; Iocca, H.; Toews, A.; Morrison, J.P.; Chen, V.S.; Gris, D.; Matsushima, G.K.; Ting, J.P.-Y. The inflammasome sensor, NLRP3, regulates CNS inflammation and demyelination via caspase-1 and interleukin-18. J. Neurosci. 2010, 30, 15811–15820.
- Breijyeh, Z.; Karaman, R. Comprehensive review on Alzheimer’s disease: Causes and treatment. Molecules 2020, 25, 5789.
- Rui, W.; Xiao, H.; Fan, Y.; Ma, Z.; Xiao, M.; Li, S.; Shi, J. Systemic inflammasome activation and pyroptosis associate with the progression of amnestic mild cognitive impairment and Alzheimer’s disease. J. Neuroinflammation 2021, 18, 280.
- Liang, T.; Zhang, Y.; Wu, S.; Chen, Q.; Wang, L. The role of NLRP3 inflammasome in Alzheimer’s disease and potential therapeutic targets. Front. Pharmacol. 2022, 13, 845185.
- Van Zeller, M.; Dias, D.; Sebastião, A.M.; Valente, C.A. NLRP3 inflammasome: A starring role in amyloid-β-and tau-driven pathological events in Alzheimer’s disease. J. Alzheimer’s Dis. 2021, 83, 939–961.
- Wang, W.-Y.; Tan, M.-S.; Yu, J.-T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136.
- Lai, A.Y.; McLaurin, J. Clearance of amyloid-β peptides by microglia and macrophages: The issue of what, when and where. Future Neurol. 2012, 7, 165–176.
- Cornell, J.; Salinas, S.; Huang, H.-Y.; Zhou, M. Microglia regulation of synaptic plasticity and learning and memory. Neural Regen. Res. 2022, 17, 705.
- Sutinen, E.M.; Pirttilä, T.; Anderson, G.; Salminen, A.; Ojala, J.O. Pro-inflammatory interleukin-18 increases Alzheimer’s disease-associated amyloid-β production in human neuron-like cells. J. Neuroinflammation 2012, 9, 199.
- Sita, G.; Graziosi, A.; Hrelia, P.; Morroni, F. NLRP3 and Infections: β-Amyloid in Inflammasome beyond Neurodegeneration. Int. J. Mol. Sci. 2021, 22, 6984.
- Lu, R.; Zhang, L.; Yang, X. Interaction between autophagy and the NLRP3 inflammasome in Alzheimer’s and Parkinson’s disease. Front. Aging Neurosci. 2022, 14, 1018848.
- Iqbal, K.; Liu, F.; Gong, C.-X.; Grundke-Iqbal, I. Tau in Alzheimer disease and related tauopathies. Curr. Alzheimer Res. 2010, 7, 656–664.
- Liu, L.; Chan, C. The role of inflammasome in Alzheimer’s disease. Ageing Res. Rev. 2014, 15, 6–15.
- Arumugam, S.; Qin, Y.; Liang, Z.; Han, S.-N.; Boodapati, S.T.; Li, J.; Lu, Q.; Flavell, R.A.; Mehal, W.Z.; Ouyang, X. GSK3β mediates the spatiotemporal dynamics of NLRP3 inflammasome activation. Cell Death Differ. 2022, 29, 2060–2069.
- Kang, S.-W.; Kim, S.J.; Kim, M.-S. Oxidative stress with tau hyperphosphorylation in memory impaired 1, 2-diacetylbenzene-treated mice. Toxicol. Lett. 2017, 279, 53–59.
- Biasizzo, M.; Kopitar-Jerala, N. Interplay between NLRP3 inflammasome and autophagy. Front. Immunol. 2020, 11, 591803.
- Liu, Z.; Li, T.; Li, P.; Wei, N.; Zhao, Z.; Liang, H.; Ji, X.; Chen, W.; Xue, M.; Wei, J. The ambiguous relationship of oxidative stress, tau hyperphosphorylation, and autophagy dysfunction in Alzheimer’s disease. Oxidative Med. Cell. Longev. 2015, 2015, 352723.
