 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Matthew Philip Hardy | -- | 3444 | 2023-11-15 10:14:56 | | | |
| 2 | Lindsay Dong | -11 word(s) | 3433 | 2023-11-17 02:00:01 | | |
Video Upload Options
Human complement receptor 1 (CR1) is a membrane-bound regulator of complement that has been the subject of attempts to generate soluble therapeutic compounds comprising different fragments of its extracellular domain.
1. Introduction
CR1 is a type I membrane glycoprotein expressed on the surface of erythrocytes (E-CR1) and immune cells that acts as a central regulator of the classical, lectin, and alternative pathways of the complement system [1][2][3][4]. The complement system is an integral part of the innate immune response and has been extensively reviewed [5][6][7][8][9][10]. The predominant allelic variant of CR1 (CR1*1) has a large, flexible extracellular domain comprised of 30 highly homologous domains called short consensus repeats (SCRs), followed by a transmembrane domain and a short, 43-amino acid cytoplasmic tail [11][12]. SCR domains 1–28 are arranged in groups of seven to form four larger units: long homologous repeat (LHR) domains -A to -D (Figure 1A) [11][13][14][15]. Including SCR domains 29–30 into an expanded definition of LHR-D has been performed routinely [16][17][18].
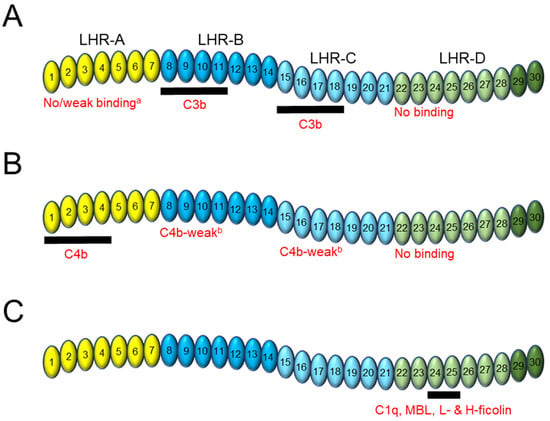
2. CR1 Binding to C3b and C4b
2.1. General and Comparative
2.2. Domain Contribution
3. CR1 Binding to Other Ligands
4. Structural Data
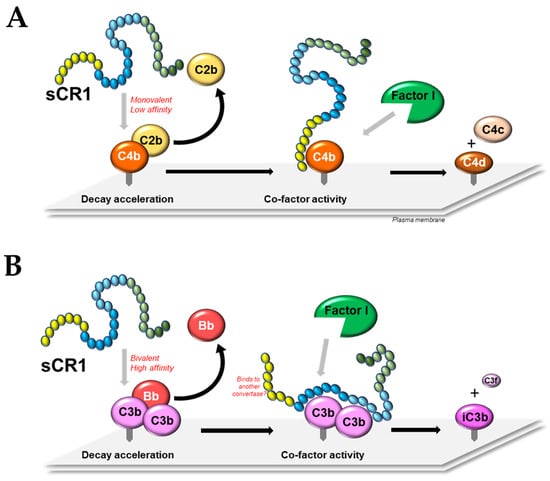
5. Decay Acceleration Activity of CR1
5.1. Classical/Lectin Pathway C3 and C5 Convertases
5.2. Alternative Pathway C3 and C5 Convertases
6. Co-Factor Activity of CR1
6.1. General and Comparative
6.2. Domain Contribution
7. Domain Contribution to CR1-Mediated Complement Pathway Inhibition
8. CSL040 and Its Mechanism of Action
9. Conclusions
References
- Fearon, D.T. Identification of the membrane glycoprotein that is the C3b receptor of the human erythrocyte, polymorphonuclear leukocyte, B lymphocyte, and monocyte. J. Exp. Med. 1980, 152, 20–30.
- Ross, G.D.; Lambris, J.D. Identification of a C3bi-specific membrane complement receptor that is expressed on lymphocytes, monocytes, neutrophils, and erythrocytes. J. Exp. Med. 1982, 155, 96–110.
- Ahearn, J.M.; Fearon, D.T. Structure and function of the complement receptors, CR1 (CD35) and CR2 (CD21). Adv. Immunol. 1989, 46, 183–219.
- Lublin, D.M.; Griffith, R.C.; Atkinson, J.P. Influence of glycosylation on allelic and cell-specific Mr variation, receptor processing, and ligand binding of the human complement C3b/C4b receptor. J. Biol. Chem. 1986, 261, 5736–5744.
- Lachmann, P.J. Biological functions of the complement system. Biochem. Soc. Trans. 1990, 18, 1143–1145.
