Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Stavros P. Papadakos | -- | 2549 | 2023-11-08 18:33:14 | | | |
| 2 | Camila Xu | Meta information modification | 2549 | 2023-11-09 02:22:32 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Papadakos, S.P.; Arvanitakis, K.; Stergiou, I.E.; Vallilas, C.; Sougioultzis, S.; Germanidis, G.; Theocharis, S. The Molecular Mechanisms of Lipopolysaccharides Tolerance. Encyclopedia. Available online: https://encyclopedia.pub/entry/51308 (accessed on 25 July 2026).
Papadakos SP, Arvanitakis K, Stergiou IE, Vallilas C, Sougioultzis S, Germanidis G, et al. The Molecular Mechanisms of Lipopolysaccharides Tolerance. Encyclopedia. Available at: https://encyclopedia.pub/entry/51308. Accessed July 25, 2026.
Papadakos, Stavros P., Konstantinos Arvanitakis, Ioanna E. Stergiou, Christos Vallilas, Stavros Sougioultzis, Georgios Germanidis, Stamatios Theocharis. "The Molecular Mechanisms of Lipopolysaccharides Tolerance" Encyclopedia, https://encyclopedia.pub/entry/51308 (accessed July 25, 2026).
Papadakos, S.P., Arvanitakis, K., Stergiou, I.E., Vallilas, C., Sougioultzis, S., Germanidis, G., & Theocharis, S. (2023, November 08). The Molecular Mechanisms of Lipopolysaccharides Tolerance. In Encyclopedia. https://encyclopedia.pub/entry/51308
Papadakos, Stavros P., et al. "The Molecular Mechanisms of Lipopolysaccharides Tolerance." Encyclopedia. Web. 08 November, 2023.
Copy Citation
Lipopolysaccharides (LPSs) are complex molecules found in the outer membrane of Gram-negative bacteria, which are composed of three main regions: lipid A, core oligosaccharide, and O antigen.
PAMPs
DAMPs
TLR4
exosomes
extracellular vesicles
1. Introduction
1.1. Hepatocellular Carcinoma Epidemiology and Management
Hepatocellular carcinoma (HCC) is the most common type of primary liver cancer and a major cause of cancer-related mortality worldwide [1]. According to GLOBOCAN, HCC is the sixth most common cancer worldwide and the third leading cause of cancer-related deaths. It is estimated that approximately 840,000 new cases of liver cancer were diagnosed globally in 2020, and HCC accounted for approximately 75% to 85% of all primary liver cancers [2]. The incidence of HCC varied significantly by geographic region and underlying risk factors. HCC is more common in men than women, with a male-to-female ratio of approximately 2.8:1 [3]. The prognosis for HCC is generally poor, with a 5-year survival rate of approximately 18%. The mortality rate is also high. with estimated 800,000 deaths per year worldwide [2]. Nevertheless, early identification and intervention can enhance the results and augment the chances of survival. Prevention measures for HCC include reducing exposure to risk factors such as hepatitis B (HBV) and C viruses (HCV), avoiding alcohol and tobacco use, and implementing vaccination programs for hepatitis B [3]. The National Comprehensive Cancer Network’s (NCCN) guidelines for managing HCC adapted to different resource settings suggest screening individuals at high risk using both liver ultrasonography and α-fetoprotein (AFP) [4].
1.2. Extracellular Vesicles: Biogenesis and Functions
Extracellular vesicles (EVs) constitute vesicles originating from cell membranes and encompassing cargo such as proteins, nucleic acids, lipids, metabolites, and even organelles from parental cells [5][6][7]. The categorization of EVs includes exosomes, microvesicles, and apoptotic bodies, as stipulated by the International Society of Extracellular Vesicles [8]. These entities lack a self-replication ability and emerge through distinct mechanisms of biogenesis [9]. Exosomes, with dimensions ranging from 30 to 120 nm, are formed intracellularly within multivesicular bodies (MVBs). The release of exosomes into the extracellular space occurs through the fusion of MVBs with the plasma membrane [10]. Microvesicles, spanning from 50 to 1000 nm, are generated when cells directly bud vesicles from the plasma membrane [9]. Apoptotic bodies, measuring 50 to 5000 nm, emerge from the cell membrane and are primarily released by cells undergoing apoptosis [9]. The main differences in cargo composition among these various extracellular vesicles are related to their biogenesis and size [11]. Exosomes are typically smaller and enriched in specific proteins and various RNA species, making them important mediators of intercellular communication. Microvesicles, while also carrying proteins and nucleic acids, are larger and have a broader range of cargo. Apoptotic bodies, on the other hand, mainly contain cellular debris from dying cells. These differences in cargo composition reflect the distinct roles these vesicles play in cell communication and physiology [11]. The notions discussed earlier are depicted in Figure 1.
