 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Niyati Seshagiri Sharma | -- | 4856 | 2023-11-15 06:45:17 | | | |
| 2 | Peter Tang | + 2 word(s) | 4858 | 2023-11-15 09:41:33 | | |
Video Upload Options
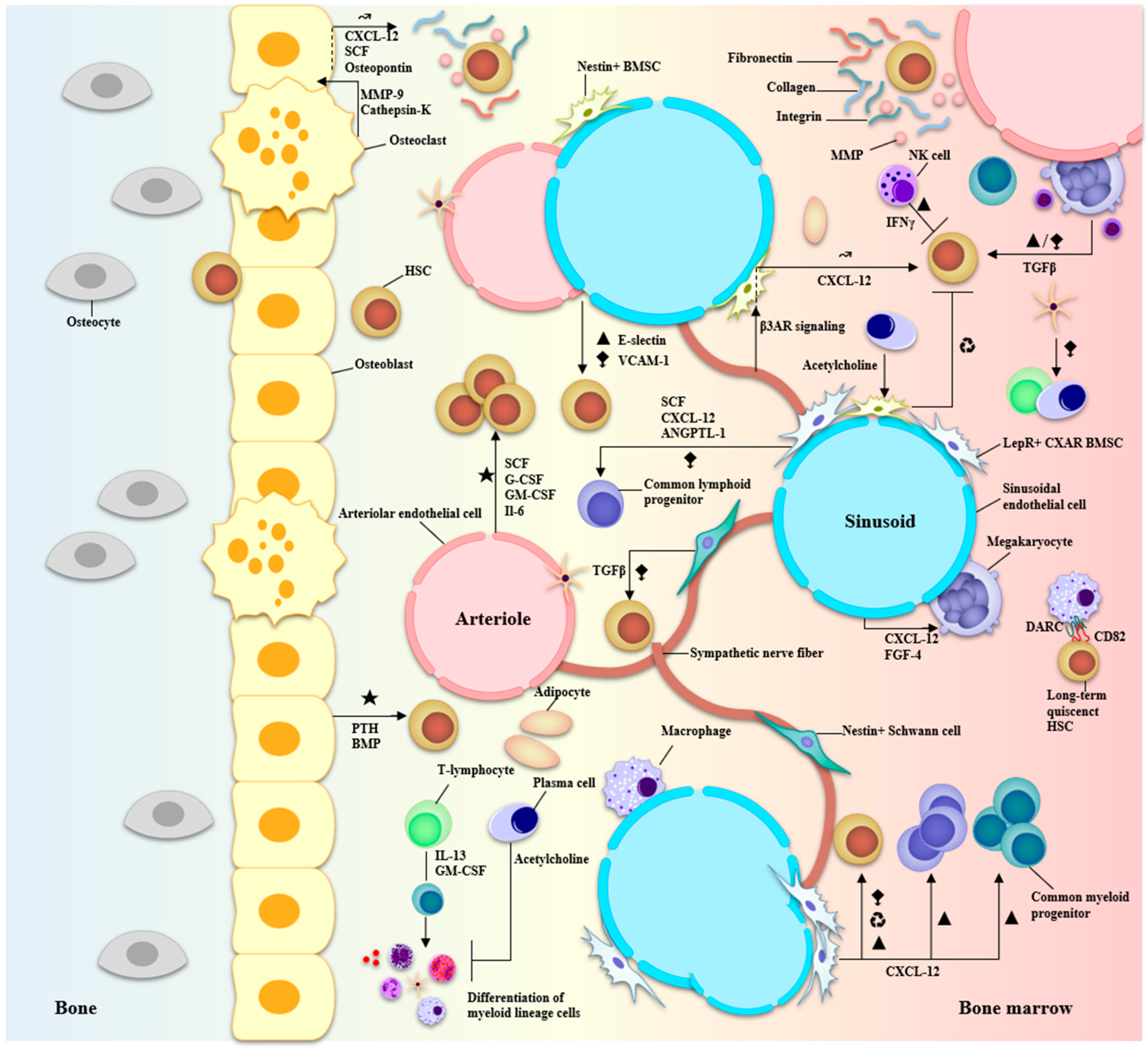
Multiple myeloma (MM) is a dyscrasia of plasma cells (PCs) characterized by abnormal immunoglobulin (Ig) production. The disease remains incurable due to a multitude of mutations and structural abnormalities in MM cells, coupled with a favorable microenvironment and immune suppression that eventually contribute to the development of drug resistance. The bone marrow microenvironment (BMME) is composed of a cellular component comprising stromal cells, endothelial cells, osteoclasts, osteoblasts, and immune cells, and a non-cellular component made of the extracellular matrix (ECM) and the liquid milieu, which contains cytokines, growth factors, and chemokines. The bone marrow stromal cells (BMSCs) are involved in the adhesion of MM cells, promote the growth, proliferation, invasion, and drug resistance of MM cells, and are also crucial in angiogenesis and the formation of lytic bone lesions. Classical immunophenotyping in combination with advanced immune profiling using single-cell sequencing technologies has enabled immune cell-specific gene expression analysis in MM to further elucidate the roles of specific immune cell fractions from peripheral blood and bone marrow (BM) in myelomagenesis and progression, immune evasion and exhaustion mechanisms, and development of drug resistance and relapse.
