Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Oscar Brenes | -- | 4320 | 2023-11-01 23:44:33 | | | |
| 2 | Lindsay Dong | Meta information modification | 4320 | 2023-11-02 02:06:24 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Brenes, O.; Pusch, M.; Morales, F. ClC-1 Chloride Channel Structure and Myotonia-Causing Mutations Update. Encyclopedia. Available online: https://encyclopedia.pub/entry/51057 (accessed on 24 July 2026).
Brenes O, Pusch M, Morales F. ClC-1 Chloride Channel Structure and Myotonia-Causing Mutations Update. Encyclopedia. Available at: https://encyclopedia.pub/entry/51057. Accessed July 24, 2026.
Brenes, Oscar, Michael Pusch, Fernando Morales. "ClC-1 Chloride Channel Structure and Myotonia-Causing Mutations Update" Encyclopedia, https://encyclopedia.pub/entry/51057 (accessed July 24, 2026).
Brenes, O., Pusch, M., & Morales, F. (2023, November 01). ClC-1 Chloride Channel Structure and Myotonia-Causing Mutations Update. In Encyclopedia. https://encyclopedia.pub/entry/51057
Brenes, Oscar, et al. "ClC-1 Chloride Channel Structure and Myotonia-Causing Mutations Update." Encyclopedia. Web. 01 November, 2023.
Copy Citation
Myotonia congenita is a hereditary muscle disease mainly characterized by muscle hyperexcitability, which leads to a sustained burst of discharges that correlates with the magnitude and duration of involuntary aftercontractions, muscle stiffness, and hypertrophy. Mutations in the chloride voltage-gated channel 1 (CLCN1) gene that encodes the skeletal muscle chloride channel (ClC-1) are responsible for this disease, which is commonly known as myotonic chloride channelopathy. The structure of the channel has been updated and the biophysical properties of the mutated channel have been explored and analyzed, providing important clues to the general function/dysfunction of the wild-type and mutated channels.
:myotonia
chloride channel
electrophysiology
channelopathy
mutation
1. Introduction
Myotonic conditions are hereditary skeletal muscle diseases mainly characterized by muscle hyperexcitability, which leads to a sustained burst of discharges that correlates with the magnitude and duration of involuntary aftercontractions [1][2], a sign known as myotonia. Electrical hyperexcitability is the primary alteration that underlies myotonia and causes a delay in muscle relaxation after muscle action. Most patients affected by these conditions show both electrical (detected in the electromyography) and clinical myotonia [3][4]. Myotonia is the hallmark of several human inherited diseases commonly grouped into 1—non-dystrophic myotonias (NDM), as they show a non-dystrophic muscle phenotype [5]; 2—myotonic dystrophies, either type 1 (DM1) or type 2 (DM2), both showing a highly variable clinical and dystrophic phenotype [6]. In the NDM group, there are two main human conditions whose clinical presentation includes myotonia: 1—the sodium channel myotonias that include several disorders with dominant inheritance and are caused by mutations in the sodium voltage-gated channel alpha subunit 4 (SCN4A) gene, which encodes the skeletal muscle voltage-gated sodium channel NaV4.1 [1]; 2—the chloride channel myotonias that includes the myotonia congenita (MC), which can be either dominantly (Thomsen’s disease, DMC) or recessively (Becker generalized myotonia, RMC) inherited, and caused by mutations in the chloride voltage-gated channel 1 (CLCN1) gene that encodes the skeletal muscle chloride channel (ClC-1) [7][8].
Mutations in the CLCN1 gene result in reduced Cl− conductance in skeletal muscle, which is the primary cause of MC. This channelopathy is characterized by muscle hyperexcitability, muscle stiffness, and hypertrophy [9][10]. The clinical picture depends on whether the disease is present in the dominant or the recessive form. The latter is more common and clinically more severe [11]. The two disorders differ by age-at-onset (in infancy or childhood and earlier in DMC), spreading of the myotonia, and a typical transient muscular weakness only present in the recessive trait [7][12][13].