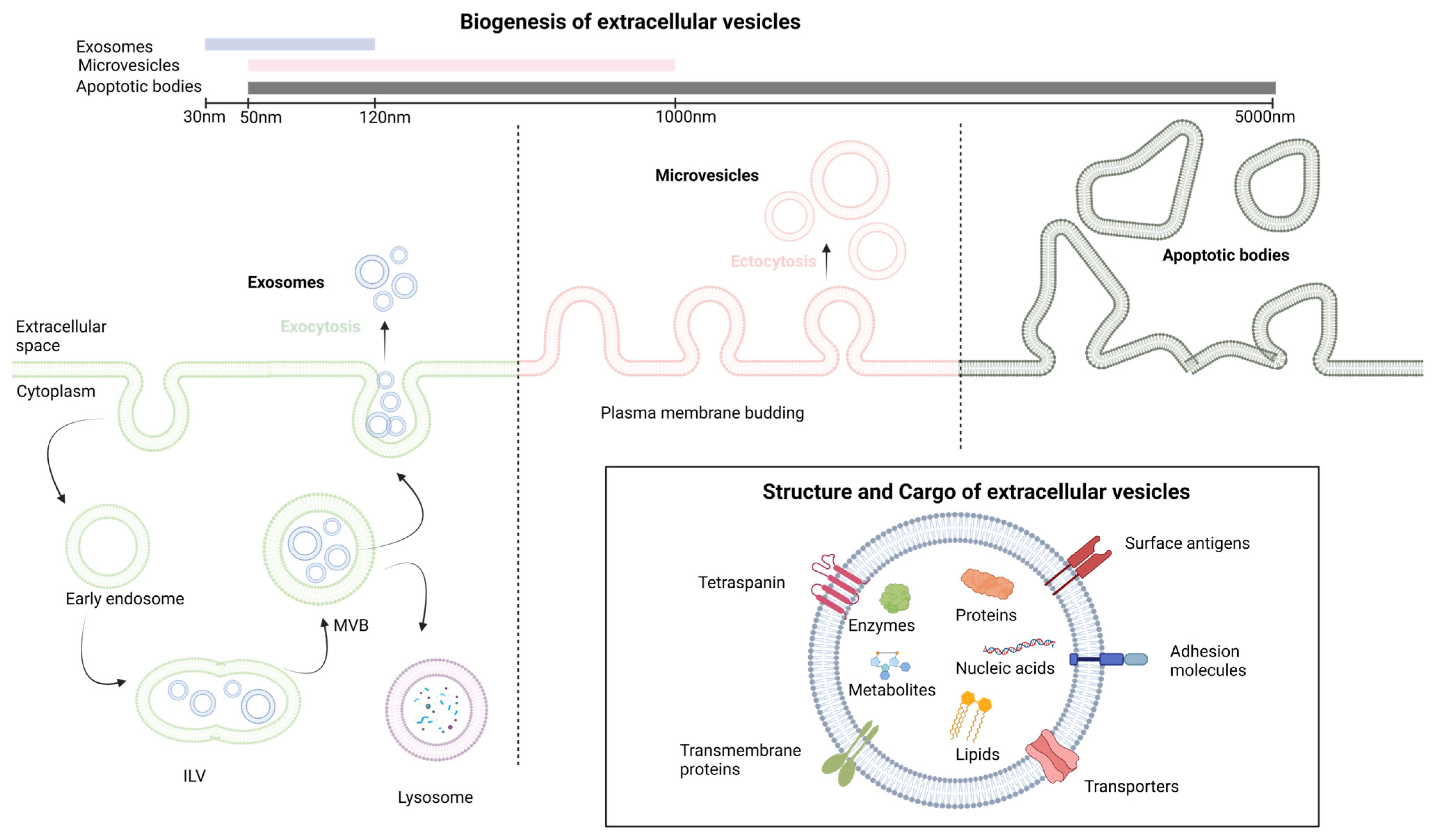
Figure 1. The fundamental aspects of extracellular vesicles structure and biogenesis. ILV, intralluminal vesicle; MVB, multivesicular body. Created with BioRender.com.
EVs fulfill a wide spectrum of biological functions, participating in various physiological and pathological processes [12][13]. When facilitating intercellular communication by transferring a diverse array of molecules between cells, EVs play pivotal roles in intricate biological events like tumorigenesis [6], the formation of pre-metastatic niches [14], inflammation, and immune modulation [15]. Their composition reflecting the state of the parent cell during production renders them promising candidates for diagnostics [16][17]. As they remain stable in numerous biological fluids and are abundant, EVs offer significant potential as biomarker reservoirs [7]. Circulating EVs within liquid biopsies could enable prognosis monitoring, disease progression tracking, and therapy response assessment [7][18]. Recent state-of-the-art reviews emphasize the significant contribution of EVs produced by HCC cells to the progression and spread of HCC [7]. Glycolytic enzymes like Rab20 and triosephosphate isomerase 1 (TPI1), as well as enzymes like caspase-3 and neutral sphingomyelinase 1 (NSMase1), have been shown to have a significant impact on HCC growth and invasion [19][20]. p120ctn and Lysyl Oxidase Like 4 (LOXL4)-containing EVs influence cell migration and proliferation [21]. Additionally, the 14-3-3ζ protein in EVs impairs tumor-infiltrating T lymphocyte functions, while complement factor H (CFH) and circular RNAs (circRNAs) regulate immune evasion and cell signaling [22][23].
1.3. The Toll-like Receptor 4 Signaling Pathway
Toll-like receptor 4 (TLR4) is a transmembrane protein that plays a crucial role in innate immune response by recognizing and responding primarily to lipopolysaccharides (LPSs) from bacterial cell walls [24]. LPSs are complex molecules found in the outer membrane of Gram-negative bacteria, which are composed of three main regions: lipid A, core oligosaccharide, and O antigen [25]. The lipid A moiety is a biologically active component of LPS. In addition to LPS, TLR4 can also respond to other molecules derived from both pathogens and host cells [26]. This includes lipoteichoic acid (LTA), which is a component of the cell wall of Gram-positive bacteria that activates TLR2 and TLR4 signaling, viral envelope proteins such as the respiratory syncytial virus (RSV) fusion protein and the HCV envelope protein alongside endogenous ligands released during tissue damage or inflammation such as heat shock proteins, the high mobility group box 1 (HMGB1) protein, and extracellular matrix components such as hyaluronan and synthetic ligands including monophosphoryl lipid A (MPLA) and glucopyranosyl lipid A (GLA) [26].
1.3.1. Ligand Recognition and TLR4 Activation
LPS recognition by TLR4 requires the TLR4 co-receptor myeloid differentiation factor 2 (MD2) [27]. The extracellular LPS binding protein (LBP) interacts with the bacterial outer membrane, leading to an alteration that facilitates the extraction of single LPS molecules via CD14, which, in turn, transfers a single LPS molecule to the MD2 protein. Once LPS is transferred to MD2 via CD14, TLR4 dimerization takes place. A functional LPS receptor comprises TLR4-MD2 heterodimers. This complex then recruits adapter proteins, initiating TLR4 downstream signaling via two main pathways [27].
