Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Asterios Tsiftsoglou | -- | 2821 | 2023-11-06 19:55:03 | | | |
| 2 | Camila Xu | -1 word(s) | 2820 | 2023-11-07 02:23:41 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Georgiou-Siafis, S.K.; Tsiftsoglou, A.S. The Biological Functions of Glutathione. Encyclopedia. Available online: https://encyclopedia.pub/entry/51214 (accessed on 24 July 2026).
Georgiou-Siafis SK, Tsiftsoglou AS. The Biological Functions of Glutathione. Encyclopedia. Available at: https://encyclopedia.pub/entry/51214. Accessed July 24, 2026.
Georgiou-Siafis, Sofia K., Asterios S. Tsiftsoglou. "The Biological Functions of Glutathione" Encyclopedia, https://encyclopedia.pub/entry/51214 (accessed July 24, 2026).
Georgiou-Siafis, S.K., & Tsiftsoglou, A.S. (2023, November 06). The Biological Functions of Glutathione. In Encyclopedia. https://encyclopedia.pub/entry/51214
Georgiou-Siafis, Sofia K. and Asterios S. Tsiftsoglou. "The Biological Functions of Glutathione." Encyclopedia. Web. 06 November, 2023.
Copy Citation
Glutathione (GSH) is a ubiquitous tripeptide that is biosynthesized in situ at high concentrations (1–5 mM) and involved in the regulation of cellular homeostasis via multiple mechanisms.
GSH redox cycle
GSH S-conjugates
GSH adducts
GSH metabolism
xenobiotics
1. Structure, Biosynthesis of GSH and Key Enzymes Involved in GSH Homeostasis
Glutathione (GSH) (PubChem CID:124886) is a ubiquitous tripeptide (of L-gamma-Glutamyl Acid-L-Cysteinyl-glycine), which is found in all aerobic organisms. In mammals, GSH accounts for >90% of non-protein thiols. GSH was discovered in 1888 by De-Rey Pailhade in different sources, like blood, sheep brain, fish, and vegetables, and named “philothion” (a substance that loves sulfur) [1][2]. It is highly important to note that this work had been prepared before the peptide bond had been formally proposed [3]. Thirty-three years later, philothion was renamed glutathione by Hopkins and Hunter, who discovered its non-enzymatic reducing power, water solubility, and its high reactivity with dyes and several other chemicals [4][5][6]. Meister, who contributed a great deal to elucidating the chemical structure of GSH, wrote a detailed historical review [7]. Thereafter, the research on GSH homeostasis was extrapolated, and nowadays, there are nearly 59,000 retrievals in PubMed (research articles, reviews, and others) referenced under the term “GSH”.
The main organs involved in the homeostasis of GSH are the liver, kidneys, lungs, and skeletal muscle [8]. In addition, red blood cells (RBCs) are a rich source of antioxidants (mainly GSH and ascorbic acid), counterbalancing the potential harmful effect associated with hemoglobin carrying [9]. Structurally, GSH possesses a gamma–peptide bond, employing the side chain of carbonyl from glutamic acid, rendering GSH resistant to peptidases. The functional group of GSH is the thiol of the cysteinyl moiety. Due to its prominent role in tissues, GSH is found in several forms, with its reduced form the main one. A small quantity of GSH is oxidized, reacting with cellular reactive oxygen species (ROS), yielding GSSG (disulfide GSH) (normal ratio of GSH/GSSG around 100:1). However, GSH is continually being regenerated by GSSG reductase, a mainly cytosolic but also mitochondrial and nuclear enzyme, using NAPDH as a co-factor [10]. The subcellular localization of GSSG reductase coincides with organelles that accumulate high levels of ROS, such as mitochondria, where mainly superoxide radicals are produced [11]. In eukaryotes, the use of alternative initiation codons deciphers whether the produced GSSG reductase will be cytosolic or mitochondrial [12]. A thiol reductase also exists in lysosomes but lacks the specificity of GSSG reductase [13]. In fact, this specificity is exploited for accurate experimental measurements of GSH content [14]. Moreover, there are the mixed disulfides of GSH with protein thiols, which form a major part of total hepatic GSH, as uncovered using GSH-tagged mono-bromobimane [15]. Last but not least, GSH has been found in the form of the covalent GSH S-conjugates of both xenobiotics and endogenous substances, incrementally increasing the human metabolome library.