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A key system for immune surveillance and homeostasis. Nat. Immunol. 2010, 11, 785–797.
- Kolev, M.; Le Friec, G.; Kemper, C. Complement—Tapping into new sites and effector systems. Nat. Rev. Immunol. 2014, 14, 811–820.
- Lachmann, P.J. The amplification loop of the complement pathways. Adv. Immunol. 2009, 104, 115–149.
- Pangburn, M.K. Initiation of the alternative pathway of complement and the history of “tickover”. Immunol. Rev. 2022, 313, 64–70.
- Wagner, E.; Frank, M.M. Therapeutic potential of complement modulation. Nat. Rev. Drug Discov. 2010, 9, 43–56.
- Krych-Goldberg, M.; Atkinson, J.P. Structure-function relationships of complement receptor type 1. Immunol. Rev. 2001, 180, 112–122.
- Furtado, P.B.; Huang, C.Y.; Ihyembe, D.; Hammond, R.A.; Marsh, H.C.; Perkins, S.J. The partly folded back solution structure arrangement of the 30 SCR domains in human complement receptor type 1 (CR1) permits access to its C3b and C4b ligands. J. Mol. Biol. 2008, 375, 102–118.
- Weis, J.H.; Morton, C.C.; Bruns, G.A.; Weis, J.J.; Klickstein, L.B.; Wong, W.W.; Fearon, D.T. A complement receptor locus: Genes encoding C3b/C4b receptor and C3d/Epstein-Barr virus receptor map to 1q32. J. Immunol. 1987, 138, 312–315.
- Klickstein, L.B.; Wong, W.W.; Smith, J.A.; Weis, J.H.; Wilson, J.G.; Fearon, D.T. Human C3b/C4b receptor (CR1). Demonstration of long homologous repeating domains that are composed of the short consensus repeats characteristics of C3/C4 binding proteins. J. Exp. Med. 1987, 165, 1095–1112.
- Liu, D.; Niu, Z.X. The structure, genetic polymorphisms, expression and biological functions of complement receptor type 1 (CR1/CD35). Immunopharmacol. Immunotoxicol. 2009, 31, 524–535.
- Kalli, K.R.; Hsu, P.H.; Bartow, T.J.; Ahearn, J.M.; Matsumoto, A.K.; Klickstein, L.B.; Fearon, D.T. Mapping of the C3b-binding site of CR1 and construction of a (CR1)2-F(ab’)2 chimeric complement inhibitor. J. Exp. Med. 1991, 174, 1451–1460.
- Mqadmi, A.; Abdullah, Y.; Yazdanbakhsh, K. Characterization of complement receptor 1 domains for prevention of complement-mediated red cell destruction. Transfusion 2005, 45, 234–244.
- Wymann, S.; Dai, Y.; Nair, A.G.; Cao, H.; Powers, G.A.; Schnell, A.; Martin-Roussety, G.; Leong, D.; Simmonds, J.; Lieu, K.G.; et al. A novel soluble complement receptor 1 fragment with enhanced therapeutic potential. J. Biol. Chem. 2021, 296, 100200.
- Fearon, D.T. Regulation of the amplification C3 convertase of human complement by an inhibitory protein isolated from human erythrocyte membrane. Proc. Natl. Acad. Sci. USA 1979, 76, 5867–5871.
- Karthikeyan, G.; Baalasubramanian, S.; Seth, S.; Das, N. Low levels of plasma soluble complement receptor type 1 in patients receiving thrombolytic therapy for acute myocardial infarction. J. Thromb. Thrombolysis 2007, 23, 115–120.
- Weisman, H.F.; Bartow, T.; Leppo, M.K.; Marsh, H.C., Jr.; Carson, G.R.; Concino, M.F.; Boyle, M.P.; Roux, K.H.; Weisfeldt, M.L.; Fearon, D.T. Soluble human complement receptor type 1: In vivo inhibitor of complement suppressing post-ischemic myocardial inflammation and necrosis. Science 1990, 249, 146–151.
- Makrides, S.C.; Scesney, S.M.; Ford, P.J.; Evans, K.S.; Carson, G.R.; Marsh, H.C., Jr. Cell surface expression of the C3b/C4b receptor (CR1) protects Chinese hamster ovary cells from lysis by human complement. J. Biol. Chem. 1992, 267, 24754–24761.
- Schramm, E.C.; Roumenina, L.T.; Rybkine, T.; Chauvet, S.; Vieira-Martins, P.; Hue, C.; Maga, T.; Valoti, E.; Wilson, V.; Jokiranta, S.; et al. Mapping interactions between complement C3 and regulators using mutations in atypical hemolytic uremic syndrome. Blood 2015, 125, 2359–2369.