-
The MyD88-Dependent Pathway:
The MyD88-dependent pathway is the first and most well-known pathway activated by TLR4 [28]. Upon recruitment to the TLR4-MD2 complex, MyD88 interacts with the cytoplasmic domain of TLR4, leading to the recruitment and activation of interleukin (IL)-1 receptor-associated kinase 1 (IRAK1) and IRAK4. This results in the phosphorylation and activation of tumor necrosis factor receptor-associated factor 6 (TRAF6), which, in turn, activates the downstream transcription factor nuclear factor-kappa B (NF-κB). NF-κB then translocates to the nucleus and induces the expression of pro-inflammatory cytokines, such as tumor necrosis factor (TNF)-α, IL-1β, and IL-6.
-
The MyD88-Independent Pathway:
The MyD88-independent pathway is also known as the toll-interleukin-1 receptor (TIR)-domain-containing adaptor protein that induces the interferon-β (TRIF)-dependent pathway as it requires TRIF for downstream signaling [28]. Upon ligand binding, the TLR4-MD2 complex recruits TRIF, which activates a series of kinases, including TANK-binding kinase 1 (TBK1) and the inhibitor of nuclear factor kappa-B kinase ε (IKKε). These kinases then phosphorylate and activate the transcription factor interferon regulatory factor 3 (IRF3), which translocates to the nucleus and induces the expression of type I interferons (IFN-α and IFN-β). Figure 2 provides a concise illustration of the aforementioned content.
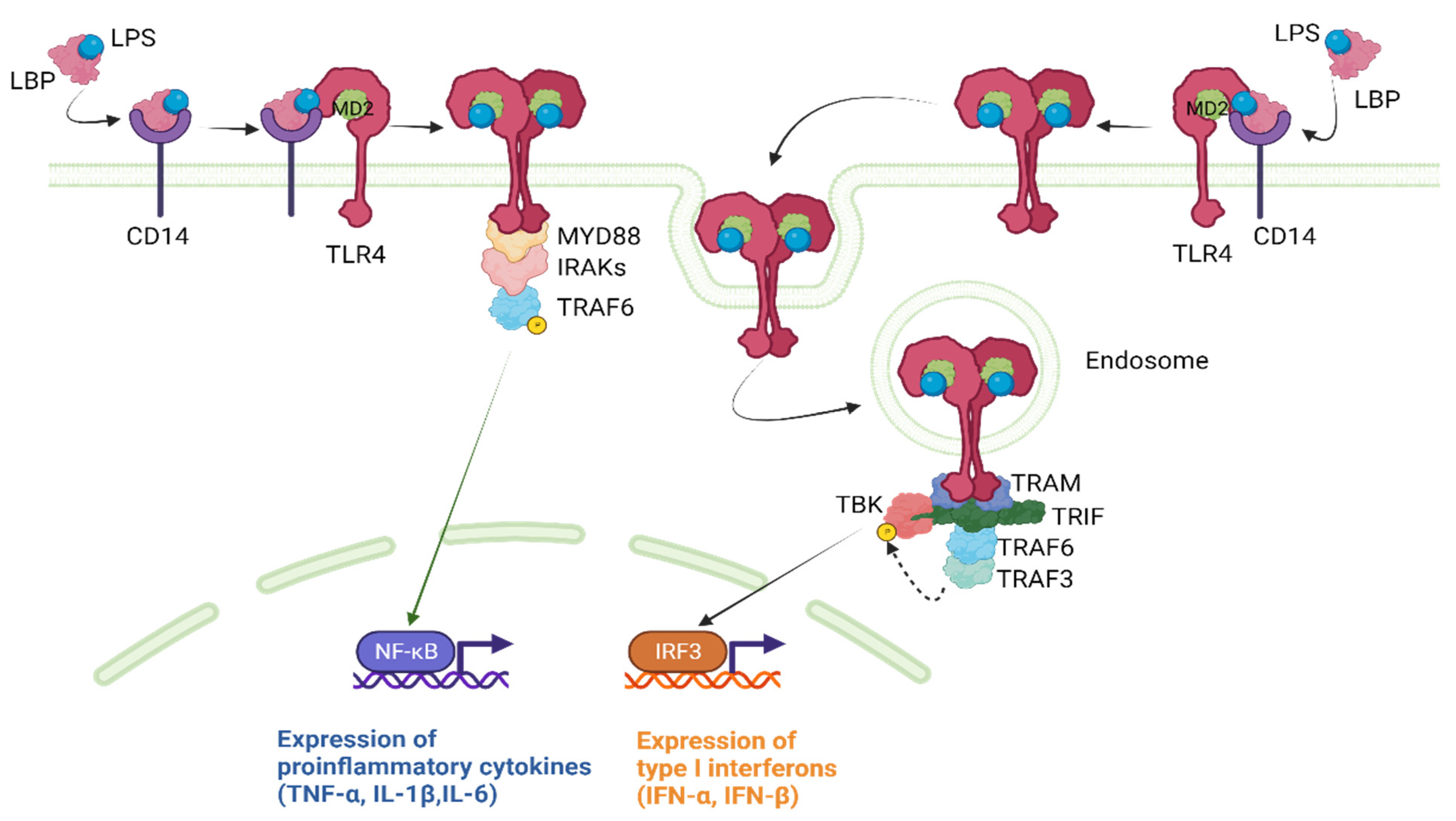
Figure 2. TLR4 downstream signaling pathways. IFN, interferon; IL, interleukin; IRAKs, IL-1 receptor-associated kinases; IRF3, interferon regulatory factor 3; LBP, lipopolysaccharide binding protein; LPS, lipopolysaccharide; MD2, myeloid differentiation factor 2; NF-κΒ, nuclear factor-kappa B; TBK1, TANK-binding kinase; TLR4, Toll-like receptor 4; TNF-α, tumor necrosis factor α; TRAF, tumor necrosis factor receptor-associated factor; TRAM, TRIF-related adaptor molecule; TRIF, TIR-domain-containing adaptor protein inducing interferon-β. Created with BioRender.com.