The severity of myotonia in MC is clinically highly variable, ranging from myotonic discharges only detectable during an electromyography test (electrical myotonia) to disabling muscle stiffness at an early age (clinical myotonia) [11]. In MC, almost every skeletal muscle in the body might show muscular stiffness, which can be ameliorated by exercise (warm-up phenomenon) [14]. In addition, MC can be associated with transient weakness during quick movements, lasting only seconds or as long as thirty minutes in Becker’s disease [12].
The biophysical properties of the mutated channel have been explored and analyzed through in vitro approaches, providing important clues to the general function/dysfunction of the wild-type and mutated channels. Despite this, functional analyses have only been carried out for less than half of the mutations identified in the CLCN1 gene, making it impossible to have a clear and robust genotype–function–structure–phenotype relationship in MC [2].
Even before the full ClC-1 structure was described, several studies had already tried to implement new approaches in order to analyze the effect of the mutation on the structure of the channel and, therefore, its dysfunction [15][16][17][18][19][20][21]. Approaches such as homology models, molecular dynamics, or the analysis of structural changes (by other means) caused by CLCN1 mutations will cope with the improvement in the structure–function relationships in MC and will provide a better explanation of the effect of some mutations on the function of the channel, especially for those mutations that do not seem to differ functionally from the wild-type, show a dual inheritance pattern, or trigger an unusual MC phenotype.
2. Relevance of the ClC-1 Channel in Skeletal Muscle Physiology
Like most excitable cells, human skeletal muscle cells exhibit a quite large negative electrical potential across the membrane in the absence of stimulating signals, which is called resting membrane potential (Em). The negative value of Em is related to the high K+ conductance, mostly due to the presence in the membrane of the K+ channel from subfamilies like the K+ leak 2-P-domain (K2P) [22][23]. However, in the adult muscle cells, both the K2P and the inward rectifier K+ channels (Kir) contribute to Em [24][25]. In addition, muscle cells are unique because of the significant contribution of Cl− to the Em stability, mainly due to a Cl− conductance four-fold larger than K+ conductance [26]. Cl− conductance is about 80% of the total resting membrane conductance thanks to the robust expression of the ClC-1 channel [1][27]. ClC-1 is a doubled-barreled pore channel with an open probability of about 20–40% at the resting potential [1][28].
Interestingly, although the Cl− conductance is larger than the K+ conductance at resting conditions, K+ is the principal factor setting the resting potential, because K+ ions are actively imported by the Na-K-ATPase, while the Cl− concentration is “passively” adjusted according to the negative membrane potential. This leads to the fact that the intracellular Cl− concentration is low and that the Cl− equilibrium potential is very close to Em (about −85 mV) [29]. The large Cl− conductance thereby stabilizes Em close to the K+ equilibrium potential (Figure 1A).
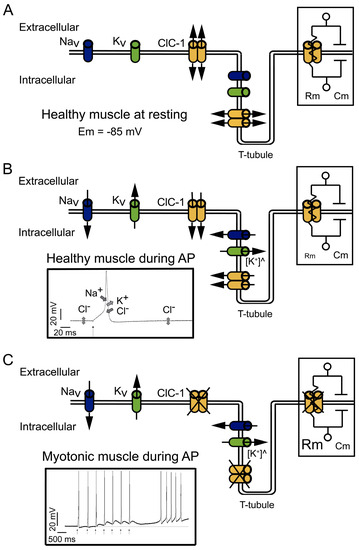
Figure 1. Physiological functions and effects of dysfunction of ClC-1. (A) At physiological resting conditions, ClC-1 doubled-barreled channels dominate sarcolemmal conductance, stabilizing the resting membrane potential (Em) at −85 mV. The right insert shows part of the equivalent circuit model of the plasma membrane; the membrane resistance (Rm) is directly dependent on the open ClC-1 channels, and the membrane capacitance (Cm) is dependent on the membrane area. Potassium (K+) “leak” channels, opened at rest, are not depicted. (B) At physiological conditions, during action potential (AP) firing, sodium (Na+) influx through voltage-dependent Na+ channels (NaV) drive the upstroke of the action potential. Repolarization occurs through K+ efflux through voltage-dependent K+ channels (KV) and the influx of Cl− through ClC-1. Upon repetitive action potential firing, K+ slowly accumulates in T-tubules ([K+]^), causing transient changes in the K+ equilibrium potential at a level that can lead to spontaneous (i.e., not neurotransmitter triggered) action