GSH is biosynthesized through two sequential ATP-dependent enzymatic reactions. At first, glutamic acid is bonded via gamma–peptide linkage to cysteine, which is catalyzed by gamma-glutamyl-cysteine synthetase or ligase (GCS or GCL). GCL is the rate-limiting enzyme in the biosynthesis of GSH, being competitively inhibited by GSH itself [16]. Among glutamic acid, cysteine, and glycine, cysteine is found at a lower concentration in plasma and tissues, and cysteine is thus the rate-limiting substrate in GSH biosynthesis. The GCL enzyme is a heterodimer composed of a catalytic and a regulatory subunit. Buthionine sulfoximide (BSO), the gold standard in the experimental depletion of GSH, inhibits this enzyme by forming transitional state analogs [17]. The second raw enzyme is GSH synthetase (GSS), condensing the gamma-glutamyl-cysteine moiety with glycine, via a regular peptide bond. Both enzymes are characterized by a low Km for ATP (at low μΜ level) [18], thus permitting GSH biosynthesis to continue in hypoxic and/or energy-depleted conditions, and highlighting its significance for body homeostasis. Cysteine is supplied to the organism via nutrition, protein catabolism and via the cystathionine pathway, acting in the liver to synthesize cysteine form methionine [19]. However, in extracellular fluids, such as plasma, cysteine is easily oxidized in the dipeptide cysteine. Both the enzymes of GSH biosynthesis are expressed in multiple tissues, such as liver, kidney, muscle, brain, and RBCs [8]. In fact, the gene encoding the catalytic subunit of GCL (GCLC) is under the control of the antioxidant-responsive element (ARE), promoting the binding of the major detoxification-involved transcription factor (TF), nuclear factor erythroid 2-related factor 2 (Nrf2) [20]. To this end, multiple factors, such as H2O2, ionizing radiation, smoke, and chemotherapeutics, induce the transcription of GCLC [8].
Free heme is known to be a major, intrinsic oxidant promoting severe damage to nearby tissues when released under hemolytic conditions [21][22]. Hemin (an oxidized, experimental form of heme) has been recently found, by the research group, to stabilize the Nrf2 TF via action at the protein level without affecting its mRNA levels in pro-erythroid K562 cells [23]. Mechanistically, hemin leads to inhibition of the Kelch-like ECH-associated protein 1 (Keap1)-dependent ubiquitination and degradation of Nrf2. At the same time, Keap1 becomes the substrate of the E3 ligase, attached to the holo-complex of Keap1-Nrf2 [24], and leads to the ubiquitination and proteasomal degradation of Keap1. Nrf2 is, thus, free to translocate into the nucleus and induces, by activating the transcription of GSH biosynthesis-related genes, GCLC and the gene of cystine glutamate antiporter (xCT), along with the gene of glutathione S-transferase P1 (GSTP1), to a lesser degree. The net result recorded is an increase in GSH content associated with the cytotoxic actions of hemin. There is indeed a discrepancy here, since the pro-oxidant effect of hemin diminishes GSH at the intracellular level in both RBCs and astrocytes [25][26]. In this cell model, an assumption would be that the concomitant induction of the xCT leads to a continuous supply of cystine, thus replenishing GSH.
The plasma concentration of GSH has been proposed as an indicator of the body’s health status [27]. Plasma GSH (calculated at 3.6 µM) is representative of total GSH [28]. This plasma concentration is at least 10-fold lower that the concentration of GSH found in the tissues. RBCs’ concentration in GSH is among the higher, at 0.4 to 3.0 mM, characterized by interindividual variations [29]. Liver concentration is the higher in the body, reaching 10 mΜ [30]. Lungs are charged with the role to detoxify air pollutants and ROS and thus contain 10-fold more GSH than plasma (200–400μΜ) [31]. In the brain, the GSH concentrations reach 8 mM, as measured in astrocytes [32] (see [33] for a detailed analysis of organs’ concentration in GSH). Plasma GSH comes from the liver, skeletal muscle, and RBCs’ excretion [34]. To this end, it has become clear that GSH has to be transported not only between the different organs but also from the cytoplasm being synthesized to mitochondria, the endoplasmic reticulum [35], and nucleus. There are indeed transporters of GSH, which are mainly members of the multidrug-associated resistance protein (MRP) family of ABC transporters and also of transporters named organic anion transporting polypeptides (OATPs) [36][37]. In mitochondria, porins are exploited for the transport of GSH [10] as well as the glyoxylases’ system [38]. Half of the quantity of GSH exported by the liver is driven to bile (GSH at 8–10 mM) [39] via mainly MRPs.
GSH homeostasis is thus a complicated process depending on the biosynthesis, transport, and utilization (as at GSH-S conjugates) as well as catabolism and regeneration of GSH (Figure 1). The membranous enzyme gamma-glutamyl transpeptidase (gamma-GT) bound in liver and bile ducts catabolizes the gamma–peptide bond of GSH, releasing the cysteinyl–glycine dipeptide cleaved by normal peptidases [40]. In fact, this is the major pathway used also for the catabolism of GSH S-conjugates. Regarding the key enzymes involved in the metabolism of GSH, there are the GSH peroxidases (GPx), involved in ROS detoxification, and GSTs, implicated in the covalent conjunction of GSH to mainly detoxify toxic compounds.