1. Introduction
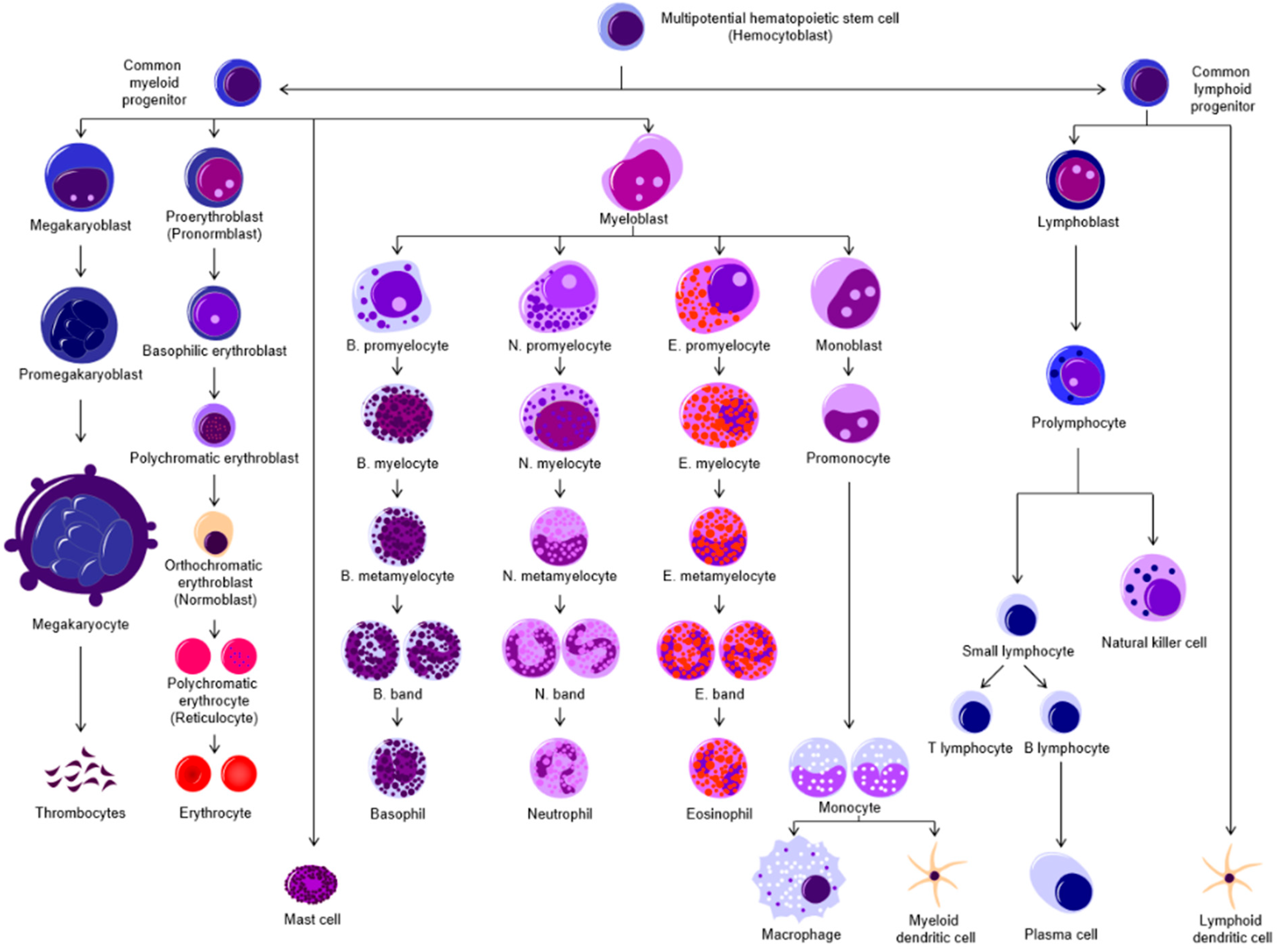

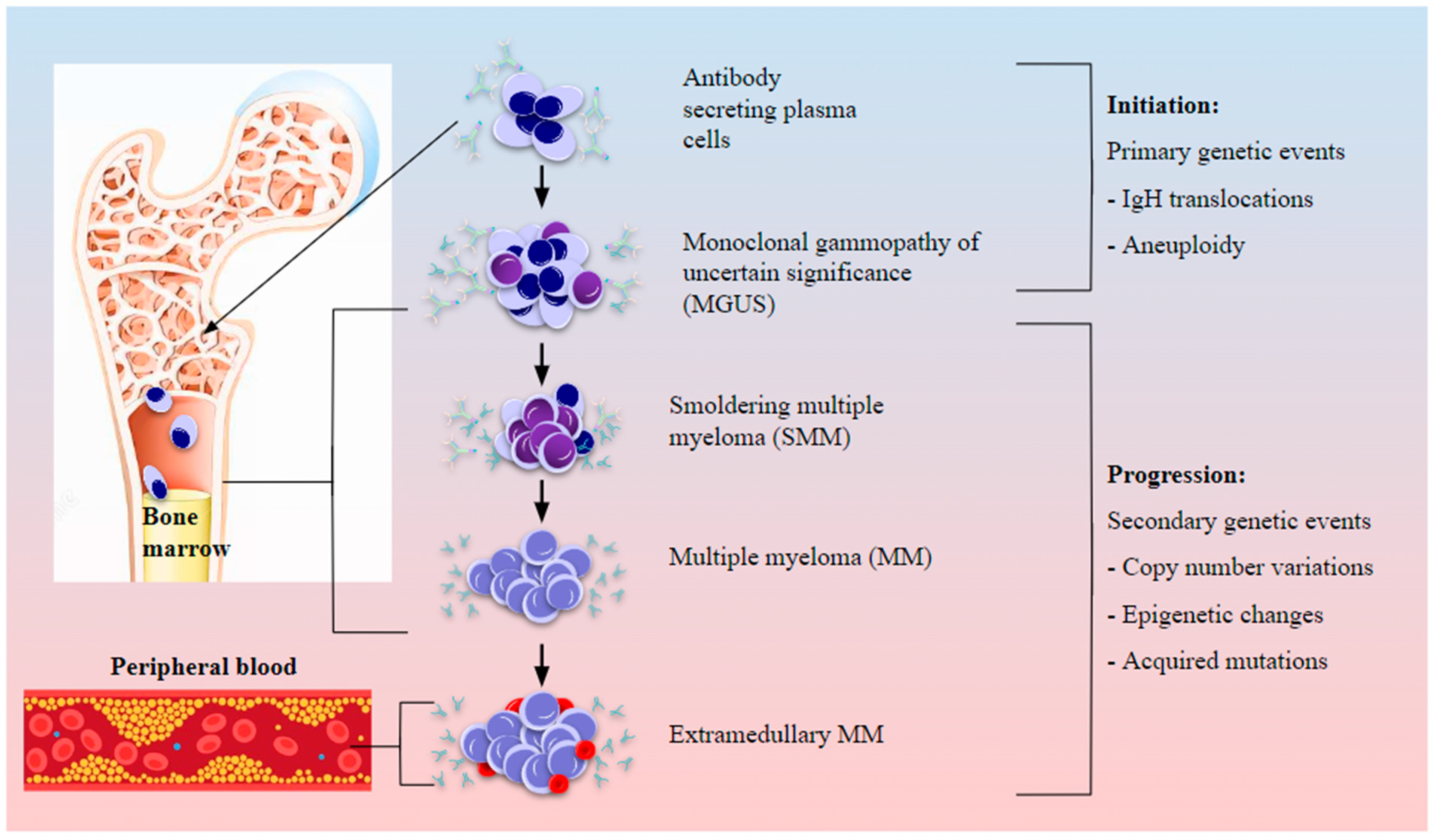
Epidemiology, Diagnosis, and Cytogenetics in Multiple Myeloma
|
Myeloma Stage |
Clinical Parameters |
Cytogenetic Abnormalities |
Prognosis |
|---|---|---|---|
|
MGUS |
Serum M-protein ≤ 30 mg/l, <10% BM clonal PC, no organ damage evidence (CRAB *) |
Hyperdiploidy (HD) (chromosomes 3, 5, 7, 9, 11, 15, 17) |
SR |
|
IgH translocations (IgHT)—t(11;14), t(4;14), t(6;14), t(14;16) and t(14;20) and involved partner genes—4p16: FGFR3/MMSET, 11q13: CCND1, 16q23: MAF, 6p21: CCND3, 20q11: MAFB |
HR, AP |
||
|
monosomy 13/del(13q) |
SR/IR |
||
|
SMM |
Serum M-protein ≥ 30 mg/l (IgG or IgA), >10% BM clonal PC, no organ damage evidence (CRAB) |
MGUS abnormalities |
SR |
|
MUGUS + secondary abnormalities—monosomy17/del(17p) (gene P53), gain 1q21 |
HR |
||
|
MM |
Serum M-protein ≥ 30 mg/l (IgG or IgA), >10% BM clonal PC, and organ damage evidence (CRAB) |
HD, HD (chromosomes 3, 5) + gain 5q31 |
IS |
|
Hypoploidy, IgHT + non-HD |
HR, AP |
||
|
del(17p), gain 1q21 (gene CSK1B), MYC translocations, del(1p) |
AP |
||
|
Extramedullary MM (not including solitary/bone plasmacytoma) |
MM criteria and involvement of skeleton or soft tissue or lymph node(s), BM-independence, drug resistance |
del(17p13), del(13q14), MYC-over-expression, t(4;14) |
AP |
|
Mutations in TP53, RB1, KRAS, FAK |
HR |
||
|
del(17p) + non-HD |
AP |
||
|
RRMM |
Reappearance of M-protein, ≥5% BMPCs, new lytic bone lesions and/or soft tissue plasmacytomas, increase in size of residual bone lesions, and/or development of hypercalcemia > 11.5 mg/dL not attributable to another cause |
del(17p), t(4;14) or t(14;16) |
HR, AP |
|
gain 1q21 |
AP |
||
|
t(4;14): overexpression of FGFR3, t(14:16): overexpression of c-maf, t(14:20): overexpression of c-maf, del(17p): deletion of p53 |
AP |
2. Marrow, Microenvironment, and Multiple Myeloma
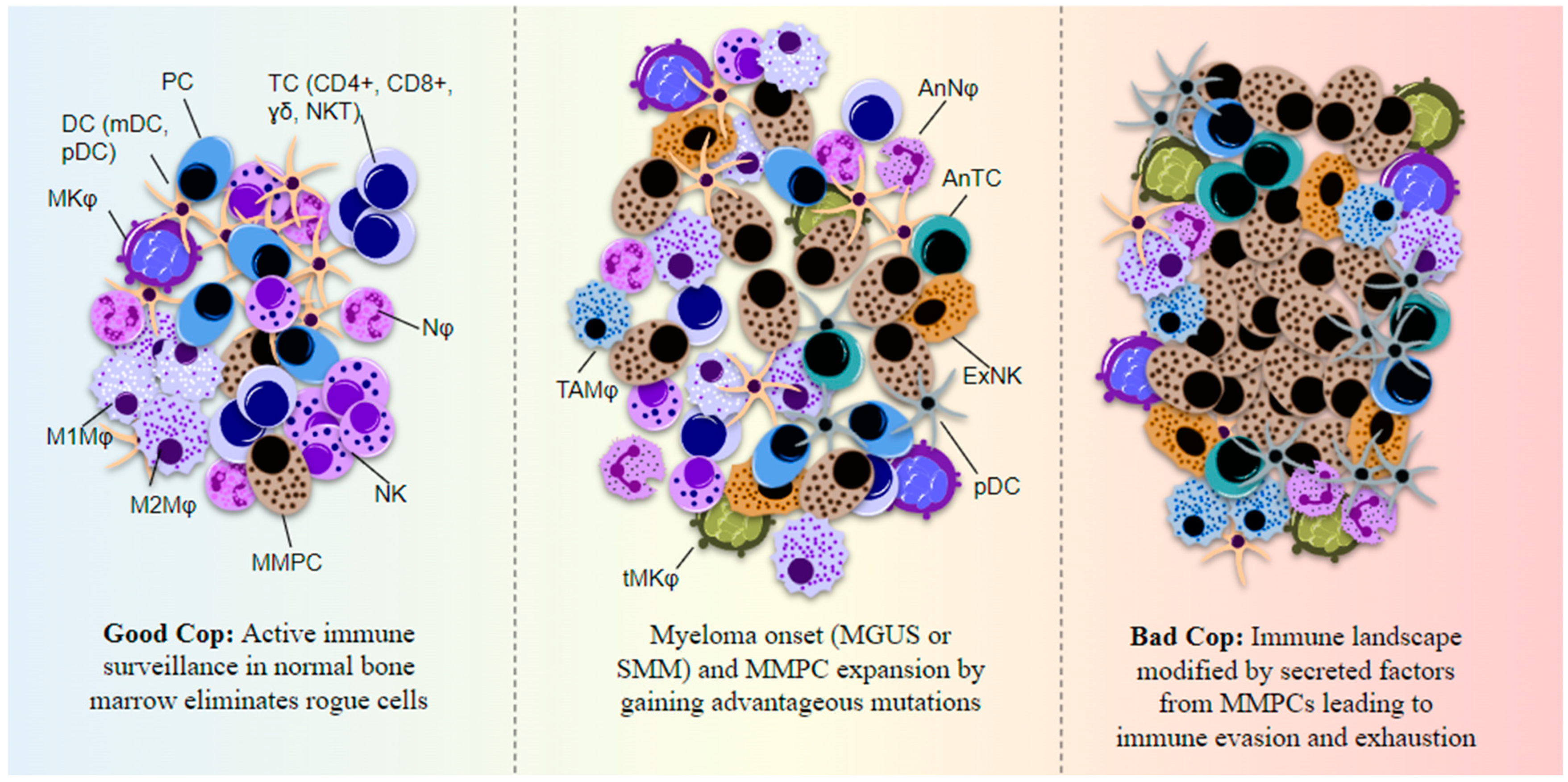
3. Immune Profiling Using Single-Cell Transcriptome Sequencing in Multiple Myeloma
|
Disease Stage |
Type of Samples and Number of Cells |
Number of Samples |
Type of Sequencing |
Key Findings |
Year and Reference |
|---|---|---|---|---|---|
|
MGUS, SMM, MM, primary light chain amyloidosis (AL) |
PCs from peripheral blood (3540) and BM (20,586) |
MGUS = 7, SMM = 6, MM = 12, AL = 4, controls = 11 |
scRNA-seq |
Use of single cell transcriptomics for prognostic prediction and personalized therapy based on unique subclonal structures within individuals, detection of tumor cell populations in MRD stages matching those from active MM. |
2018 [54] |
|
5TGM1 murine myeloma |
NA |
NA |
scRNA-seq |
Identification of myeloma-specific transcriptome signature enriched for genes associated with immune function and myeloid cell differentiation, loss of dormancy related gene expression such as AXL |
2019 [55] |
|
MGUS, SMM |
Patient BM aspirates, 19,000 CD45+/CD138− cells |
MGUS = 5, LR-SMM = 3, HR-SMM = 8, NDMM = 7, healthy donors = 9 |