potential firing and eventually to inactivation of sodium channels. The contribution of ClC-1 to action potential repolarization reduces K+ accumulation effects. In addition, K+ building up in T-tubules can induce a depolarization in Em able to spread through the sarcolemma; however, the increased opening of ClC-1 channels decreases Rm (right insert), which in turn decreases the length constant (λ), hampering the spreading of Em changes. The left insert shows the contribution and direction of NaV, KV, and ClC-1 current during action potentials; K+ “leak” channels’ contribution to Em is not shown. (C) In myotonic muscles with dysfunctional ClC-1, during action potential firing, Cl− contribution to the repolarization and stabilization of Em is lost, causing a larger depolarization during K+ accumulation in T-tubules. The increased Rm (right insert) helps to propagate the depolarization through the sarcolemma. The left insert shows the action potential induced by a stimulus (black arrows) during voluntary contractions and action potentials spontaneously triggered after voluntary contractions.
Cl− currents become even more significant during muscle activity (Figure 1B). During stimulation, the influx of Na+ through the voltage-gated 1.4 sodium channel (NaV1.4) is responsible for driving the electrical potential away from its resting value toward positive values, a phase called depolarization (left insert in Figure 1B). The subsequent repolarization phase of the action potential is usually due to the inactivation of NaV1.4 channels in conjunction with the opening of voltage-gated delayed rectifier K+ channels [25][30].
The currents through ClC-1 channels are believed to be especially important in the muscular transverse tubule system called T-tubules [15][31]. K+ efflux could be enough to control the repolarization phase of the action potentials on the surface of the cells (Figure 1B), but not in the T-tubules, which are a narrow reticular network with tubules having a diameter of about 40 nm. The network extends across a 30 µm fiber radius with a high surface area/volume ratio (equivalent to 106 cm2/mL). This narrow space limits the equilibrium of ions by simple diffusion between its lumen and the surrounding interstitial fluid.
Moreover, during high-frequency stimulation, it has been shown in cultured muscle cells that in the absence of Cl−, the accumulation of K+ can induce depolarization of about 1 mV for each action potential. Therefore, after a few spikes, the depolarization shift in the resting membrane potential is enough to create a depolarization-driven series of after-discharges of self-sustained action potential (a phenomenon called myotonic run), characteristic of myotonic muscles [31] (left insert in Figure 1C).
In myotonic muscles, the hyperexcitability can be counteracted by the warm-up phenomenon, and the hypotheses explaining this phenomenon are related to an activity-dependent reduction in cellular pH and the depolarization induced by K+ accumulation. The proposed mechanisms implying K+ accumulation in T-tubules during activity imply that the depolarization induced can be sufficiently large enough to impair sodium channel recovery after inactivation, producing the accumulation of inactive NaV1.4 channels and reducing cellular excitability and myotonic runs [1].
In addition, during intense muscle activity, if the K+ accumulation in the T-tubules induces depolarization of the resting potential, this depolarization can spread along the sarcolemma. However, the presence of ClC-1 channels and the high Cl− conductance of skeletal muscle reduces the membrane resistance (Rm) (right insert in Figure 1B). Small Rm reduces sarcolemmal length constant (λ) and prevents the local propagation of depolarizations [26]. However, in the absence of functional ClC-1 channels, it is plausible that the increased Rm (right insert in Figure 1C) favors depolarization spreading.
3. ClC-1 Localization Controversy: T-Tubule System and/or Sarcolemma
Several studies have been carried out to identify the exact localization of ClC-1, but thus far, there is no consensus among research groups. The controversy started many years ago when, with elegant experiments, Hodgkin and Horowicz [32] indicated for the first time that the muscle chloride conductance was localized in the sarcolemma, findings that were also found by Gurnett et al. in 1995 [33].