- Ren, P.; Chen, J.; Li, B.; Zhang, M.; Yang, B.; Guo, X.; Chen, Z.; Cheng, H.; Wang, P.; Wang, S. Nrf2 ablation promotes Alzheimer’s disease-like pathology in APP/PS1 transgenic mice: The role of neuroinflammation and oxidative stress. Oxidative Med. Cell. Longev. 2020, 2020, 3050971.
- Dempsey, C.; Araiz, A.R.; Bryson, K.; Finucane, O.; Larkin, C.; Mills, E.; Robertson, A.; Cooper, M.; O’Neill, L.; Lynch, M. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-β and cognitive function in APP/PS1 mice. Brain Behav. Immun. 2017, 61, 306–316.
- Flores, J.; Noël, A.; Foveau, B.; Beauchet, O.; LeBlanc, A.C. Pre-symptomatic Caspase-1 inhibitor delays cognitive decline in a mouse model of Alzheimer disease and aging. Nat. Commun. 2020, 11, 4571.
- Shukla, P.K.; Delotterie, D.F.; Xiao, J.; Pierre, J.F.; Rao, R.; McDonald, M.P.; Khan, M.M. Alterations in the gut-microbial-inflammasome-brain axis in a mouse model of Alzheimer’s disease. Cells 2021, 10, 779.
- Liu, S.; Gao, J.; Zhu, M.; Liu, K.; Zhang, H.-L. Gut microbiota and dysbiosis in Alzheimer’s disease: Implications for pathogenesis and treatment. Mol. Neurobiol. 2020, 57, 5026–5043.
- Luan, J.; Ju, D. Inflammasome: A double-edged sword in liver diseases. Front. Immunol. 2018, 9, 2201.
- de Brito Toscano, E.C.; Rocha, N.P.; Lopes, B.N.; Suemoto, C.K.; Teixeira, A.L. Neuroinflammation in Alzheimer’s disease: Focus on NLRP1 and NLRP3 inflammasomes. Curr. Protein Pept. Sci. 2021, 22, 584–598.
- Wu, P.-J.; Hung, Y.-F.; Liu, H.-Y.; Hsueh, Y.-P. Deletion of the inflammasome sensor Aim2 mitigates Aβ deposition and microglial activation but increases inflammatory cytokine expression in an Alzheimer disease mouse model. Neuroimmunomodulation 2017, 24, 29–39.
- Saadi, M.; Karkhah, A.; Pourabdolhossein, F.; Ataie, A.; Monif, M.; Nouri, H.R. Involvement of NLRC4 inflammasome through caspase-1 and IL-1β augments neuroinflammation and contributes to memory impairment in an experimental model of Alzheimer’s like disease. Brain Res. Bull. 2020, 154, 81–90.
- DeMaagd, G.; Philip, A. Parkinson’s disease and its management: Part 1: Disease entity, risk factors, pathophysiology, clinical presentation, and diagnosis. Pharm. Ther. 2015, 40, 504.
- Przedborski, S. Inflammation and Parkinson’s disease pathogenesis. Mov. Disord. 2010, 25, S55–S57.
- Nguyen, L.T.N.; Nguyen, H.D.; Kim, Y.J.; Nguyen, T.T.; Lai, T.T.; Lee, Y.K.; Ma, H.-i.; Kim, Y.E. Role of NLRP3 inflammasome in Parkinson’s disease and therapeutic considerations. J. Park. Dis. 2022, 12, 2117–2133.
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489.
- Murta, V.; Ferrari, C. Peripheral inflammation and demyelinating diseases. Adv. Exp. Med. Biol. 2016, 949, 263–285.
- Jewell, S.; Herath, A.M.; Gordon, R. Inflammasome activation in Parkinson’s disease. J. Park. Dis. 2022, 12, S113–S128.
- Versele, R.; Sevin, E.; Gosselet, F.; Fenart, L.; Candela, P. TNF-α and IL-1β modulate blood-brain barrier permeability and decrease amyloid-β peptide efflux in a human blood-brain barrier model. Int. J. Mol. Sci. 2022, 23, 10235.
- Faustini, G.; Bono, F.; Valerio, A.; Pizzi, M.; Spano, P.; Bellucci, A. Mitochondria and α-synuclein: Friends or foes in the pathogenesis of Parkinson’s disease? Genes 2017, 8, 377.