scRNA-seq |
Immune profile-based patient risk stratification: increased NK cell abundance in early stages, loss of cytotoxic T cells in SMM, MHC-II dysregulation in CD14+ monocytes causing T-cell suppression. |
2020 [56] |
|
NDMM to relapse follow-up samples |
Patient BM aspirates, CD138+ sorted and unsorted cells: 17,267 PCs and 57,719 immune cells |
14 patients across stages for a total of 29 samples |
scRNA-seq and 10x WGS |
Three distinct patterns identified with stability from precancer to diagnosis and gain or loss from diagnosis to relapse involving B-cell type PCs, inflammation regulated gene expression changes in IL6 and IL1B |
2021 [57] |
|
Across MM stages |
Patient BM aspirates. CD138+ and CD138− sorted BM cells |
18 patients across stages |
scRNA-seq, CyTOF, and CITE-seq |
Advanced stage MM patients had reduced CD4+ T/CD8+ T cells ratio, overexpression of RAC2 and PSMB9 observed in NK cells of progressors compared to non-progressors, rapid progression-specific markers identified for MM |
2022 [58] |
|
MM before and after 2 cycles of bortezomib-cyclophosphamide-dexamethasone (VCD) treatment |
Patient and control BM and peripheral blood samples, 25,231 plasma cells and 216,209 immune cells |
MM patients = 10, healthy controls = 3 |
scRNA-seq |
In response to drug treatment, reduced unfolded protein response and metabolic signaling, increased stress and immune-reactive signaling; reduced checkpoint molecules expression, T-cell, NK cell and monocyte exhaustion in MM indicating immunosuppression in the BMME and related to poor prognosis |
2023 [59] |
|
Across MM stages |
NA |
NA |
scRNA-seq |
Progression associated with immunosuppressive tumor microenvironment, with increased levels of exhausted CD8+ T-cells, NK cells and reprogrammed TAMs showing both M1 and higher M2 phenotypes with impaired phagocytic activity demonstrated in vitro |
2023 [60] |
|
NDMM followed by 2 cycles of VCD |
BM and peripheral blood samples, DCs = 1429, monocytes = 42,464 |
MM patients = 10, healthy volunteers = 3 |
scRNA-seq |
Dysfunctional conventional DC2 (cDC2), mono-DC, and intermediate monocytes through downregulation of interferon regulatory factor (IRF)-1 signaling with reduced antigen presentation capacity in MM compared to controls |
2023 [61] |
4. Immunotherapy in Multiple Myeloma
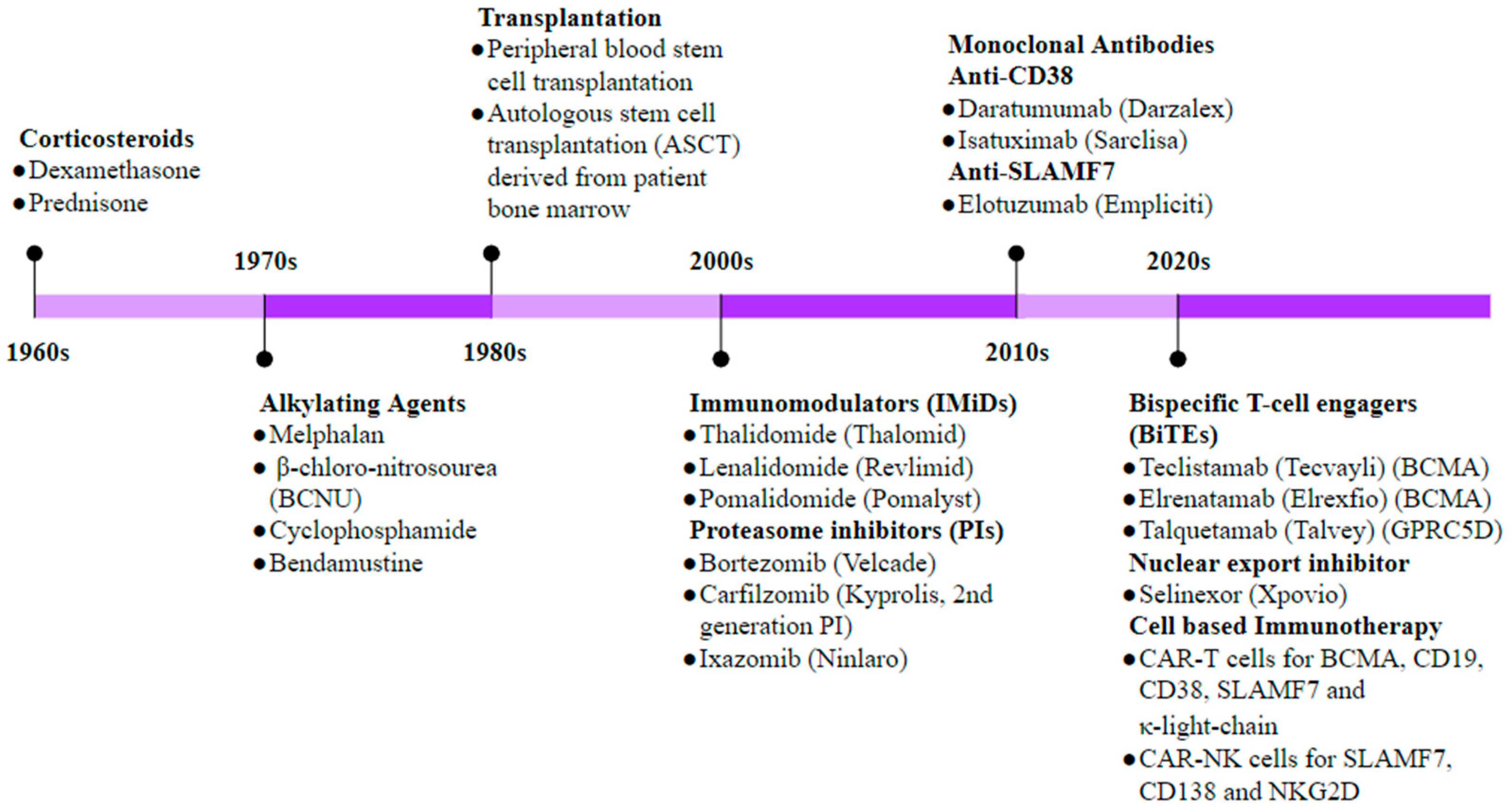
|
NCT Number |
Study Title |
Conditions |
Targets |
Type of Immunotherapy |
Expected Outcome |
Phases |
|---|---|---|---|---|---|---|
|
NCT00090493 |
Study of MAGE-A3 and NY-ESO-1 Immunotherapy in Combo with DTPACE Chemo and Auto Transplantation in Multiple Myeloma |
MM |
Myeloma cells expressing MAGE-A3 and NY-ESP-1 proteins |
Peptide vaccine against MAGE-A and NY-ESP-1 |
Generation of anti-myeloma T cells to kill myeloma cells |
Phase 2, Phase 3 |
|
NCT00006244 |
Melphalan, Peripheral Stem Cell Transplantation, and Interleukin-2 Followed by Interferon Alfa in Treating Patients with Advanced Multiple Myeloma |