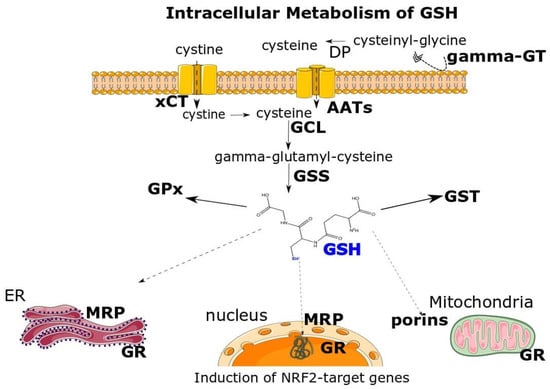
Figure 1. Multifaceted intracellular metabolism of GSH. Cysteine, as the rate-limiting substrate for GSH’s biosynthesis, can be supplied to cells indirectly by the cystine/glutamate antiporter (xCT). Subsequently, the intracellularly imported cystine is reduced to cysteine. Moreover, GSH can itself supply cysteine by the sequential action of extracellular gamma-glutamyltranspeptidase (gamma-GT) and dipeptidases (DP) releasing cysteine that is transported inside cells by common amino-acid transporters (AATs). Glutamate cysteine ligase (GCL) and GSH synthetase (GSS) act in the cytosol to synthesize GSH, serving the coenzyme to glutathione S-transferases (GSTs) and glutathione peroxidase (GPx). GSH is transported in the endoplasmic reticulum (ER), nucleus and mitochondria via multidrug-associated resistance protein (MRP) and porins, respectively. In addition, GSSG reductase (GR) regenerates the GSH pool. Different oxidants, xenobiotics and anti-oxidative compounds induce the GSH biosynthesis, via the NRF2 transcription factor, by upregulating GCL and/or xCT. Parts of the figure have been acquired by Smart Servier Medical Art.
2. The Biological Functions of GSH
GSH is indeed contained inside cells in concentrations higher than most metabolites [41]. It is not an exaggeration to say that GSH possesses multiple and highly crucial functions in the body [42] (summarized in Figure 2).
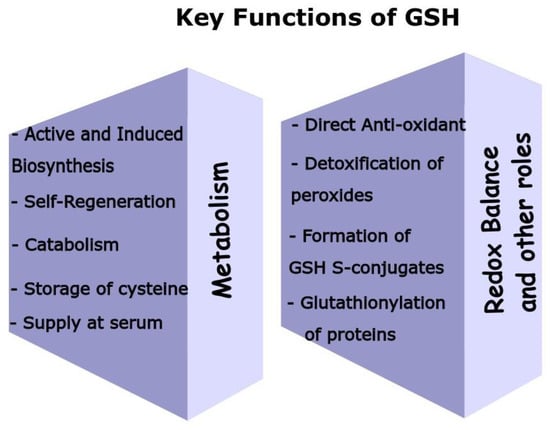
Figure 2. GSH employs multiple biochemical pathways to support key roles in mammalian cells and maintain the redox cycle. By taking into account all these functions and enzymes involved, GSH is indeed a key metabolite. GSH scavenges various ROS either directly or via the action of GSH peroxidases, where GSH is a co-factor. Moreover, GSH serves as a stable storage of the sulfhydryl group, while the self-regenerative capacity of GSH, by the GSH reductase, contributes to maintain its cellular levels. GSH biosynthesis occurs at high rates at many organs induced by oxidative stress or other stimuli. RBCs, liver, and skeletal muckle support plasma GSH’s levels, while liver (via the gamma-GT) is able to accelerate the uptake of GSH’s precursors. Finally, GSH forms various GSH S-conjugates with many different structurally compounds acting as the substrates, which is an event of vast importance in cell protection.
2.1. Antioxidant Role
The antioxidant property of GSH is probably the most known of its multilevel actions [43]. GSH is able to directly scavenge several ROS (hydrogen peroxide, superoxide and others) [44] and RNS (as peroxynitrite) [45]. GSH, reacting in vitro even at very low concentrations (10 μM), scavenges these reactive species, as evidenced by electron paramagnetic resonance spectrometry [46], and it decomposes hydrogen peroxide [47]. The pKa of the thiol moiety of GSH is at 9.6, providing a low reactivity [48], but this is not the only parameter that counts (see Eh below). In these reactions, GSH is converted to intermediate products like glutathiyl radicals and sulfenic acids. A detailed review regarding the reactions’ kinetics can be found elsewhere [49]. Given the relative intracellular quantities of these oxidants at the nM level, even under oxidative stress, it can be drawn than GSH can efficiently react with them [50]. This powerful antioxidant action of GSH led to the conclusion that it is the main mechanism of the cytoprotective action of the N-acetyl-cysteine (NAC), a rather weak antioxidant, acting as GSH’s precursor [43][46]. GSH can form a barrier to intracellular ROS propagation and be an important gatekeeper of mitochondrial DNA integrity [51], as shown in human lymphocytes [52]. Moreover, during the phase I detoxification reaction in liver cytochromes P450 enzymes, ROS are produced, such as in alcohol-induced liver damage [53].