1.3.2. Negative Regulators of TLR4 Signaling
To prevent the excessive or prolonged activation of TLR4 signaling, several negative regulators have been identified [28][29]. These include the single immunoglobulin (Ig) IL-1 receptor-related molecule (SIGIRR), which acts as a decoy receptor and inhibits TLR4 signaling, the suppressor of cytokine signaling 1 (SOCS1) which negatively regulates the MyD88-dependent pathway, A20 which is a protein that can inhibit the activation of TLR4 signaling by removing ubiquitin chains from signaling molecules such as TRAF6, the toll-interacting protein (TOLLIP) which binds to the TLR4 receptor and prevents the recruitment of downstream signaling molecules and IRAK-M which is an inhibitor of IRAK1 and IRAK4 [29]. In addition, several microRNAs, such as microRNA-146b, have been identified to target TLR4 signaling components and modulate the magnitude and duration of the response [30].
In the context of HCC, mounting evidence has emerged, pointing toward an intricate interplay between EVs and TLR4 pathways [31]. TLR4 signaling is a multifaceted player in HCC, influencing metastasis, drug resistance, epigenetic regulation, proliferation, apoptosis, and angiogenesis [31]. TLR4 signaling in HCC immunotherapy involves enhancing the effectiveness of treatments like atezolizumab and bevacizumab or tremelimumab and durvalumab. It has shown promise in reshaping the tumor microenvironment and promoting anti-tumor immune responses [31]. TLR4 signaling can improve HCC cancer vaccines, regulate the immune landscape of the HCC microenvironment, and impact various immune cells, including T cells, B cells, dendritic cells, neutrophils, myeloid-derived suppressor cells, and macrophages [31]. Targeting TLR4 signaling may enhance the efficacy of the PD-1 blockade and other immunotherapies in HCC treatment [31]. Utilizing extracellular vesicles as a platform for drug delivery is a burgeoning area of research and development [18]. The field of EVs has seen rapid evolution, spanning from their discovery in 1967 to their current applications in diagnostics, therapeutics, and drug delivery systems. Future prospects for EVs in drug delivery are promising, with standardized procedures and new insights into donor cell types and drug-loading techniques expected to drive clinical successes [32]. The use of EVs in HCC treatment and the delivery of nucleic acids and small molecule drugs are on the rise [32]. For example, hepatocyte-derived EVs enriched with saturated fatty acids activate TLR4 signaling, promoting pro-inflammatory responses and hepatocyte insulin resistance [33][34]. In a similar way, the exploitation of EVs for the delivery of various molecules that could interfere with TLR4 signaling has increased interest in the field of HCC.
2. The Molecular Mechanisms of LPS Tolerance
The liver in humans receives 1.5 L of blood every minute, originating from two sources: the portal vein and the hepatic artery. This blood supply carries an enormous antigenic load, consisting of harmless dietary and commensal products that the hepatic immune system must tolerate [35]. Simultaneously, the immune system in the liver must respond to a variety of blood-borne pathogens, such as viruses, bacteria, and parasites, as well as metastatic cells that frequently target the liver [36]. To address this challenge, the liver requires tight immune regulation, or the so-called LPS/endotoxin tolerance [37]. In essence, the liver has a complex and unique system that enables it to manage the dual challenge of tolerating harmless antigens and responding appropriately to pathogenic challenges [36][38]. Several cellular mechanisms, such as immunosuppressive cells and molecules, including cytokines and ligands, are present in abundance in the liver to ensure that pathogen products and antigens typically do not stimulate immune responses.
To begin with, numerous non-parenchymal cells (NPCs) including liver sinusoidal endothelial cells (LSECs), Kupffer cells (KCs) and dendritic cells (DCs) have the ability to present antigens to T cells in ways that promote exhaustion [39][40], leading to the expression of inhibitory receptors on T cells such as programmed cell death protein 1 (PD-1) and T-cell Ig mucin domain-containing protein 3 (Tim-3) [41]. Secondly, another key mechanism of immune tolerance in the liver involves liver DCs. The liver contains multiple subsets of DCs, including myeloid DCs (mDCs) and plasmacytoid DCs (pDCs). These cells are specialized for antigen presentation and can induce either a state of tolerance or effective immunity [42]. In the liver, mDCs express programmed cell death ligand-1 (PD-L1), which drives the activation and expansion of classic FoxP3+ CD25+ CD4+ T regulatory (Treg) cells [43], which have been shown to suppress liver allograft rejection. DCs can also promote Treg development through their expression of