Refractory MM, Stage I, Stage II and Stage III MM |
Activating patient WBC response |
Recombinant IL-2 (Aldsleukin), IFN-α |
Stimulate patient WBCs with IL-2 to kill myeloma cells and arrest proliferation with IFN-α |
Phase 2 |
|
NCT03525678 |
A Study to Investigate the Efficacy and Safety of Two Doses of GSK2857916 in Participants with Multiple Myeloma Who Have Failed Prior Treatment with an Anti-CD38 Antibody |
MM |
BCMA on myeloma cells |
Antibody-drug conjugate (ADC)—belantamab mafodotin |
Anti-BCMA antibody belantamab will target myeloma cells and the conjugate mafodotin is a cytotoxic agent to effect cell cycle arrest and ADCC of myeloma cells |
Phase 2 |
|
NCT02336815 |
Selinexor Treatment of Refractory Myeloma |
MM |
Exportin-1 (XPO-1)—overexpressed in myeloma cells |
Inhibitor of nuclear export |
Arrest cell cycle and proliferation of myeloma cells by blocking nuclear to cytoplasmic transport and signaling |
Phase 2 |
|
NCT03958656 |
T-cells Expressing an Anti-SLAMF7 CAR for Treating Multiple Myeloma |
MM, plasma cell (PC) MM |
SLAMF7 |
Anti-SLAMF7 CAR-T cell |
Targeting myeloma cell SLAMF7 using CAR-T cell and T cells along with a stop gene to limit toxicity |
Phase 1 |
|
NCT03602612 |
T Cells Expressing a Novel Fully Human Anti-BCMA CAR for Treating Multiple Myeloma |
MM, PCMM |
BCMA on myeloma cells |
Anti-BCMA CAR-T cells |
Patient- derived T-cells cultured to express anti-BCMA CAR to kill BCMA expressing myeloma cells |
Phase 1 |
|
NCT02215967 |
Study of T Cells Targeting B-Cell Maturation Antigen for Previously Treated Multiple Myeloma |
PCMM, MM |
BCMA on myeloma cells of treated patients |
Anti-BCMA CAR-T cells |
Patient- derived T-cells cultured to express anti-BCMA CAR to kill BCMA expressing myeloma cells |
Phase 1 |
|
NCT03338972 |
Immunotherapy with BCMA CAR-T Cells in Treating Patients with BCMA Positive Relapsed or Refractory Multiple Myeloma |
Recurrent PCMM, refractory PCMM |
BCMA on myeloma cells of relapsed or refractory MM patients |
Autologous Anti-BCMA-CAR-expressing CD4+/CD8+ T-lymphocytes |
Patient- derived T-cells cultured to express anti-BCMA CAR to kill BCMA expressing myeloma cells |
Phase 1 |
|
NCT00566098 |
Activated White Blood Cells with ASCT for Newly Diagnosed Multiple Myeloma |
MM and PC neoplasm |
Newly diagnosed MM cells |
Activated patient-derived WBCs |
Use patient-derived, cultured WBCs to study MM cell cytotoxicity and side effects |
Phase 1, Phase 2 |
|
NCT01245673 |
Combination Immunotherapy and Autologous Stem Cell Transplantation for Myeloma |
MM |
Myeloma cells |
MAGE-A3 vaccine + activated T-cells |
To test if this combination: (i) provides “immunity” against myeloma cells and (ii) prevents progression |
Phase 2 |
|
NCT00439465 |
Adoptive Cellular Immunotherapy Following Autologous Peripheral Blood Stem Cell Transplantation (APBSCT) for Multiple Myeloma |
Transplant eligible MM patients |
Myeloma cells in post-APBSCT patients |