- Stefanis, L. α-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399.
- Li, Y.; Xia, Y.; Yin, S.; Wan, F.; Hu, J.; Kou, L.; Sun, Y.; Wu, J.; Zhou, Q.; Huang, J. Targeting microglial α-synuclein/TLRs/NF-kappaB/NLRP3 inflammasome axis in Parkinson’s disease. Front. Immunol. 2021, 12, 719807.
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687.
- Su, Q.; Ng, W.L.; Goh, S.Y.; Gulam, M.Y.; Wang, L.-F.; Tan, E.-K.; Ahn, M.; Chao, Y.-X. Targeting the inflammasome in Parkinson’s disease. Front. Aging Neurosci. 2022, 14, 957705.
- Kaushal, V.; Dye, R.; Pakavathkumar, P.; Foveau, B.; Flores, J.; Hyman, B.; Ghetti, B.; Koller, B.; LeBlanc, A. Neuronal NLRP1 inflammasome activation of Caspase-1 coordinately regulates inflammatory interleukin-1-beta production and axonal degeneration-associated Caspase-6 activation. Cell Death Differ. 2015, 22, 1676–1686.
- Bido, S.; Muggeo, S.; Massimino, L.; Marzi, M.J.; Giannelli, S.G.; Melacini, E.; Nannoni, M.; Gambarè, D.; Bellini, E.; Ordazzo, G. Microglia-specific overexpression of α-synuclein leads to severe dopaminergic neurodegeneration by phagocytic exhaustion and oxidative toxicity. Nat. Commun. 2021, 12, 6237.
- Wolf, D.; Ley, K. Immunity and inflammation in atherosclerosis. Circ. Res. 2019, 124, 315–327.
- Alfaddagh, A.; Martin, S.S.; Leucker, T.M.; Michos, E.D.; Blaha, M.J.; Lowenstein, C.J.; Jones, S.R.; Toth, P.P. Inflammation and cardiovascular disease: From mechanisms to therapeutics. Am. J. Prev. Card. 2020, 4, 100130.
- Packard, R.R.; Lichtman, A.H.; Libby, P. Innate and adaptive immunity in atherosclerosis. Semin. Immunopathol. 2009, 31, 5–22.
- Ilhan, F.; Kalkanli, S.T. Atherosclerosis and the role of immune cells. World J. Clin. Cases 2015, 3, 345.
- Piscopiello, M.; Sessa, M.; Anzalone, N.; Castellano, R.; Maisano, F.; Ferrero, E.; Chiesa, R.; Alfieri, O.; Comi, G.; Ferrero, M.E. P2X7 receptor is expressed in human vessels and might play a role in atherosclerosis. Int. J. Cardiol. 2013, 168, 2863–2866.
- Peng, K.; Liu, L.; Wei, D.; Lv, Y.; Wang, G.; Xiong, W.; Wang, X.; Altaf, A.; Wang, L.; He, D. P2X7R is involved in the progression of atherosclerosis by promoting NLRP3 inflammasome activation. Int. J. Mol. Med. 2015, 35, 1179–1188.
- Ganesan, R.; Henkels, K.M.; Wrenshall, L.E.; Kanaho, Y.; Di Paolo, G.; Frohman, M.A.; Gomez-Cambronero, J. Oxidized LDL phagocytosis during foam cell formation in atherosclerotic plaques relies on a PLD2–CD36 functional interdependence. J. Leukoc. Biol. 2018, 103, 867–883.
- Wang, R.; Wu, W.; Li, W.; Huang, S.; Li, Z.; Liu, R.; Shan, Z.; Zhang, C.; Li, W.; Wang, S. Activation of NLRP3 inflammasome promotes foam cell formation in vascular smooth muscle cells and atherogenesis via HMGB1. J. Am. Heart Assoc. 2018, 7, e008596.
- Varghese, J.F.; Patel, R.; Yadav, U.C. Sterol regulatory element binding protein (SREBP)-1 mediates oxidized low-density lipoprotein (oxLDL) induced macrophage foam cell formation through NLRP3 inflammasome activation. Cell. Signal. 2019, 53, 316–326.