GSH serves as a co-factor of the antioxidant-related enzymes, the glutathione peroxidases (GPxs) [49]. These enzymes accelerate the antioxidant potential of GSH, detoxifying mainly peroxides including, but not limited to, hydrogen peroxide. Eight different isoenzymes of GPxs exist, differing in their localization and specificity for phospholipid peroxides, cholesterol peroxides and others. Toxic peroxides are converted to non-toxic alcohols in a process coupled to GSH dimerization [54]. In addition, GSH aids in the regeneration of both vitamin C and E, being oxidized via reacting with ROS and lipid peroxides [55]. The advantage that GSH possesses is its enzymatic recycling by GSSG reductase, as analyzed in .
2.2. Cell Redox Status
In starting to analyze the other key roles of GSH, one should consider that GSH is the safe body’s alternative for storage of the unstable cysteine. Once needed, GSH will be degraded by the gamma-glutamyltranspeptidase (gamma-GT). The gamma-glutamyl cycle acts in organs, like the liver and kidneys, as a carrier transport system for amino acids [56]. Intracellularly, GSH has the prominent role of acting as the redox buffer to protect the proper folding of proteins. Cysteines are the highly conserved residues in proteins that are implicated in key functions, such as catalysis and metal binding [57]. Upon the deprotonation of cysteine to the thiolate ion, it is vulnerable to oxidation and adduction by electrophiles, disrupting the proper folding and function. For instance, a hallmark in hemin-induced cytotoxicity (HIC) is the perturbation of proper folding of proteins [58] and the abnormal cross-linking of RBCs’ major cytoskeletal proteins [59]. GSH, in such a high intracellular concentration, contributes to preventing these abnormal effects. To gain further insight into the high reactivity of thiols, it is worth considering Keap1, which acts as a sensor for electrophiles through its highly reactive cysteines, bearing a suitable environment to lower the pKa [60]. This binding leads to the inhibition of the Keap1-dependent ubiquitination of Nrf2 TF, stabilizing it, to induce on time a battery of cytoprotective genes.
The GSH/GSSG along with the couples of NADPH/NADP+ and thioredoxin (Trx)-SH/Trx-SS are essential in maintaining the cellular redox state [61]. In vivo, the redox potential of the GSH/GSSG couple (Eh, as calculated by Nernst equation [62]) is between −260 and−150 mV in different tissues [63]; the more negative the Eh value, the more potent the reductant. Under mild oxidative conditions, cellular signaling is affected, as key TFs, Nrf2, Nuclear Factor-kB, and Activator Protein (AP-1) are redox dependent [64]. Upon oxidative stress, the GSH/GSSG ratio significantly decreases, since GSH is consumed due to covalent conjugation to electrophiles, the formation of mixed disulfides with proteins, and the export of GSH and GSSG to reach places with high oxidative milieu [65].
The covalent conjugation of GSH to cysteinyl residues of proteins is an important post-translational modification (S-glutathionylation) [66]. One of its roles is the protection of this critical residue from oxidative stress-induced damage. Moreover, this modification has a regulatory roles in the function of proteins, as in the case of Heat Shock Protein 70 (HSP70), where the S-glutathionylated form mimics a substrate-binding condition [67], or in glyceraldeyde-3 phosphate dehydrogenase (GAPDH) where the GSH-mixed enzyme is enzymatically inactive [68]. S-glutathionylation is supposed to occur directly by GSH, but it is also accelerated by the enzymatic action of GSTs and at the same time be reversible by glutaredoxins.
One should not forget to mention that GSH is an important antioxidant keeping the redox balance in the endoplasmic reticulum (ER) [69]. As evidenced by the pioneering work of Hwang et al., using a small, radiolabeled peptide, susceptible to oxidation, the GSH/GSSG ratio in ER is remarkably low, ranging from 1:1 to 3:1. This oxidative environment is permissive for the action of protein disulfide isomerases (PDIs) toward the correct folding of proteins. To this end, the concentration of ER in GSH was significantly decreased by the expression of a chimeric γ-transpeptidase, while several proteins, used as the control, still properly folded [70].