Cytotoxic T-cells + IL-2 + recombinant GM-CSF |
Combination of Cytotoxic T-cells + IL-2 + recombinant GM-CSF to enhance anti-tumor immune reconstitution and improve outcome of MM patients. |
Phase 2 |
|
NCT05652530 |
Clinical Study of the Safety and Efficacy of BCMA CAR-NK |
MM |
BCMA on myeloma cells |
Anti-BCMA CAR-NK cells |
Targeting myeloma cells expressing BCMA using CAR-NK cells in relapsed/refractory MM patients |
Early Phase 1 |
|
NCT03940833 |
Clinical Research of Adoptive BCMA CAR-NK Cells on Relapse/Refractory MM |
MM |
BCMA on myeloma cells |
Anti-BCMA CAR-NK 92 cells |
To kill myeloma cells expressing BCMA using NK 92 cells in MM patients |
Phase 1, Phase 2 |
|
NCT05182073 |
FT576 in Subjects with Multiple Myeloma |
MM |
BCMA on myeloma cells |
FT576 (Allogeneic BCMA CAR-NK cells) |
FT576 as monotherapy and in combination with the monoclonal antibody daratumumab in MM |
Phase 1 |
|
NCT06045091 |
To Evaluate the Safety and Efficacy of Human BCMA Targeted CAR-NK Cells Injection for Subjects With R/R MM or PCL |
MM, PC leukemia |
BCMA on myeloma cells |
Anti-BCMA CAR-NK cells |
Targeting myeloma cells expressing BCMA using CAR-NK cells in MM and PC leukemia patients |
Early Phase 1 |
|
NCT05008536 |
Anti-BCMA CAR-NK Cell Therapy for the Relapsed or Refractory Multiple Myeloma |
Refractory MM |
BCMA on myeloma cells |
Umbilical and cord blood derived, anti-BCMA engineered CAR-NK cells |
Targeting malignant myeloma cells expressing BCMA using engineered CAR-NK cells in refractory MM patients |
Early Phase 1 |
References
- Rieger, M.A.; Schroeder, T. Hematopoiesis. Cold Spring Harb. Perspect. Biol. 2012, 4, a008250.
- Elsaid, R.; Soares-Da-Silva, F.; Peixoto, M.; Amiri, D.; Mackowski, N.; Pereira, P.; Bandeira, A.; Cumano, A. Hematopoiesis: A Layered Organization Across Chordate Species. Front. Cell Dev. Biol. 2020, 8, 606642.
- González, D.; van der Burg, M.; García-Sanz, R.; Fenton, J.A.; Langerak, A.W.; González, M.; van Dongen, J.J.M.; Miguel, J.F.S.; Morgan, G.J. Immunoglobulin gene rearrangements and the pathogenesis of multiple myeloma. Blood 2007, 110, 3112–3121.
- Godin, I.; Cumano, A. Hematopoietic Stem Cell Development; Springer Science & Business Media: Berlin, Germany, 2010; 178p.
- Barwick, B.G.; Gupta, V.A.; Vertino, P.M.; Boise, L.H. Cell of Origin and Genetic Alterations in the Pathogenesis of Multiple Myeloma. Front. Immunol. 2019, 10, 1121.
- Cowan, A.J.; Green, D.J.; Kwok, M.; Lee, S.; Coffey, D.G.; Holmberg, L.A.; Tuazon, S.; Gopal, A.K.; Libby, E.N. Diagnosis and Management of Multiple Myeloma: A Review. JAMA 2022, 327, 464–477.
- Rustad, E.H.; Yellapantula, V.; Leongamornlert, D.; Bolli, N.; Ledergor, G.; Nadeu, F.; Angelopoulos, N.; Dawson, K.J.; Mitchell, T.J.; Osborne, R.J.; et al. Timing the initiation of multiple myeloma. Nat. Commun. 2020, 11, 1917.
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The genetic architecture of multiple myeloma. Nat. Rev. Cancer 2012, 12, 335–348.