- Maiolino, G.; Rossitto, G.; Caielli, P.; Bisogni, V.; Rossi, G.P.; Calò, L.A. The role of oxidized low-density lipoproteins in atherosclerosis: The myths and the facts. Mediat. Inflamm. 2013, 2013, 714653.
- Grebe, A.; Hoss, F.; Latz, E. NLRP3 inflammasome and the IL-1 pathway in atherosclerosis. Circ. Res. 2018, 122, 1722–1740.
- Karasawa, T.; Takahashi, M. Role of NLRP3 inflammasomes in atherosclerosis. J. Atheroscler. Thromb. 2017, 24, 443–451.
- Doran, A.C.; Meller, N.; McNamara, C.A. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 812–819.
- Kaplanski, G. Interleukin-18: Biological properties and role in disease pathogenesis. Immunol. Rev. 2018, 281, 138–153.
- Mallat, Z.; Corbaz, A.; Scoazec, A.; Besnard, S.; Lesèche, G.; Chvatchko, Y.; Tedgui, A. Expression of interleukin-18 in human atherosclerotic plaques and relation to plaque instability. Circulation 2001, 104, 1598–1603.
- Rezaieyazdi, Z.; AkbariRad, M.; Saadati, N.; Salari, M.; Orang, R.; Sedighi, S.; Esmaily, H.; Azarpazhooh, M.R.; Firoozi, A.; Akbarpour, E. Serum interleukin-18 and its relationship with subclinical atherosclerosis in systemic lupus erythematosus. ARYA Atheroscler. 2021, 17, 1–6.
- Yu, L.; Wang, L.; Chen, S. Endogenous toll-like receptor ligands and their biological significance. J. Cell. Mol. Med. 2010, 14, 2592–2603.
- Vargas, A.M.; Rivera-Rodriguez, D.E.; Martinez, L.R. Methamphetamine alters the TLR4 signaling pathway, NF-κB activation, and pro-inflammatory cytokine production in LPS-challenged NR-9460 microglia-like cells. Mol. Immunol. 2020, 121, 159–166.
- Čokić, V.P.; Mitrović-Ajtić, O.; Beleslin-Čokić, B.B.; Marković, D.; Buač, M.; Diklić, M.; Kraguljac-Kurtović, N.; Damjanović, S.; Milenković, P.; Gotić, M. Proinflammatory cytokine IL-6 and JAK-STAT signaling pathway in myeloproliferative neoplasms. Mediat. Inflamm. 2015, 2015, 453020.
- Xu, J.; Lu, X.; Shi, G.-P. Vasa vasorum in atherosclerosis and clinical significance. Int. J. Mol. Sci. 2015, 16, 11574–11608.
- Hameed, I.; Masoodi, S.R.; Mir, S.A.; Nabi, M.; Ghazanfar, K.; Ganai, B.A. Type 2 diabetes mellitus: From a metabolic disorder to an inflammatory condition. World J. Diabetes 2015, 6, 598.
- Galicia-Garcia, U.; Benito-Vicente, A.; Jebari, S.; Larrea-Sebal, A.; Siddiqi, H.; Uribe, K.B.; Ostolaza, H.; Martín, C. Pathophysiology of type 2 diabetes mellitus. Int. J. Mol. Sci. 2020, 21, 6275.
- Sjöholm, Å.; Nyström, T. Inflammation and the etiology of type 2 diabetes. Diabetes. Metab. Res. Rev. 2006, 22, 4–10.
- Chen, X.; Zhang, D.; Li, Y.; Wang, W.; Bei, W.; Guo, J. NLRP3 inflammasome and IL-1β pathway in type 2 diabetes and atherosclerosis: Friend or foe? Pharmacol. Res. 2021, 173, 105885.
- Lee, H.-M.; Kim, J.-J.; Kim, H.J.; Shong, M.; Ku, B.J.; Jo, E.-K. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes 2013, 62, 194–204.
- Masters, S.L.; Dunne, A.; Subramanian, S.L.; Hull, R.L.; Tannahill, G.M.; Sharp, F.A.; Becker, C.; Franchi, L.; Yoshihara, E.; Chen, Z. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat. Immunol. 2010, 11, 897–904.