2.3. Formation of GSH S-Conjugates
The conjugation of GSH to toxic electrophilic compounds is an underestimated function of GSH, taking into account its merit. As eloquently pointed out by Pizzorno, GSH does not merely scavenge ROS produced by the harmful oxidant but deals with the “problem” itself [41]. An example will be given with HIC, where a molar extracellular, excess of GSH (or other thiol, such as NAC and cysteine) conjugates with hemin, leading to a significant decrease in hemin’s intracellular accumulation [71][72]. To this end, ROS accumulation, impaired proteostasis, cell-cycle arrest, and non-classical apoptotic cell death, all molecular signs of HIC [23], were withdrawn in a dose-dependent manner proportional to the thiols’ quantity.
In fact, it has been long known that conjugation reactions are Phase II biotransformation reactions that occur mainly in the liver. The addition of GSH moiety takes place primarily by GSTs decreasing both the hydrophobicity and toxicity as well as promoting the detoxification and elimination of the unwanted compounds [73]. In some cases, the conjugation renders the resulting metabolites chemically inert or destabilized. There is also the case that GSTs are recruited for the biosynthesis of biologically important molecules, such as in leukotrienes. The substrates for these reactions are both endogenous (intrinsic) and exogenous (xenobiotics), being mainly electrophiles, Michael acceptors, epoxides, electron-withdrawing structures and carbonyls, which are able to react through nucleophilic additions with GSH. Moreover, nucleophilic substitutions and displacements are also important conjugation reactions. Over the years, many such novel metabolites were uncovered, referred to as GSH S-conjugates or GSH adducts, which were assigned their name by the chemical entity adducted.
Multiple families and classes of GSTs exist in mammals, including soluble (alpha, mu, pi, theta, zeta, omega, sigma and kappa) and six-membered membranous (microsomal) named membrane-associated proteins in eicosanoid and glutathione metabolism (MAPEG), which all have a well-conserved G-(Glutathione-binding) site. An important step in the catalysis is the activation of GSH to the thiyl radical. A critical residue (tyrosine, or serine, or cysteine) is responsible for GSH activation in the G-site [74]. The H-site composed of mainly hydrophobic residues is responsible for the substrate binding [75]. Overlapping substrates, serial reactions of nucleophilic addition, isomerization, and the stabilization of transitions states are common between the different GSTs [54][76].
References
- Rey-Pailhade, D. Sur un corps d’origine organique hydrogénant le soufre á froid. Hebd. Séances Acad. Sci. 1888, 106, 1683–1684.
- Rey-Pailhade, D. Sur un nouveau principe immédiat organique. le philothion. Bull. Soc. Hist Nat. Toulouse 1888, 173–180.
- Fruton, J.S. Contrasts in Scientific Style. Emil Fischer and Franz Hofmeister: Their Research Groups and Their Theory of Protein Structure. Proc. Am. Philos. Soc. JSTOR 1985, 129, 313–370.
- Hopkins, F.G. On an Autoxidisable Constituent of the Cell. Biochem. J. 1921, 15, 286–305.
- Hunter, G.; Eagles, B.A. Glutathione. A critical Study. J. Biol. Chem. 1927, 72, 147–166.
- Robert, D.; Simoni, R.L.H. Martha Vaughan. The Discovery of Glutathione by F. Gowland Hopkins and the Beginning of Biochemistry at Cambridge University. J. Biol. Chem. 2002, 277, 27–28.
- Meister, A. On the discovery of glutathione. Trends Biochem. Sci. 1988, 13, 185–188.
- Kretzschmar, M. Regulation of hepatic glutathione metabolism and its role in hepatotoxicity. Exp. Toxicol. Pathol. Off. J. Ges. Fur. Toxikol. Pathol. 1996, 48, 439–446.
- Franco, R.; Navarro, G.; Martinez-Pinilla, E. Antioxidant Defense Mechanisms in Erythrocytes and in the Central Nervous System. Antioxidants 2019, 8, 46.
- Couto, N.; Wood, J.; Barber, J. The role of glutathione reductase and related enzymes on cellular redox homoeostasis network. Free Radic. Biol. Med. 2016, 95, 27–42.
- Kowaltowski, A.J.; de Souza-Pinto, N.C.; Castilho, R.F.; Vercesi, A.E. Mitochondria and reactive oxygen species. Free Radic. Biol. Med. 2009, 47, 333–343.
- Couto, N.; Malys, N.; Gaskell, S.J.; Barber, J. Partition and turnover of glutathione reductase from Saccharomyces cerevisiae: A proteomic approach. J. Proteome Res. 2013, 12, 2885–2894.
- Chiang, H.S.; Maric, M. Lysosomal thiol reductase negatively regulates autophagy by altering glutathione synthesis and oxidation. Free Radic. Biol. Med. 2011, 51, 688–699.