- Padala, S.A.; Barsouk, A.; Barsouk, A.; Rawla, P.; Vakiti, A.; Kolhe, R.; Kota, V.; Ajebo, G.H. Epidemiology, Staging, and Management of Multiple Myeloma. Med. Sci. 2021, 9, 3.
- Röllig, C.; Knop, S.; Bornhäuser, M. Multiple myeloma. Lancet 2015, 385, 2197–2208.
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249.
- Zhou, L.; Yu, Q.; Wei, G.; Wang, L.; Huang, Y.; Hu, K.; Hu, Y.; Huang, H. Measuring the global, regional, and national burden of multiple myeloma from 1990 to 2019. BMC Cancer 2021, 21, 606.
- Turesson, I.; Bjorkholm, M.; Blimark, C.H.; Kristinsson, S.; Velez, R.; Landgren, O. Rapidly changing myeloma epidemiology in the general population: Increased incidence, older patients, and longer survival. Eur. J. Haematol. 2018, 101, 237–244.
- Kazandjian, D. Multiple myeloma epidemiology and survival: A unique malignancy. Semin. Oncol. 2016, 43, 676–681.
- International Myeloma Foundation. Durie-Salmon Staging System. Available online: https://www.myeloma.org/ (accessed on 8 October 2022).
- Filonzi, G.; Mancuso, K.; Zamagni, E.; Nanni, C.; Spinnato, P.; Cavo, M.; Fanti, S.; Salizzoni, E.; Bazzocchi, A. A Comparison of Different Staging Systems for Multiple Myeloma: Can the MRI Pattern Play a Prognostic Role? AJR Am. J. Roentgenol. 2017, 209, 152–158.
- International Myeloma Foundation. International Staging System (ISS) and Revised ISS (R-ISS). Available online: https://www.myeloma.org/ (accessed on 8 October 2022).
- International Myeloma Foundation. International Myeloma Working Group (IMWG) criteria for the diagnosis of multiple myeloma. Available online: https://www.myeloma.org/ (accessed on 8 October 2022).
- Sawyer, J.R. The prognostic significance of cytogenetics and molecular profiling in multiple myeloma. Cancer Genet. 2011, 204, 3–12.
- Liebisch, P.; Viardot, A.; Baßermann, N.; Wendl, C.; Roth, K.; Goldschmidt, H.; Einsele, H.; Straka, C.; Stilgenbauer, S.; Döhner, H.; et al. Value of comparative genomic hybridization and fluorescence in situ hybridization for molecular diagnostics in multiple myeloma. Br. J. Haematol. 2003, 122, 193–201.
- Tassone, P.; Tagliaferri, P.; Rossi, M.; Gaspari, M.; Terracciano, R.; Venuta, S. Genetics and molecular profiling of multiple myeloma: Novel tools for clinical management? Eur. J. Cancer 2006, 42, 1530–1538.
- Bergsagel, P.L.; Kuehl, W.M. Molecular Pathogenesis and a Consequent Classification of Multiple Myeloma. J. Clin. Oncol. 2005, 23, 6333–6338.
- Hideshima, T.; Mitsiades, C.; Tonon, G.; Richardson, P.G.; Anderson, K.C. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat. Rev. Cancer 2007, 7, 585–598.
- Fairfield, H.; Falank, C.; Avery, L.; Reagan, M.R. Multiple myeloma in the marrow: Pathogenesis and treatments. Ann. N. Y. Acad. Sci. 2016, 1364, 32–51.
- Kuehl, W.M.; Bergsagel, P.L. Multiple myeloma: Evolving genetic events and host interactions. Nat. Rev. Cancer 2002, 2, 175–187.
- Bergsagel, P.L.; Kuehl, W.M. Chromosome translocations in multiple myeloma. Oncogene 2001, 20, 5611–5622.
- Rajan, A.M.; Rajkumar, S.V. Interpretation of cytogenetic results in multiple myeloma for clinical practice. Blood Cancer J. 2015, 5, e365.
- Hillengass, J.; Moehler, T.; Hundemer, M. Monoclonal gammopathy and smoldering multiple myeloma: Diagnosis, staging, prognosis, management. Recent Results Cancer Res. 2011, 183, 113–131.
- Korde, N.; Kristinsson, S.Y.; Landgren, O. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM): Novel biological insights and development of early treatment strategies. Blood 2011, 117, 5573–5581.
- Seong, C.; Delasalle, K.; Hayes, K.; Weber, D.; Dimopoulos, M.; Swantkowski, J.; Huh, Y.; Glassman, A.; Champlin, R.; Alexanian, R. Prognostic value of cytogenetics in multiple myeloma. Br. J. Haematol. 1998, 101, 189–194.
- Kumar, S.; Fonseca, R.; Ketterling, R.P.; Dispenzieri, A.; Lacy, M.Q.; Gertz, M.A.; Hayman, S.R.; Buadi, F.K.; Dingli, D.; Knudson, R.A.; et al. Trisomies in multiple myeloma: Impact on survival in patients with high-risk cytogenetics. Blood 2012, 119, 2100–2105.
- Sonneveld, P.; Avet-Loiseau, H.; Lonial, S.; Usmani, S.; Siegel, D.; Anderson, K.C.; Chng, W.-J.; Moreau, P.; Attal, M.; Kyle, R.A.; et al. Treatment of multiple myeloma with high-risk cytogenetics: A consensus of the International Myeloma Working Group. Blood 2016, 127, 2955–2962.
- Billecke, L.; Penas, E.M.M.; May, A.M.; Engelhardt, M.; Nagler, A.; Leiba, M.; Schiby, G.; Kröger, N.; Zustin, J.; Marx, A.; et al. Cytogenetics of extramedullary manifestations in multiple myeloma. Br. J. Haematol. 2013, 161, 87–94.
- Besse, L.; Sedlarikova, L.; Greslikova, H.; Kupska, R.; Almasi, M.; Penka, M.; Jelinek, T.; Pour, L.; Adam, Z.; Kuglik, P.; et al. Cytogenetics in multiple myeloma patients progressing into extramedullary disease. Eur. J. Haematol. 2016, 97, 93–100.
- Harrison, S.J.; Perrot, A.; Alegre, A.; Simpson, D.; Wang, M.C.; Spencer, A.; Delimpasi, S.; Hulin, C.; Sunami, K.; Facon, T.; et al. Subgroup analysis of ICARIA-MM study in relapsed/refractory multiple myeloma patients with high-risk cytogenetics. Br. J. Haematol. 2021, 194, 120–131.
- Dimopoulos, M.A.; Kastritis, E.; Christoulas, D.; Migkou, M.; Gavriatopoulou, M.; Gkotzamanidou, M.; Iakovaki, M.; Matsouka, C.; Mparmparoussi, D.; Roussou, M.; et al. Treatment of patients with relapsed/refractory multiple myeloma with lenalidomide and dexamethasone with or without bortezomib: Prospective evaluation of the impact of cytogenetic abnormalities and of previous therapies. Leukemia 2010, 24, 1769–1778.