- Luo, B.; Li, B.; Wang, W.; Liu, X.; Xia, Y.; Zhang, C.; Zhang, M.; Zhang, Y.; An, F. NLRP3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model. PLoS ONE 2014, 9, e104771.
- Kim, Y.; Wang, W.; Okla, M.; Kang, I.; Moreau, R.; Chung, S. Suppression of NLRP3 inflammasome by γ-tocotrienol ameliorates type 2 diabetes. J. Lipid Res. 2016, 57, 66–76.
- Jourdan, T.; Godlewski, G.; Cinar, R.; Bertola, A.; Szanda, G.; Liu, J.; Tam, J.; Han, T.; Mukhopadhyay, B.; Skarulis, M.C. Activation of the Nlrp3 inflammasome in infiltrating macrophages by endocannabinoids mediates beta cell loss in type 2 diabetes. Nat. Med. 2013, 19, 1132–1140.
- Sokolova, M.; Sahraoui, A.; Høyem, M.; Øgaard, J.; Lien, E.; Aukrust, P.; Yndestad, A.; Ranheim, T.; Scholz, H. NLRP3 inflammasome mediates oxidative stress-induced pancreatic islet dysfunction. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E912–E923.
- Marchetti, P.; Bugliani, M.; De Tata, V.; Suleiman, M.; Marselli, L. Pancreatic beta cell identity in humans and the role of type 2 diabetes. Front. Cell Dev. Biol. 2017, 5, 55.
- Eguchi, N.; Vaziri, N.D.; Dafoe, D.C.; Ichii, H. The role of oxidative stress in pancreatic β cell dysfunction in diabetes. Int. J. Mol. Sci. 2021, 22, 1509.
- Tsalamandris, S.; Antonopoulos, A.S.; Oikonomou, E.; Papamikroulis, G.-A.; Vogiatzi, G.; Papaioannou, S.; Deftereos, S.; Tousoulis, D. The role of inflammation in diabetes: Current concepts and future perspectives. Eur. Cardiol. Rev. 2019, 14, 50.
- Chen, L.; Chen, R.; Wang, H.; Liang, F. Mechanisms linking inflammation to insulin resistance. Int. J. Endocrinol. 2015, 2015, 508409.
- Lee, Y.H.; Giraud, J.; Davis, R.J.; White, M.F. c-Jun N-terminal kinase (JNK) mediates feedback inhibition of the insulin signaling cascade. J. Biol. Chem. 2003, 278, 2896–2902.
- Goldfine, A.B.; Shoelson, S.E. Therapeutic approaches targeting inflammation for diabetes and associated cardiovascular risk. J. Clin. Investig. 2017, 127, 83–93.
- Wang, Y.; Yu, B.; Wang, L.; Yang, M.; Xia, Z.; Wei, W.; Zhang, F.; Yuan, X. Pioglitazone ameliorates glomerular NLRP3 inflammasome activation in apolipoprotein E knockout mice with diabetes mellitus. PLoS ONE 2017, 12, e0181248.
- Song, S.; Guo, R.; Mehmood, A.; Zhang, L.; Yin, B.; Yuan, C.; Zhang, H.; Guo, L.; Li, B. Liraglutide attenuate central nervous inflammation and demyelination through AMPK and pyroptosis-related NLRP3 pathway. CNS Neurosci. Ther. 2022, 28, 422–434.
- McArdle, M.A.; Finucane, O.M.; Connaughton, R.M.; McMorrow, A.M.; Roche, H.M. Mechanisms of obesity-induced inflammation and insulin resistance: Insights into the emerging role of nutritional strategies. Front. Endocrinol. 2013, 4, 52.
- Manna, P.; Jain, S.K. Obesity, oxidative stress, adipose tissue dysfunction, and the associated health risks: Causes and therapeutic strategies. Metab. Syndr. Relat. Disord. 2015, 13, 423–444.
- Welsh, P.; Polisecki, E.; Robertson, M.; Jahn, S.; Buckley, B.M.; de Craen, A.J.; Ford, I.; Jukema, J.W.; Macfarlane, P.W.; Packard, C.J. Unraveling the directional link between adiposity and inflammation: A bidirectional Mendelian randomization approach. J. Clin. Endocrinol. Metab. 2010, 95, 93–99.