- Rahman, I.; Kode, A.; Biswas, S.K. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat. Protoc. 2006, 1, 3159–3165.
- Bass, R.; Ruddock, L.W.; Klappa, P.; Freedman, R.B. A major fraction of endoplasmic reticulum-located glutathione is present as mixed disulfides with protein. J. Biol. Chem. 2004, 279, 5257–5262.
- Griffith, O.W. Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free Radic. Biol. Med. 1999, 27, 922–935.
- Crook, T.R.; Souhami, R.L.; Whyman, G.D.; McLean, A.E. Glutathione depletion as a determinant of sensitivity of human leukemia cells to cyclophosphamide. Cancer Res. 1986, 46, 5035–5038.
- Meierjohann, S.; Walter, R.D.; Muller, S. Glutathione synthetase from Plasmodium falciparum. Biochem. J. 2002, 363, 833–838.
- Zuhra, K.; Augsburger, F.; Majtan, T.; Szabo, C. Cystathionine-beta-Synthase: Molecular Regulation and Pharmacological Inhibition. Biomolecules 2020, 10, 697.
- Mani, M.; Khaghani, S.; Gol Mohammadi, T.; Zamani, Z.; Azadmanesh, K.; Meshkani, R.; Pasalar, P.; Mostafavi, E. Activation of Nrf2-Antioxidant Response Element Mediated Glutamate Cysteine Ligase Expression in Hepatoma Cell line by Homocysteine. Hepat. Mon. 2013, 13, e8394.
- Balla, G.; Jacob, H.S.; Eaton, J.W.; Belcher, J.D.; Vercellotti, G.M. Hemin: A possible physiological mediator of low density lipoprotein oxidation and endothelial injury. Arterioscler. Thromb. A J. Vasc. Biol. 1991, 11, 1700–1711.
- Kumar, S.; Bandyopadhyay, U. Free heme toxicity and its detoxification systems in human. Toxicol. Lett. 2005, 157, 175–188.
- Georgiou-Siafis, S.K.; Tsiftsoglou, A.S. Activation of KEAP1/NRF2 stress signaling involved in the molecular basis of hemin-induced cytotoxicity in human pro-erythroid K562 cells. Biochem. Pharmacol. 2020, 175, 113900.
- Suzuki, T.; Yamamoto, M. Molecular basis of the Keap1-Nrf2 system. Free Radic. Biol. Med. 2015, 88, 93–100.
- Atamna, H.; Ginsburg, H. Heme degradation in the presence of glutathione. A proposed mechanism to account for the high levels of non-heme iron found in the membranes of hemoglobinopathic red blood cells. J. Biol. Chem. 1995, 270, 24876–24883.
- Laird, M.D.; Wakade, C.; Alleyne, C.H., Jr.; Dhandapani, K.M. Hemin-induced necroptosis involves glutathione depletion in mouse astrocytes. Free Radic. Biol. Med. 2008, 45, 1103–1114.
- Richie, J.P., Jr.; Abraham, P.; Leutzinger, Y. Long-term stability of blood glutathione and cysteine in humans. Clin. Chem. 1996, 42, 1100–1105.
- Perez, L.M.; Hooshmand, B.; Mangialasche, F.; Mecocci, P.; Smith, A.D.; Refsum, H.; Inzitari, M.; Fratiglioni, L.; Rizzuto, D.; Calderon-Larranaga, A. Glutathione Serum Levels and Rate of Multimorbidity Development in Older Adults. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2020, 75, 1089–1094.
- van‘t Erve, T.J.; Wagner, B.A.; Ryckman, K.K.; Raife, T.J.; Buettner, G.R. The concentration of glutathione in human erythrocytes is a heritable trait. Free Radic. Biol. Med. 2013, 65, 742–749.
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153.
- Rahman, I.; MacNee, W. Lung glutathione and oxidative stress: Implications in cigarette smoke-induced airway disease. Am. J. Physiol. 1999, 277, L1067–L1088.
- Dringen, R.; Hamprecht, B. Glutathione restoration as indicator for cellular metabolism of astroglial cells. Dev. Neurosci. 1998, 20, 401–407.
- Vazquez-Meza, H.; Vilchis-Landeros, M.M.; Vazquez-Carrada, M.; Uribe-Ramirez, D.; Matuz-Mares, D. Cellular Compartmentalization, Glutathione Transport and Its Relevance in Some Pathologies. Antioxidants 2023, 12, 834.
- Giustarini, D.; Milzani, A.; Dalle-Donne, I.; Rossi, R. Red blood cells as a physiological source of glutathione for extracellular fluids. Blood Cells Mol. Dis. 2008, 40, 174–179.
- Montero, D.; Tachibana, C.; Rahr Winther, J.; Appenzeller-Herzog, C. Intracellular glutathione pools are heterogeneously concentrated. Redox Biol. 2013, 1, 508–513.