- Bhutani, M.; Foureau, D.M.; Atrash, S.; Voorhees, P.M.; Usmani, S.Z. Extramedullary multiple myeloma. Leukemia 2019, 34, 1–20.
- Lonial, S.; Mitsiades, C.S.; Richardson, P.G. Treatment Options for Relapsed and Refractory Multiple Myeloma. Clin. Cancer Res. 2011, 17, 1264–1277.
- Shafat, M.S.; Gnaneswaran, B.; Bowles, K.M.; Rushworth, S.A. The bone marrow microenvironment—Home of the leukemic blasts. Blood Rev. 2017, 31, 277–286.
- Morrison, S.J.; Scadden, D.T. The bone marrow niche for haematopoietic stem cells. Nature 2014, 505, 327–334.
- Yu, V.W.; Scadden, D.T. Heterogeneity of the bone marrow niche. Curr. Opin. Hematol. 2016, 23, 331–338.
- Méndez-Ferrer, S.; Bonnet, D.; Steensma, D.P.; Hasserjian, R.P.; Ghobrial, I.M.; Gribben, J.G.; Andreeff, M.; Krause, D.S. Bone marrow niches in haematological malignancies. Nat. Rev. Cancer 2020, 20, 285–298.
- Orkin, S.H.; Zon, L.I. Hematopoiesis: An Evolving Paradigm for Stem Cell Biology. Cell 2008, 132, 631–644.
- Jagannathan-Bogdan, M.; Zon, L.I. Hematopoiesis. Development 2013, 140, 2463–2467.
- Pucella, J.N.; Upadhaya, S.; Reizis, B. The Source and Dynamics of Adult Hematopoiesis: Insights from Lineage Tracing. Annu. Rev. Cell Dev. Biol. 2020, 36, 529–550.
- Yokota, T.; Oritani, K.; Mitsui, H.; Aoyama, K.; Ishikawa, J.; Sugahara, H.; Matsumura, I.; Tsai, S.; Tomiyama, Y.; Kanakura, Y.; et al. Growth-supporting activities of fibronectin on hematopoietic stem/progenitor cells in vitro and in vivo: Structural requirement for fibronectin activities of CS1 and cell-binding domains. Blood 1998, 91, 3263–3272.
- Smith, C. Hematopoietic Stem Cells and Hematopoiesis. Cancer Control. 2003, 10, 9–16.
- Waterstrat, A.; Van Zant, G. Effects of aging on hematopoietic stem and progenitor cells. Curr. Opin. Immunol. 2009, 21, 408–413.
- Fröbel, J.; Landspersky, T.; Percin, G.; Schreck, C.; Rahmig, S.; Ori, A.; Nowak, D.; Essers, M.; Waskow, C.; Oostendorp, R.A.J. The Hematopoietic Bone Marrow Niche Ecosystem. Front. Cell Dev. Biol. 2021, 9, 705410.
- Krause, D.S.; Scadden, D.T. A hostel for the hostile: The bone marrow niche in hematologic neoplasms. Haematologica 2015, 100, 1376–1387.
- Kumar, R.; Godavarthy, P.S.; Krause, D.S. The bone marrow microenvironment in health and disease at a glance. J. Cell Sci. 2018, 131, jcs201707.
- Duarte, D.; Hawkins, E.D.; Celso, C.L. The interplay of leukemia cells and the bone marrow microenvironment. Blood 2018, 131, 1507–1511.
- Lyons, Y.A.; Wu, S.Y.; Overwijk, W.W.; Baggerly, K.A.; Sood, A.K. Immune cell profiling in cancer: Molecular approaches to cell-specific identification. Npj Precis. Oncol. 2017, 1, 26.
- Ledergor, G.; Weiner, A.; Zada, M.; Wang, S.-Y.; Cohen, Y.C.; Gatt, M.E.; Snir, N.; Magen, H.; Koren-Michowitz, M.; Herzog-Tzarfati, K.; et al. Single cell dissection of plasma cell heterogeneity in symptomatic and asymptomatic myeloma. Nat. Med. 2018, 24, 1867–1876.
- Khoo, W.H.; Ledergor, G.; Weiner, A.; Roden, D.L.; Terry, R.L.; McDonald, M.M.; Chai, R.C.; De Veirman, K.; Owen, K.L.; Opperman, K.S.; et al. A niche-dependent myeloid transcriptome signature defines dormant myeloma cells. Blood 2019, 134, 30–43.
- Zavidij, O.; Haradhvala, N.J.; Mouhieddine, T.H.; Sklavenitis-Pistofidis, R.; Cai, S.; Reidy, M.; Rahmat, M.; Flaifel, A.; Ferland, B.; Su, N.K.; et al. Single-cell RNA sequencing reveals compromised immune microenvironment in precursor stages of multiple myeloma. Nat. Cancer 2020, 1, 493–506.
- Liu, R.; Gao, Q.; Foltz, S.M.; Fowles, J.S.; Yao, L.; Wang, J.T.; Cao, S.; Sun, H.; Wendl, M.C.; Sethuraman, S.; et al. Co-evolution of tumor and immune cells during progression of multiple myeloma. Nat. Commun. 2021, 12, 282.
- Yao, L.; Jayasinghe, R.G.; Lee, B.H.; Bhasin, S.S.; Pilcher, W.; Doxie, D.B.; Gonzalez-Kozlova, E.; Dasari, S.; Fiala, M.A.; Pita-Juarez, Y.; et al. Comprehensive Characterization of the Multiple Myeloma Immune Microenvironment Using Integrated scRNA-seq, CyTOF, and CITE-seq Analysis. Cancer Res. Commun. 2022, 2, 1255–1265.
- Chen, M.; Wan, Y.; Li, X.; Xiang, J.; Chen, X.; Jiang, J.; Han, X.; Zhong, L.; Xiao, F.; Liu, J.; et al. Dynamic single-cell RNA-seq analysis reveals distinct tumor program associated with microenvironmental remodeling and drug sensitivity in multiple myeloma. Cell Biosci. 2023, 13, 19.
- Li, J.; Yang, Y.; Wang, W.; Xu, J.; Sun, Y.; Jiang, J.; Tan, H.; Ren, L.; Wang, Y.; Ren, Y.; et al. Single-cell atlas of the immune microenvironment reveals macrophage reprogramming and the potential dual macrophage-targeted strategy in multiple myeloma. Br. J. Haematol. 2023, 201, 917–934.
- Jiang, J.; Xiang, J.; Chen, M.; Wan, Y.; Zhong, L.; Han, X.; Chen, X.; Wang, J.; Xiao, F.; Liu, J.; et al. Distinct mechanisms of dysfunctional antigen-presenting DCs and monocytes by single-cell sequencing in multiple myeloma. Cancer Sci. 2023, 114, 2750–2760.
- Donnall Thomas, E. A history of haemopoietic cell transplantation. Br. J. Haematol. 1999, 105, 330–339.
- Lanier, O.L.; Pérez-Herrero, E.; Andrea, A.P.D.; Bahrami, K.; Lee, E.; Ward, D.M.; Ayala-Suárez, N.; Rodríguez-Méndez, S.M.; Peppas, N.A. Immunotherapy approaches for hematological cancers. iScience 2022, 25, 105326.