- Vandanmagsar, B.; Youm, Y.-H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188.
- Youm, Y.-H.; Adijiang, A.; Vandanmagsar, B.; Burk, D.; Ravussin, A.; Dixit, V.D. Elimination of the NLRP3-ASC inflammasome protects against chronic obesity-induced pancreatic damage. Endocrinology 2011, 152, 4039–4045.
- Wu, K.K.-L.; Cheung, S.W.-M.; Cheng, K.K.-Y. NLRP3 inflammasome activation in adipose tissues and its implications on metabolic diseases. Int. J. Mol. Sci. 2020, 21, 4184.
- Sokolova, M.; Yang, K.; Hansen, S.H.; Louwe, M.C.; Kummen, M.; Hov, J.E.; Sjaastad, I.; Berge, R.K.; Halvorsen, B.; Aukrust, P. NLRP3 inflammasome deficiency attenuates metabolic disturbances involving alterations in the gut microbial profile in mice exposed to high fat diet. Sci. Rep. 2020, 10, 21006.
- Javaid, H.M.A.; Sahar, N.E.; ZhuGe, D.-L.; Huh, J.Y. Exercise inhibits NLRP3 inflammasome activation in obese mice via the anti-inflammatory effect of meteorin-like. Cells 2021, 10, 3480.
- Hammerschmidt, P.; Brüning, J.C. Contribution of specific ceramides to obesity-associated metabolic diseases. Cell. Mol. Life Sci. 2022, 79, 395.
- Stienstra, R.; Van Diepen, J.A.; Tack, C.J.; Zaki, M.H.; Van De Veerdonk, F.L.; Perera, D.; Neale, G.A.; Hooiveld, G.J.; Hijmans, A.; Vroegrijk, I. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2011, 108, 15324–15329.
- Hara, H.; Tsuchiya, K.; Kawamura, I.; Fang, R.; Hernandez-Cuellar, E.; Shen, Y.; Mizuguchi, J.; Schweighoffer, E.; Tybulewicz, V.; Mitsuyama, M. Phosphorylation of the adaptor ASC acts as a molecular switch that controls the formation of speck-like aggregates and inflammasome activity. Nat. Immunol. 2013, 14, 1247–1255.
- Nagar, A.; Rahman, T.; Harton, J.A. The ASC speck and NLRP3 inflammasome function are spatially and temporally distinct. Front. Immunol. 2021, 12, 752482.
- Beckley, A.J.; Lan, L.-Q.; Aono, S.; Wang, L.; Shi, J.N. Caspase-1 activation and mature interleukin-1β release are uncoupled events in monocytes. World J. Biol. Chem. 2013, 4, 30–34.
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.-L. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid. Redox Signal. 2015, 22, 1111–1129.
- Picó, C.; Palou, M.; Pomar, C.A.; Rodríguez, A.M.; Palou, A. Leptin as a key regulator of the adipose organ. Rev. Endocr. Metab. Disord. 2022, 23, 13–30.
- Rahman, M.S.; Hossain, K.S.; Das, S.; Kundu, S.; Adegoke, E.O.; Rahman, M.A.; Hannan, M.A.; Uddin, M.J.; Pang, M.-G. Role of insulin in health and disease: An update. Int. J. Mol. Sci. 2021, 22, 6403.
- Baral, A.; Park, P.-H. Leptin Induces Apoptotic and Pyroptotic Cell Death via NLRP3 Inflammasome Activation in Rat Hepatocytes. Int. J. Mol. Sci. 2021, 22, 12589.
- Iikuni, N.; Kwan Lam, Q.L.; Lu, L.; Matarese, G.; Cava, A.L. Leptin and inflammation. Curr. Immunol. Rev. 2008, 4, 70–79.
- Ding, S.; Xu, S.; Ma, Y.; Liu, G.; Jang, H.; Fang, J. Modulatory mechanisms of the NLRP3 inflammasomes in diabetes. Biomolecules 2019, 9, 850.