indoleamine-pyrrole 2,3-dioxygenase (IDO), an enzyme that catabolizes tryptophan and generates an immunosuppressive product (kynurenine) [44]. Another important mechanism of immune tolerance in the liver is the role of KCs, which are resident macrophages of hepatic sinusoids. KCs can endocytose fragments of damaged hepatocytes, which convey transforming growth factor (TGF)-β1 and cause KCs to secrete IL-10 [45]. IL-10 acts in an autocrine manner, leading KCs to induce a variety of immunosuppressive mechanisms, including both effector T cell suppression and Treg cell promotion [40][46]. KCs also express B7-H1 (PD-L1), a ligand that engages the PD-1 receptor on activated T cells, resulting in clonal exhaustion [47]. Continuous exposure to LPS from the intestine may cause the down-regulation of TLR4-signaling pathways in KCs, resulting in LPS tolerance. This tolerance is partly induced by iIRAK-M [48]. Regarding LSECs, they respond to LPS initially by expressing TLR4/CD14, but with repeated stimulation, they become unresponsive to LPS while still retaining their scavenger activity. The LPS tolerance observed in LSECs is marked by a decrease in the nuclear translocation of NF-κB upon subsequent LPS exposure, which is intricately associated with the production of prostanoids. LPS tolerance in LSECs results in reduced leukocyte adhesion following LPS rechallenge and improved sinusoidal microcirculation in the liver, contributing to the local hepatic control of inflammation [49]. The molecular mechanisms that have been associated with the suppression of TLR4 signaling after prolonged LPS stimulation [50] involve changes in gene expression, signaling pathways, and epigenetic modifications and are analyzed in depth elsewhere [37]. These mechanisms include reduced LPS-induced MyD88-TLR4 association [51], reduced IRAK activity [51], enhanced nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor α (IkBα) degradation [52] and reduced activation of p38, extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and/or NF-κB [53]. Additionally, negative feedback regulators of TLR4 signaling, such as A20 [54][55] and IRAK-M [56], have been found to delay the onset of LPS tolerance and could contribute to the suppression of gene expression during tolerance [37]. Moreover, NF-κB subunits and other transcription factors that are activated by LPS are also altered during tolerance. In naive cells, LPS stimulation leads to the loss of histone H3K9 demethylation [57] and an increase in H4 acetylation, allowing for SWItch/sucrose non-fermentable (SWI/SNF) complex recruitment and gene transcription. However, in tolerized cells, a Trichostatin A (TSA)-sensitive histone deacetylase may prevent H4 acetylation. Sirt1 facilitates the recruitment of RelB to promoters of tolerizable genes [58][59], subsequently engaging G9a to inhibit the expression of tolerizable genes by countering H3K9 demethylation [60]. For non-tolerizable gene promoters, G9a demethylates H3K9 in naive cells through the basal association of Atf7 [61]. Subsequent to tolerization, Atf7, and G9a are eliminated from these promoters, leading to the loss of H3K9 demethylation, which permits gene expression. Furthermore, latent enhancers might allow for the rapid expression of some genes during tolerance [62]. It is important to note that these changes have been directly shown to occur for only particular subsets of tolerizable and non-tolerizable genes and specific gene subsets, which likely differ in the specifics of their regulation, although the general paradigms of histone modification and transcription factor recruitment may be broadly applicable [37]. TLR4-signaling pathway alterations during LPS tolerance are summarized in Figure 3.
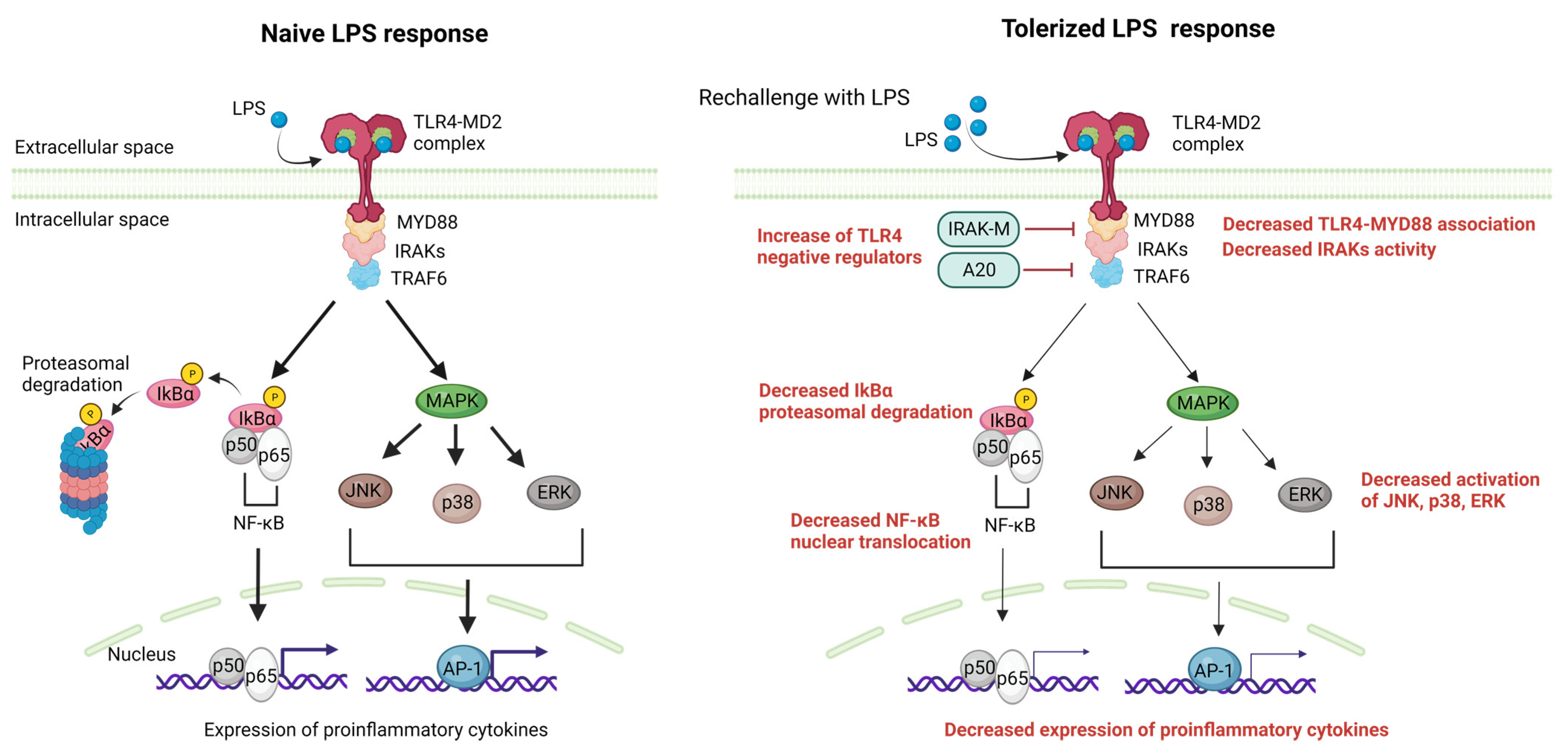
Figure 3. The liver manages a complex antigenic load from the blood, relying on immune tolerance mechanisms and mainly LPS tolerance. Myd88-dependent pathways activated after LPS engagement to the LPS-MD2 complex are negatively regulated in the case of LPS rechallenge. The main mechanisms include the increased activity of TLR4 negative regulators (i.e., IRAK-M,A20), decreased TLR4-Myd88 association, decreased IRAKs activity, decreased IkBα degradation resulting in decreased NF-κΒ nuclear translocation, and the decreased MAPK-mediated activation of JNK,p38 and ERK, resulting in decreased AP-1 induced transcription. The net result of these alterations is a reduction in the expression of proinflammatory cytokines after repeated exposure to LPS. AP-1, activator protein 1; ERKs, extracellular signal-regulated kinase; IRAKs, IL-1 receptor-associated kinases; JNK, c-Jun N-terminal kinase; LPS, lipopolysaccharide; MAPK,mitogen-activated protein kinase; MD2, myeloid differentiation factor 2; NF-κΒ, nuclear factor-kappa B; TLR4, Toll-like receptor 4; TRAF, tumor necrosis factor receptor-associated factor. Created with BioRender.com.