- Holthof, L.C.; Mutis, T. Challenges for Immunotherapy in Multiple Myeloma: Bone Marrow Microenvironment-Mediated Immune Suppression and Immune Resistance. Cancers 2020, 12, 988.
- A Shah, U.; Mailankody, S. Emerging immunotherapies in multiple myeloma. BMJ 2020, 370, m3176.
- Ntanasis-Stathopoulos, I.; Gavriatopoulou, M.; Kastritis, E.; Terpos, E.; Dimopoulos, M.A. Multiple myeloma: Role of autologous transplantation. Cancer Treat. Rev. 2020, 82, 101929.
- Mateos, M.-V.; Ludwig, H.; Bazarbachi, A.; Beksac, M.; Bladé, J.; Boccadoro, M.; Cavo, M.; Delforge, M.; Dimopoulos, M.A.; Facon, T.; et al. Insights on Multiple Myeloma Treatment Strategies. HemaSphere 2019, 3, e163.
- Gandolfi, S.; Laubach, J.P.; Hideshima, T.; Chauhan, D.; Anderson, K.C.; Richardson, P.G. The proteasome and proteasome inhibitors in multiple myeloma. Cancer Metastasis Rev. 2017, 36, 561–584.
- Palma, B.D.; Marchica, V.; Catarozzo, M.T.; Giuliani, N.; Accardi, F. Monoclonal and Bispecific Anti-BCMA Antibodies in Multiple Myeloma. J. Clin. Med. 2020, 9, 3022.
- Gogishvili, T.; Danhof, S.; Prommersberger, S.; Rydzek, J.; Schreder, M.; Brede, C.; Einsele, H.; Hudecek, M. SLAMF7-CAR T cells eliminate myeloma and confer selective fratricide of SLAMF7+ normal lymphocytes. Blood 2017, 130, 2838–2847.
- Nishida, H.; Yamada, T. Monoclonal Antibody Therapies in Multiple Myeloma: A Challenge to Develop Novel Targets. J. Oncol. 2019, 2019, 6084012.
- Bapatla, A.; Kaul, A.; Dhalla, P.S.; Armenta-Quiroga, A.S.; Khalid, R.; Garcia, J.; Khan, S. Role of Daratumumab in Combination With Standard Therapies in Patients With Relapsed and Refractory Multiple Myeloma. Cureus 2021, 13, e15440.
- Trudel, S.; Moreau, P.; Touzeau, C. Update on elotuzumab for the treatment of relapsed/refractory multiple myeloma: Patients’ selection and perspective. OncoTargets Ther. 2019, 12, 5813–5822.
- Abodunrin, F.O.; Tauseef, A.; Silberstein, P. Role of Daratumumab in Relapsed and Refractory Multiple Myeloma Patients: Meta-Analysis and Literature Review. Blood 2021, 138, 4734.
- Bruzzese, A.; Martino, E.A.; Vigna, E.; Iaccino, E.; Mendicino, F.; Lucia, E.; Olivito, V.; Filippelli, G.; Neri, A.; Morabito, F.; et al. Elotuzumab in multiple myeloma. Expert Opin. Biol. Ther. 2022, 23, 7–10.
- Grosicki, S.; Bednarczyk, M.; Barchnicka, A.; Grosicka, O. Elotuzumab in the treatment of relapsed and refractory multiple myeloma. Futur. Oncol. 2021, 17, 1581–1591.
- Carpenter, R.O.; Evbuomwan, M.O.; Pittaluga, S.; Rose, J.J.; Raffeld, M.; Yang, S.; Gress, R.E.; Hakim, F.T.; Kochenderfer, J.N. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin. Cancer Res. 2013, 19, 2048–2060.
- Ali, S.A.; Shi, V.; Maric, I.; Wang, M.; Stroncek, D.F.; Rose, J.J.; Brudno, J.N.; Stetler-Stevenson, M.; Feldman, S.A.; Hansen, B.G.; et al. T cells expressing an anti–B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood 2016, 128, 1688–1700.
- Melchers, F. Checkpoints that control B cell development. J. Clin. Investig. 2015, 125, 2203–2210.
- Raje, N.S.; Berdeja, J.G.; Lin, Y.; Munshi, N.C.; Siegel, D.S.D.; Liedtke, M.; Jagannath, S.; Madduri, D.; Rosenblatt, J.; Maus, M.V.; et al. bb2121 anti-BCMA CAR T-cell therapy in patients with relapsed/refractory multiple myeloma: Updated results from a multicenter phase I study. J. Clin. Oncol. 2018, 36 (Suppl. 15), 8007.
- Zhao, W.H.; Liu, J.; Wang, B.-Y.; Chen, Y.-X.; Cao, X.-M.; Yang, Y.; Zhang, Y.-L.; Wang, F.-X.; Zhang, P.-Y.; Lei, B.; et al. A phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. J. Hematol. Oncol. 2018, 11, 141.
- Prommersberger, S.; Reiser, M.; Beckmann, J.; Danhof, S.; Amberger, M.; Quade-Lyssy, P.; Einsele, H.; Hudecek, M.; Bonig, H.; Ivics, Z. CARAMBA: A first-in-human clinical trial with SLAMF7 CAR-T cells prepared by virus-free Sleeping Beauty gene transfer to treat multiple myeloma. Gene Ther. 2021, 28, 560–571.
- Chu, J.; Deng, Y.; Benson, D.M.; He, S.; Hughes, T.; Zhang, J.; Peng, Y.; Mao, H.; Yi, L.; Ghoshal, K.; et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia 2013, 28, 917–927.
- Wang, X.; Pan, B.; Huang, H.; Xu, K. Non-BCMA targeted CAR-T cell therapies for multiple myeloma. ImmunoMedicine 2021, 1, e1030.
- Gagelmann, N.; Riecken, K.; Wolschke, C.; Berger, C.; Ayuk, F.A.; Fehse, B.; Kröger, N. Development of CAR-T cell therapies for multiple myeloma. Leukemia 2020, 34, 2317–2332.
- Bahlis, N.J.; Costello, C.L.; Raje, N.S.; Levy, M.Y.; Dholaria, B.; Solh, M.; Tomasson, M.H.; Damore, M.A.; Jiang, S.; Basu, C.; et al. Elranatamab in relapsed or refractory multiple myeloma: The MagnetisMM-1 phase 1 trial. Nat. Med. 2023, 29, 2023.
- Caraccio, C.; Krishna, S.; Phillips, D.J.; Schürch, C.M. Bispecific Antibodies for Multiple Myeloma: A Review of Targets, Drugs, Clinical Trials, and Future Directions. Front. Immunol. 2020, 11, 501.
- CTG Labs—NCBI. Available online: https://clinicaltrials.gov/search?cond=multiple%20myeloma&viewType=Table&limit=50&aggFilters=phase:1%202%203%204,results:with,status:com%20act%20not%20rec&intr=Immunotherapy (accessed on 23 September 2023).