- Chang, Y.-W.; Hung, L.-C.; Chen, Y.-C.; Wang, W.-H.; Lin, C.-Y.; Tzeng, H.-H.; Suen, J.-L.; Chen, Y.-H. Insulin reduces inflammation by regulating the activation of the NLRP3 inflammasome. Front. Immunol. 2021, 11, 587229.
- Guo, C.; Fu, R.; Wang, S.; Huang, Y.; Li, X.; Zhou, M.; Zhao, J.; Yang, N. NLRP3 inflammasome activation contributes to the pathogenesis of rheumatoid arthritis. Clin. Exp. Immunol. 2018, 194, 231–243.
- Spel, L.; Martinon, F. Inflammasomes contributing to inflammation in arthritis. Immunol. Rev. 2020, 294, 48–62.
- Tourkochristou, E.; Aggeletopoulou, I.; Konstantakis, C.; Triantos, C. Role of NLRP3 inflammasome in inflammatory bowel diseases. World J. Gastroenterol. 2019, 25, 4796.
- Martinon, F.; Pétrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241.
- Kingsbury, S.R.; Conaghan, P.G.; McDermott, M.F. The role of the NLRP3 inflammasome in gout. J. Inflamm. Res. 2011, 4, 39–49.
- Ciążyńska, M.; Olejniczak-Staruch, I.; Sobolewska-Sztychny, D.; Narbutt, J.; Skibińska, M.; Lesiak, A. The role of NLRP1, NLRP3, and AIM2 inflammasomes in psoriasis. Int. J. Mol. Sci. 2021, 22, 5898.
- Forouzandeh, M.; Besen, J.; Keane, R.W.; de Rivero Vaccari, J.P. The inflammasome signaling proteins ASC and IL-18 as biomarkers of psoriasis. Front. Pharmacol. 2020, 11, 1238.
- Verma, D.; Fekri, S.Z.; Sigurdardottir, G.; Eding, C.B.; Sandin, C.; Enerbäck, C. Enhanced inflammasome activity in patients with psoriasis promotes systemic inflammation. J. Invest. Dermatol. 2021, 141, 586–595.e5.
- Tervaniemi, M.H.; Katayama, S.; Skoog, T.; Siitonen, H.A.; Vuola, J.; Nuutila, K.; Sormunen, R.; Johnsson, A.; Linnarsson, S.; Suomela, S. NOD-like receptor signaling and inflammasome-related pathways are highlighted in psoriatic epidermis. Sci. Rep. 2016, 6, 22745.
- Yang, Q.; Yu, C.; Yang, Z.; Wei, Q.; Mu, K.; Zhang, Y.; Zhao, W.; Wang, X.; Huai, W.; Han, L. Deregulated NLRP3 and NLRP1 inflammasomes and their correlations with disease activity in systemic lupus erythematosus. J. Rheumatol. 2014, 41, 444–452.
- Pontillo, A.; Girardelli, M.; Kamada, A.J.; Pancotto, J.A.; Donadi, E.A.; Crovella, S.; Sandrin-Garcia, P. Polimorphisms in inflammasome genes are involved in the predisposition to systemic lupus erythematosus. Autoimmunity 2012, 45, 271–278.
- da Cruz, H.L.A.; Cavalcanti, C.A.J.; de Azêvedo Silva, J.; de Lima, C.A.D.; Fragoso, T.S.; Barbosa, A.D.; Dantas, A.T.; de Ataíde Mariz, H.; Duarte, A.L.B.P.; Pontillo, A. Differential expression of the inflammasome complex genes in systemic lupus erythematosus. Immunogenetics 2020, 72, 217–224.
- Kahlenberg, J.M.; Thacker, S.G.; Berthier, C.C.; Cohen, C.D.; Kretzler, M.; Kaplan, M.J. Inflammasome activation of IL-18 results in endothelial progenitor cell dysfunction in systemic lupus erythematosus. J. Immunol. 2011, 187, 6143–6156.
- Ma, Z.-Z.; Sun, H.-S.; Lv, J.-C.; Guo, L.; Yang, Q.-R. Expression and clinical significance of the NEK7-NLRP3 inflammasome signaling pathway in patients with systemic lupus erythematosus. J. Inflamm. 2018, 15, 16.