- Ballatori, N.; Hammond, C.L.; Cunningham, J.B.; Krance, S.M.; Marchan, R. Molecular mechanisms of reduced glutathione transport: Role of the MRP/CFTR/ABCC and OATP/SLC21A families of membrane proteins. Toxicol. Appl. Pharmacol. 2005, 204, 238–255.
- Bachhawat, A.K.; Thakur, A.; Kaur, J.; Zulkifli, M. Glutathione transporters. Biochim Biophys Acta 2013, 1830, 3154–3164.
- Mari, M.; de Gregorio, E.; de Dios, C.; Roca-Agujetas, V.; Cucarull, B.; Tutusaus, A.; Morales, A.; Colell, A. Mitochondrial Glutathione: Recent Insights and Role in Disease. Antioxidants 2020, 9, 909.
- Ballatori, N.; Rebbeor, J.F. Roles of MRP2 and oatp1 in hepatocellular export of reduced glutathione. Semin. Liver Dis. 1998, 18, 377–387.
- Hanigan, M.H. Gamma-glutamyl transpeptidase: Redox regulation and drug resistance. Adv. Cancer Res. 2014, 122, 103–141.
- Pizzorno, J. Glutathione! Integr. Med. A Clin. J. 2014, 13, 8–12.
- Averill-Bates, D.A. The antioxidant glutathione. Vitam. Horm. 2023, 121, 109–141.
- Rushworth, G.F.; Megson, I.L. Existing and potential therapeutic uses for N-acetylcysteine: The need for conversion to intracellular glutathione for antioxidant benefits. Pharmacol. Ther. 2014, 141, 150–159.
- Jones, C.M.; Lawrence, A.; Wardman, P.; Burkitt, M.J. Electron paramagnetic resonance spin trapping investigation into the kinetics of glutathione oxidation by the superoxide radical: Re-evaluation of the rate constant. Free Radic. Biol. Med. 2002, 32, 982–990.
- Kirsch, M.; Lehnig, M.; Korth, H.G.; Sustmann, R.; de Groot, H. Inhibition of peroxynitrite-induced nitration of tyrosine by glutathione in the presence of carbon dioxide through both radical repair and peroxynitrate formation. Chemistry 2001, 7, 3313–3320.
- Gibson, K.R.; Neilson, I.L.; Barrett, F.; Winterburn, T.J.; Sharma, S.; MacRury, S.M.; Megson, I.L. Evaluation of the antioxidant properties of N-acetylcysteine in human platelets: Prerequisite for bioconversion to glutathione for antioxidant and antiplatelet activity. J. Cardiovasc. Pharmacol. 2009, 54, 319–326.
- Winterbourn, C.C.; Metodiewa, D. Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic. Biol. Med. 1999, 27, 322–328.
- Pirie, N.W.; Pinhey, K.G. The Titration Curve of Glutathione. J. Biol. Chem. 1929, 84, 321–333.
- Deponte, M. The Incomplete Glutathione Puzzle: Just Guessing at Numbers and Figures? Antioxid. Redox Signal. 2017, 27, 1130–1161.
- Stone, J.R.; Yang, S. Hydrogen peroxide: A signaling messenger. Antioxid. Redox Signal. 2006, 8, 243–270.
- Ribas, V.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Glutathione and mitochondria. Front. Pharmacol. 2014, 5, 151.
- Hollins, D.L.; Suliman, H.B.; Piantadosi, C.A.; Carraway, M.S. Glutathione regulates susceptibility to oxidant-induced mitochondrial DNA damage in human lymphocytes. Free Radic. Biol. Med. 2006, 40, 1220–1226.
- Cichoz-Lach, H.; Michalak, A. Oxidative stress as a crucial factor in liver diseases. World J. Gastroenterol. 2014, 20, 8082–8091.
- Deponte, M. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim. Biophys. Acta 2013, 1830, 3217–3266.
- Guo, Q.; Packer, L. Ascorbate-dependent recycling of the vitamin E homologue Trolox by dihydrolipoate and glutathione in murine skin homogenates. Free Radic. Biol. Med. 2000, 29, 368–374.
- Orlowski, M.; Meister, A. The γ-Glutamyl Cycle: A Possible Transport System for Amino Acids. Proc. Natl. Acad. Sci. USA 1970, 67, 1248–1255.
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med. 2015, 80, 148–157.
- Higdon, A.N.; Benavides, G.A.; Chacko, B.K.; Ouyang, X.; Johnson, M.S.; Landar, A.; Zhang, J.; Darley-Usmar, V.M. Hemin causes mitochondrial dysfunction in endothelial cells through promoting lipid peroxidation: The protective role of autophagy. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1394–H1409.