References
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462.
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249.
- Akinyemiju, T.; Abera, S.; Ahmed, M.; Alam, N.; Alemayohu, M.A.; Allen, C.; Al-Raddadi, R.; Alvis-Guzman, N.; Amoako, Y.; Artaman, A.; et al. The burden of primary liver cancer and underlying etiologies from 1990 to 2015 at the global, regional, and national level results from the global burden of disease study 2015. JAMA Oncol. 2017, 3, 1683–1691.
- Benson, A.B.; D’Angelica, M.I.; Abrams, T.; Abbott, D.E.; Ahmed, A.; Anaya, D.A.; Anders, R.; Are, C.; Bachini, M.; Binder, D.; et al. NCCN Guidelines® Insights: Biliary Tract Cancers, Version 2.2023. J. Natl. Compr. Canc. Netw. 2023, 21, 694–704.
- Elsharkasy, O.M.; Nordin, J.Z.; Hagey, D.W.; De Jong, O.G.; Schiffelers, R.M.; Andaloussi, S.E.; Vader, P. Extracellular vesicles as drug delivery systems: Why and how? Adv. Drug Deliv. Rev. 2020, 159, 332–343.
- Papadakos, S.P.; Dedes, N.; Pergaris, A.; Gazouli, M.; Theocharis, S. Exosomes in the Treatment of Pancreatic Cancer: A Moonshot to PDAC Treatment? Int. J. Mol. Sci. 2022, 23, 3620.
- Seay, T.W.; Suo, Z. Roles of Extracellular Vesicles on the Progression and Metastasis of Hepatocellular Carcinoma. Cells 2023, 12, 1879.
- Toh, W.S.; Yarani, R.; El Andaloussi, S.; Cho, B.S.; Choi, C.; Corteling, R.; De Fougerolles, A.; Gimona, M.; Herz, J.; Khoury, M.; et al. A report on the International Society for Cell & Gene Therapy 2022 Scientific Signature Series, “Therapeutic advances with native and engineered human extracellular vesicles”. Cytotherapy 2023, 25, 810–814.
- Willms, E.; Cabañas, C.; Mäger, I.; Wood, M.J.A.; Vader, P. Extracellular Vesicle Heterogeneity: Subpopulations, Isolation Techniques, and Diverse Functions in Cancer Progression. Front. Immunol. 2018, 9, 738.
- Cocucci, E.; Meldolesi, J. Ectosomes and exosomes: Shedding the confusion between extracellular vesicles. Trends Cell Biol. 2015, 25, 364–372.
- Kalluri, R.; McAndrews, K.M. The role of extracellular vesicles in cancer. Cell 2023, 186, 1610–1626.
- Yuana, Y.; Sturk, A.; Nieuwland, R. Extracellular vesicles in physiological and pathological conditions. Blood Rev. 2013, 27, 31–39.
- Sinha, D.; Roy, S.; Saha, P.; Chatterjee, N.; Bishayee, A. Trends in Research on Exosomes in Cancer Progression and Anticancer Therapy. Cancers 2021, 13, 326.
- Kong, J.; Tian, H.; Zhang, F.; Zhang, Z.; Li, J.; Liu, X.; Li, X.; Liu, J.; Li, X.; Jin, D.; et al. Extracellular vesicles of carcinoma-associated fibroblasts creates a pre-metastatic niche in the lung through activating fibroblasts. Mol. Cancer 2019, 18, 175.
- Papadakos, S.P.; Machairas, N.; Stergiou, I.E.; Arvanitakis, K.; Germanidis, G.; Frampton, A.E.; Theocharis, S. Unveiling the Yin-Yang Balance of M1 and M2 Macrophages in Hepatocellular Carcinoma: Role of Exosomes in Tumor Microenvironment and Immune Modulation. Cells 2023, 12, 2036.
- Kwantwi, L.B. Exosome-mediated crosstalk between tumor cells and innate immune cells: Implications for cancer progression and therapeutic strategies. J. Cancer Res. Clin. Oncol. 2023, 149, 9487–9503.
- Zeng, H.; Guo, S.; Ren, X.; Wu, Z.; Liu, S.; Yao, X. Current Strategies for Exosome Cargo Loading and Targeting Delivery. Cells 2023, 12, 1416.
- De Castilla, P.E.M.; Tong, L.; Huang, C.; Sofias, A.M.; Pastorin, G.; Chen, X.; Storm, G.; Schiffelers, R.M.; Wang, J.-W. Extracellular vesicles as a drug delivery system: A systematic review of preclinical studies. Adv. Drug Deliv. Rev. 2021, 175, 113801.
- Liu, B.H.M.; Tey, S.K.; Mao, X.; Ma, A.P.Y.; Yeung, C.L.S.; Wong, S.W.K.; Ng, T.H.; Xu, Y.; Yao, Y.; Fung, E.Y.M.; et al. TPI1-reduced extracellular vesicles mediated by Rab20 downregulation promotes aerobic glycolysis to drive hepatocarcinogenesis. J. Extracell. Vesicles 2021, 10, e12135.