- Ross, G.D. Analysis of the different types of leukocyte membrane complement receptors and their interaction with the complement system. J. Immunol. Methods 1980, 37, 197–211.
- Klickstein, L.B.; Bartow, T.J.; Miletic, V.; Rabson, L.D.; Smith, J.A.; Fearon, D.T. Identification of distinct C3b and C4b recognition sites in the human C3b/C4b receptor (CR1, CD35) by deletion mutagenesis. J. Exp. Med. 1988, 168, 1699–1717.
- Pangburn, M.K. Differences between the binding sites of the complement regulatory proteins DAF, CR1, and factor H on C3 convertases. J. Immunol. 1986, 136, 2216–2221.
- Belt, K.T.; Carroll, M.C.; Porter, R.R. The structural basis of the multiple forms of human complement component C4. Cell 1984, 36, 907–914.
- Clemenza, L.; Isenman, D.E. The C4A and C4B isotypic forms of human complement fragment C4b have the same intrinsic affinity for complement receptor 1 (CR1/CD35). J. Immunol. 2004, 172, 1670–1680.
- Gatenby, P.A.; Barbosa, J.E.; Lachmann, P.J. Differences between C4A and C4B in the handling of immune complexes: The enhancement of CR1 binding is more important than the inhibition of immunoprecipitation. Clin. Exp. Immunol. 1990, 79, 158–163.
- Reilly, B.D.; Mold, C. Quantitative analysis of C4Ab and C4Bb binding to the C3b/C4b receptor (CR1, CD35). Clin. Exp. Immunol. 1997, 110, 310–316.
- Prohaska, R.; Adolf, G.R. Characterization of the human erythrocyte complement receptor CR1 (C3b receptor) by epitope mapping. Immunobiology 1987, 174, 93–106.
- Krych, M.; Clemenza, L.; Howdeshell, D.; Hauhart, R.; Hourcade, D.; Atkinson, J.P. Analysis of the functional domains of complement receptor type 1 (C3b/C4b receptor; CD35) by substitution mutagenesis. J. Biol. Chem. 1994, 269, 13273–13278.
- Krych, M.; Hauhart, R.; Atkinson, J.P. Structure-function analysis of the active sites of complement receptor type 1. J. Biol. Chem. 1998, 273, 8623–8629.
- Scesney, S.M.; Makrides, S.C.; Gosselin, M.L.; Ford, P.J.; Andrews, B.M.; Hayman, E.G.; Marsh, H.C., Jr. A soluble deletion mutant of the human complement receptor type 1, which lacks the C4b binding site, is a selective inhibitor of the alternative complement pathway. Eur. J. Immunol. 1996, 26, 1729–1735.
- Krych, M.; Hourcade, D.; Atkinson, J.P. Sites within the complement C3b/C4b receptor important for the specificity of ligand binding. Proc. Natl. Acad. Sci. USA 1991, 88, 4353–4357.
- Yazdanbakhsh, K.; Kang, S.; Tamasauskas, D.; Sung, D.; Scaradavou, A. Complement receptor 1 inhibitors for prevention of immune-mediated red cell destruction: Potential use in transfusion therapy. Blood 2003, 101, 5046–5052.
- Wong, W.W.; Farrell, S.A. Proposed structure of the F’ allotype of human CR1. Loss of a C3b binding site may be associated with altered function. J. Immunol. 1991, 146, 656–662.
- Smith, B.O.; Mallin, R.L.; Krych-Goldberg, M.; Wang, X.; Hauhart, R.E.; Bromek, K.; Uhrin, D.; Atkinson, J.P.; Barlow, P.N. Structure of the C3b binding site of CR1 (CD35), the immune adherence receptor. Cell 2002, 108, 769–780.
- Reilly, B.D.; Makrides, S.C.; Ford, P.J.; Marsh, H.C., Jr.; Mold, C. Quantitative analysis of C4b dimer binding to distinct sites on the C3b/C4b receptor (CR1). J. Biol. Chem. 1994, 269, 7696–7701.
- Korb, L.C.; Ahearn, J.M. C1q binds directly and specifically to surface blebs of apoptotic human keratinocytes: Complement deficiency and systemic lupus erythematosus revisited. J. Immunol. 1997, 158, 4525–4528.
- Sontheimer, R.D.; Racila, E.; Racila, D.M. C1q: Its functions within the innate and adaptive immune responses and its role in lupus autoimmunity. J. Investig. Dermatol. 2005, 125, 14–23.