- Solar, I.; Muller-Eberhard, U.; Shviro, Y.; Shaklai, N. Long-term intercalation of residual hemin in erythrocyte membranes distorts the cell. Biochim. Biophys. Acta 1991, 1062, 51–58.
- Dinkova-Kostova, A.T.; Kostov, R.V.; Canning, P. Keap1, the cysteine-based mammalian intracellular sensor for electrophiles and oxidants. Arch. Biochem. Biophys. 2017, 617, 84–93.
- Aquilano, K.; Baldelli, S.; Ciriolo, M.R. Glutathione: New roles in redox signaling for an old antioxidant. Front. Pharmacol. 2014, 5, 196.
- Jones, D.P.; Liang, Y. Measuring the poise of thiol/disulfide couples in vivo. Free Radic. Biol. Med. 2009, 47, 1329–1338.
- Jones, D.P. Redox potential of GSH/GSSG couple: Assay and biological significance. Methods Enzymol. 2002, 348, 93–112.
- Sen, C.K.; Packer, L. Antioxidant and redox regulation of gene transcription. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1996, 10, 709–720.
- Suzuki, H.; Sugiyama, Y. Excretion of GSSG and glutathione conjugates mediated by MRP1 and cMOAT/MRP2. Semin. Liver Dis. 1998, 18, 359–376.
- Musaogullari, A.; Chai, Y.C. Redox Regulation by Protein S-Glutathionylation: From Molecular Mechanisms to Implications in Health and Disease. Int. J. Mol. Sci. 2020, 21, 8113.
- Yang, J.; Zhang, H.; Gong, W.; Liu, Z.; Wu, H.; Hu, W.; Chen, X.; Wang, L.; Wu, S.; Chen, C.; et al. S-Glutathionylation of human inducible Hsp70 reveals a regulatory mechanism involving the C-terminal alpha-helical lid. J. Biol. Chem. 2020, 295, 8302–8324.
- Mohr, S.; Hallak, H.; de Boitte, A.; Lapetina, E.G.; Brune, B. Nitric oxide-induced S-glutathionylation and inactivation of glyceraldehyde-3-phosphate dehydrogenase. J. Biol. Chem. 1999, 274, 9427–9430.
- Hwang, C.; Sinskey, A.J.; Lodish, H.F. Oxidized redox state of glutathione in the endoplasmic reticulum. Science 1992, 257, 1496–1502.
- Tsunoda, S.; Avezov, E.; Zyryanova, A.; Konno, T.; Mendes-Silva, L.; Pinho Melo, E.; Harding, H.P.; Ron, D. Intact protein folding in the glutathione-depleted endoplasmic reticulum implicates alternative protein thiol reductants. Elife 2014, 3, e03421.
- Georgiou-Siafis, S.K.; Samiotaki, M.K.; Demopoulos, V.J.; Panayotou, G.; Tsiftsoglou, A.S. Formation of novel N-acetylcysteine-hemin adducts abrogates hemin-induced cytotoxicity and suppresses the NRF2-driven stress response in human pro-erythroid K562 cells. Eur. J. Pharmacol. 2020, 880, 173077.
- Georgiou-Siafis, S.K.; Samiotaki, M.K.; Demopoulos, V.J.; Panayotou, G.; Tsiftsoglou, A.S. Glutathione-Hemin/Hematin Adduct Formation to Disintegrate Cytotoxic Oxidant Hemin/Hematin in Human K562 Cells and Red Blood Cells’ Hemolysates: Impact of Glutathione on the Hemolytic Disorders and Homeostasis. Antioxidants 2022, 11, 1959.
- Almazroo, O.A.; Miah, M.K.; Venkataramanan, R. Drug Metabolism in the Liver. Clin. Liver Dis. 2017, 21, 1–20.
- Angelucci, F.; Baiocco, P.; Brunori, M.; Gourlay, L.; Morea, V.; Bellelli, A. Insights into the catalytic mechanism of glutathione S-transferase: The lesson from Schistosoma haematobium. Structure 2005, 13, 1241–1246.
- Yu, S.J. Substrate specificity of glutathione S-transferases from the fall armyworm. Pestic. Biochem. Physiol. 2002, 74, 41–51.
- Ji, X.; Johnson, W.W.; Sesay, M.A.; Dickert, L.; Prasad, S.M.; Ammon, H.L.; Armstrong, R.N.; Gilliland, G.L. Structure and function of the xenobiotic substrate binding site of a glutathione S-transferase as revealed by X-ray crystallographic analysis of product complexes with the diastereomers of 9-(S-glutathionyl)-10-hydroxy-9,10-dihydrophenanthrene. Biochemistry 1994, 33, 1043–1052.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.9K
Revisions:
2 times
(View History)
Update Date:
07 Nov 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No