- Lin, M.; Liao, W.; Dong, M.; Zhu, R.; Xiao, J.; Sun, T.; Chen, Z.; Wu, B.; Jin, J. Exosomal neutral sphingomyelinase 1 suppresses hepatocellular carcinoma via decreasing the ratio of sphingomyelin/ceramide. FEBS J. 2018, 285, 3835–3848.
- Cheng, Z.; Lei, Z.; Yang, P.; Si, A.; Xiang, D.; Tang, X.; Guo, G.; Zhou, J.; Hüser, N. Exosome-transmitted p120-catenin suppresses hepatocellular carcinoma progression via STAT3 pathways. Mol. Carcinog. 2019, 58, 1389–1399.
- Wang, X.; Shen, H.; Zhangyuan, G.; Huang, R.; Zhang, W.; He, Q.; Jin, K.; Zhuo, H.; Zhang, Z.; Wang, J.; et al. 14-3-3ζ delivered by hepatocellular carcinoma-derived exosomes impaired anti-tumor function of tumor-infiltrating T lymphocytes. Cell Death Dis. 2018, 9, 159.
- Laskowski, J.; Renner, B.; Pickering, M.C.; Serkova, N.J.; Smith-Jones, P.M.; Clambey, E.T.; Nemenoff, R.A.; Thurman, J.M. Complement factor H–deficient mice develop spontaneous hepatic tumors. J. Clin. Investig. 2020, 130, 4039–4054.
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384.
- Needham, B.D.; Trent, M.S. Fortifying the barrier: The impact of lipid A remodelling on bacterial pathogenesis. Nat. Rev. Microbiol. 2013, 11, 467–481.
- Rock, K.L.; Kono, H. The Inflammatory Response to Cell Death. Annu. Rev. Pathol. Mech. Dis. 2008, 3, 99–126.
- O’Neill, L.A.J.; Bowie, A.G. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 2007, 7, 353–364.
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511.
- Liew, F.Y.; Xu, D.; Brint, E.K.; O’Neill, L.A.J. Negative regulation of Toll-like receptor-mediated immune responses. Nat. Rev. Immunol. 2005, 5, 446–458.
- Curtale, G.; Mirolo, M.; Renzi, T.A.; Rossato, M.; Bazzoni, F.; Locati, M. Negative regulation of Toll-like receptor 4 signaling by IL-10–dependent microRNA-146b. Proc. Natl. Acad. Sci. USA 2013, 110, 11499–11504.
- Papadakos, S.P.; Arvanitakis, K.; Stergiou, I.E.; Lekakis, V.; Davakis, S.; Christodoulou, M.-I.; Germanidis, G.; Theocharis, S. The Role of TLR4 in the Immunotherapy of Hepatocellular Carcinoma: Can We Teach an Old Dog New Tricks? Cancers 2023, 15, 2795.
- Zeng, Y.; Hu, S.; Luo, Y.; He, K. Exosome Cargos as Biomarkers for Diagnosis and Prognosis of Hepatocellular Carcinoma. Pharmaceutics 2023, 15, 2365.
- Arvanitakis, K.; Papadakos, S.P.; Lekakis, V.; Koufakis, T.; Lempesis, I.G.; Papantoniou, E.; Kalopitas, G.; Georgakopoulou, V.E.; Stergiou, I.E.; Theocharis, S.; et al. Meeting at the Crossroad between Obesity and Hepatic Carcinogenesis: Unique Pathophysiological Pathways Raise Expectations for Innovative Therapeutic Approaches. Int. J. Mol. Sci. 2023, 24, 14704.
- Garcia-Martinez, I.; Alen, R.; Pereira, L.; Povo-Retana, A.; Astudillo, A.M.; Hitos, A.B.; Gomez-Hurtado, I.; Lopez-Collazo, E.; Boscá, L.; Francés, R.; et al. Saturated fatty acid-enriched small extracellular vesicles mediate a crosstalk inducing liver inflammation and hepatocyte insulin resistance. JHEP Rep. 2023, 5, 100756.
- Racanelli, V.; Rehermann, B. The liver as an immunological organ. Hepatology 2006, 43, S54–S62.
- Doherty, D.G. Immunity, tolerance and autoimmunity in the liver: A comprehensive review. J. Autoimmun. 2016, 66, 60–75.
- Seeley, J.J.; Ghosh, S. Molecular mechanisms of innate memory and tolerance to LPS. J. Leukoc. Biol. 2017, 101, 107–119.
- Calne, R. Immunological tolerance: The liver effect. J. Gastroenterol. Hepatol. 2002, 17, S488–S490.
- Liaskou, E.; Hirschfield, G.M.; Gershwin, M.E. Mechanisms of tissue injury in autoimmune liver diseases. Semin. Immunopathol. 2014, 36, 553–568.
- Zhang, M.; Xu, S.; Han, Y.; Cao, X. Apoptotic cells attenuate fulminant hepatitis by priming Kupffer cells to produce interleukin-10 through membrane-bound TGF-β. Hepatology 2011, 53, 306–316.
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687.
- Lutz, M.B.; Schuler, G. Immature, semi-mature and fully mature dendritic cells: Which signals induce tolerance or immunity? Trends Immunol. 2002, 23, 445–449.
- Kelly, A.; Fahey, R.; Fletcher, J.M.; Keogh, C.; Carroll, A.G.; Siddachari, R.; Geoghegan, J.; Hegarty, J.E.; Ryan, E.J.; O’farrelly, C. CD141+ myeloid dendritic cells are enriched in healthy human liver. J. Hepatol. 2014, 60, 135–142.
- Sun, X.; Gong, Z.-J.; Wang, Z.-W.; Li, T.; Zhang, J.-Y.; Sun, H.-C.; Liu, S.; Huang, L.; Huang, C.; Peng, Z.-H. IDO-competent-DCs induced by IFN-γ attenuate acute rejection in rat liver transplantation. J. Clin. Immunol. 2012, 32, 837–847.