- Klickstein, L.B.; Barbashov, S.F.; Liu, T.; Jack, R.M.; Nicholson-Weller, A. Complement receptor type 1 (CR1, CD35) is a receptor for C1q. Immunity 1997, 7, 345–355.
- Tas, S.W.; Klickstein, L.B.; Barbashov, S.F.; Nicholson-Weller, A. C1q and C4b bind simultaneously to CR1 and additively support erythrocyte adhesion. J. Immunol. 1999, 163, 5056–5063.
- Ghiran, I.; Barbashov, S.F.; Klickstein, L.B.; Tas, S.W.; Jensenius, J.C.; Nicholson-Weller, A. Complement receptor 1/CD35 is a receptor for mannan-binding lectin. J. Exp. Med. 2000, 192, 1797–1808.
- Forneris, F.; Wu, J.; Xue, X.; Ricklin, D.; Lin, Z.; Sfyroera, G.; Tzekou, A.; Volokhina, E.; Granneman, J.C.; Hauhart, R.; et al. Regulators of complement activity mediate inhibitory mechanisms through a common C3b-binding mode. EMBO J. 2016, 35, 1133–1149.
- Iida, K.; Nussenzweig, V. Complement receptor is an inhibitor of the complement cascade. J. Exp. Med. 1981, 153, 1138–1150.
- Takata, Y.; Kinoshita, T.; Kozono, H.; Takeda, J.; Tanaka, E.; Hong, K.; Inoue, K. Covalent association of C3b with C4b within C5 convertase of the classical complement pathway. J. Exp. Med. 1987, 165, 1494–1507.
- Krych-Goldberg, M.; Hauhart, R.E.; Subramanian, V.B.; Yurcisin, B.M., 2nd; Crimmins, D.L.; Hourcade, D.E.; Atkinson, J.P. Decay accelerating activity of complement receptor type 1 (CD35). Two active sites are required for dissociating C5 convertases. J. Biol. Chem. 1999, 274, 31160–31168.
- Krych-Goldberg, M.; Hauhart, R.E.; Porzukowiak, T.; Atkinson, J.P. Synergy between two active sites of human complement receptor type 1 (CD35) in complement regulation: Implications for the structure of the classical pathway C3 convertase and generation of more potent inhibitors. J. Immunol. 2005, 175, 4528–4535.
- Kinoshita, T.; Takata, Y.; Kozono, H.; Takeda, J.; Hong, K.S.; Inoue, K. C5 convertase of the alternative complement pathway: Covalent linkage between two C3b molecules within the trimolecular complex enzyme. J. Immunol. 1988, 141, 3895–3901.
- Ross, G.D.; Lambris, J.D.; Cain, J.A.; Newman, S.L. Generation of three different fragments of bound C3 with purified factor I or serum. I. Requirements for factor H vs. CR1 cofactor activity. J. Immunol. 1982, 129, 2051–2060.
- Medof, M.E.; Iida, K.; Mold, C.; Nussenzweig, V. Unique role of the complement receptor CR1 in the degradation of C3b associated with immune complexes. J. Exp. Med. 1982, 156, 1739–1754.
- Smith, R.A. Targeting anticomplement agents. Biochem. Soc. Trans. 2002, 30, 1037–1041.
- Mossakowska, D.; Dodd, I.; Pindar, W.; Smith, R.A. Structure-activity relationships within the N-terminal short consensus repeats (SCR) of human CR1 (C3b/C4b receptor, CD35): SCR 3 plays a critical role in inhibition of the classical and alternative pathways of complement activation. Eur. J. Immunol. 1999, 29, 1955–1965.
- Hardy, M.P.; Rowe, T.; Wymann, S. Soluble Complement Receptor 1 Therapeutics. J. Immunol. Sci. 2022, 6, 1–17.
- Bongoni, A.K.; Vikstrom, I.B.; McRae, J.L.; Salvaris, E.J.; Fisicaro, N.; Pearse, M.J.; Wymann, S.; Rowe, T.; Morelli, A.B.; Hardy, M.P.; et al. A potent truncated form of human soluble CR1 is protective in a mouse model of renal ischemia-reperfusion injury. Sci. Rep. 2021, 11, 21873.
- Wymann, S.; Mischnik, M.; Leong, D.; Ghosh, S.; Tan, X.; Cao, H.; Kuehnemuth, B.; Powers, G.A.; Halder, P.; de Souza, M.J.; et al. Sialylation-dependent pharmacokinetics and differential complement pathway inhibition are hallmarks of CR1 activity in vivo. Biochem. J. 2022, 479, 1007–1030.