- Erhardt, A.; Biburger, M.; Papadopoulos, T.; Tiegs, G. IL-10, regulatory T cells, and Kupffer cells mediate tolerance in concanavalin A-induced liver injury in mice. Hepatology 2007, 45, 475–485.
- Yan, M.-L. Inhibition of allogeneic T-cell response by Kupffer cells expressing indoleamine 2,3-dioxygenase. World J. Gastroenterol. 2010, 16, 636–640.
- Perez, R.V.; Swanson, C.; Morgan, M.; Erickson, K.; Hubbard, N.E.; German, J.B. Portal venous transfusion up-regulates kupffer cell cyclooxygenase activity. Transplantation 1997, 64, 135–139.
- Liu, Z.-J.; Yan, L.-N.; Li, X.-H.; Xu, F.-L.; Chen, X.-F.; You, H.-B.; Gong, J.-P. Up-Regulation of IRAK-M is Essential for Endotoxin Tolerance Induced by a Low Dose of Lipopolysaccharide in Kupffer Cells. J. Surg. Res. 2008, 150, 34–39.
- Uhrig, A.; Banafsche, R.; Kremer, M.; Hegenbarth, S.; Hamann, A.; Neurath, M.; Gerken, G.; Limmer, A.; Knolle, P.A.; Knolle, P.A. Development and functional consequences of LPS tolerance in sinusoidal endothelial cells of the liver. J. Leukoc. Biol. 2005, 77, 626–633.
- Nomura, F.; Akashi, S.; Sakao, Y.; Sato, S.; Kawai, T.; Matsumoto, M.; Nakanishi, K.; Kimoto, M.; Miyake, K.; Takeda, K.; et al. Cutting edge: Endotoxin tolerance in mouse peritoneal macrophages correlates with down-regulation of surface toll-like receptor 4 expression. J. Immunol. 2000, 164, 3476–3479.
- Medvedev, A.E.; Lentschat, A.; Wahl, L.M.; Golenbock, D.T.; Vogel, S.N. Dysregulation of LPS-induced Toll-like receptor 4-MyD88 complex formation and IL-1 receptor-associated kinase 1 activation in endotoxin-tolerant cells. J. Immunol. 2002, 169, 5209–5216.
- Medvedev, A.E.; Kopydlowski, K.M.; Vogel, S.N. Inhibition of Lipopolysaccharide-Induced Signal Transduction in Endotoxin-Tolerized Mouse Macrophages: Dysregulation of Cytokine, Chemokine, and Toll-Like Receptor 2 and 4 Gene Expression. J. Immunol. 2000, 164, 5564–5574.
- Medvedev, A.E.; Henneke, P.; Schromm, A.; Lien, E.; Ingalls, R.; Fenton, M.J.; Golenbock, D.T.; Vogel, S.N. Induction of tolerance to lipopolysaccharide and mycobacterial components in Chinese hamster ovary/CD14 cells is not affected by overexpression of Toll-like receptors 2 or 4. J. Immunol. 2001, 167, 2257–2267.
- Xiong, Y.; Qiu, F.; Piao, W.; Song, C.; Wahl, L.M.; Medvedev, A.E. Endotoxin tolerance impairs IL-1 receptor-associated kinase (IRAK) 4 and TGF-beta-activated kinase 1 activation, K63-linked polyubiquitination and assembly of IRAK1, TNF receptor-associated factor 6, and IkappaB kinase gamma and increases A20 expression. J. Biol. Chem. 2011, 286, 7905–7916.
- Xiong, Y.; Medvedev, A.E. Induction of endotoxin tolerance in vivo inhibits activation of IRAK4 and increases negative regulators IRAK-M, SHIP-1, and A20. J. Leukoc. Biol. 2011, 90, 1141–1148.
- Kobayashi, K.; Hernandez, L.D.; Galán, J.E.; Janeway, C.A.; Medzhitov, R.; Flavell, R.A. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell 2002, 110, 191–202.
- El Gazzar, M.; Yoza, B.K.; Hu, J.Y.-Q.; Cousart, S.L.; McCall, C.E. Epigenetic silencing of tumor necrosis factor alpha during endotoxin tolerance. J. Biol. Chem. 2007, 282, 26857–26864.
- Liu, T.F.; Yoza, B.K.; El Gazzar, M.; Vachharajani, V.T.; McCall, C.E. NAD+-dependent SIRT1 deacetylase participates in epigenetic reprogramming during endotoxin tolerance. J. Biol. Chem. 2011, 286, 9856–9864.
- Yoza, B.K.; Hu, J.Y.-Q.; Cousart, S.L.; Forrest, L.M.; McCall, C.E. Induction of RelB Participates in Endotoxin Tolerance. J. Immunol. 2006, 177, 4080–4085.
- Chen, X.; El Gazzar, M.; Yoza, B.K.; McCall, C.E. The NF-κB factor RelB and histone H3 lysine methyltransferase G9a directly interact to generate epigenetic silencing in endotoxin tolerance. J. Biol. Chem. 2009, 284, 27857–27865.
- Yoshida, K.; Maekawa, T.; Zhu, Y.; Renard-Guillet, C.; Chatton, B.; Inoue, K.; Uchiyama, T.; Ishibashi, K.-I.; Yamada, T.; Ohno, N.; et al. The transcription factor ATF7 mediates lipopolysaccharide-induced epigenetic changes in macrophages involved in innate immunological memory. Nat. Immunol. 2015, 16, 1034–1043.
- Ostuni, R.; Piccolo, V.; Barozzi, I.; Polletti, S.; Termanini, A.; Bonifacio, S.; Curina, A.; Prosperini, E.; Ghisletti, S.; Natoli, G. Latent enhancers activated by stimulation in differentiated cells. Cell 2013, 152, 157–171.
More
Information
Subjects:
Gastroenterology & Hepatology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
532
Revisions:
2 times
(View History)
Update Date:
09 Nov 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No