Interestingly, depending on the assay used to identify its location, the location appeared to be different. For instance, Papponen et al., 2005 [34] localized ClC-1 in the interfibrillar spaces from muscle cryosections with prominent sarcolemmal localization in a non-uniform way, being present even in the neuromuscular junction. However, when fibers were isolated and cultured in vitro, the ClC-1 was absent at the sarcolemmal level [34]. Thus, Papponen’s group proposed that the isolation process of myofibers and the resultant denervation of the myofibers changed the distribution pattern of ClC-1, explaining the lack of ClC-1 in the sarcolemma [34].
4. ClC-1 Structure–Function Relationship Overview
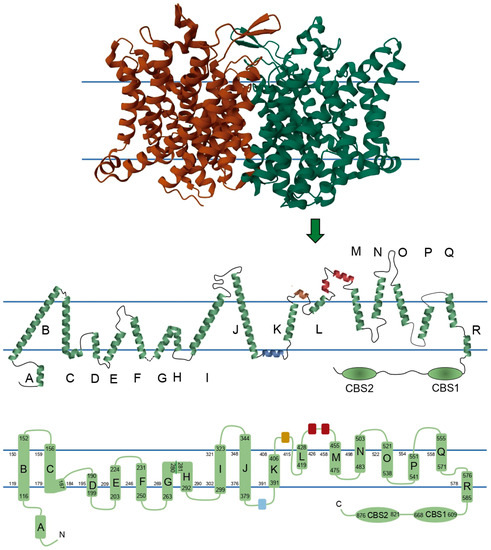
ClC-1 is composed of two identical subunits of 988 amino acid residues. Traditionally, it has been thought that each of the monomers consists of 18 α-helices: 17 α-helices expanding the membrane, named from B to R, 1 intracellular N-terminal helix named A, and 2 intracellular C-terminal cystathionin-β-synthase (CBS) segments. However, recent structures showed some subtle differences. In order to show these differences, scholars reconstructed a 2D topology of the channel based on the 3D structure published by Park and MacKinnon (2018) (Figure 2) [35]. Some of the biggest differences are 1—the absence of a loop connecting helices G and H, where instead there is a torsion in the helix; 2—the linker between helix J and K includes a small helix (colored blue in Figure 2) running parallel to the membrane, apparently being intracellular or in close association with the intracellular side of the membrane; 3—in a similar way, the linker between K and L harbors a small extracellular helix (colored brown in Figure 2), and additionally, this region exits and reenters the membrane before the beginning of helix L; and 4—there are two small helices between the helices L and M (colored red in Figure 2). Because of differences 2, 3, and 4, scholars propose calling the segments between helices J-K, K-L, and L-M as helical stretch J-K, helical stretch K-L, and helical stretch L-M, respectively, instead of loops.
Figure 2. Two-dimensional topology of ClC-1 with schematic representations of a ClC-1 monomer. Upper panel shows each ClC-1 monomer with different color. In the central panel, the size and orientation of each helix, loop, and helical stretch are based on the 3D structure described in the Protein Data Bank under the entry ID 6COY, its membrane topology, segment localizations, and secondary structure. Monomers are composed of seventeen helices embedded in the membrane, while two helices and the two CBS domains are intracellular, and three helices are extracellular. Blue parallel lines in central and bottom panels represent the position of the membrane. In central and bottom panels the blue, brown, and red segments represent the helical stretch segments identified in human ClC-1, and green segments represent the helices classically reported in the literature with the nomenclature A to R and the CBS domains. Bottom panel shows a 2D topology where the α-helices are drawn as cylinders, the numbers limiting the cylinders represent the amino acids that enclose helices B to R and CBS segments, and the numbers lining each membrane monolayer represent the amino acids that limit each intramembrane segment.
4.1. Channel Disruption
A possible modification affecting channel function in muscle physiology is the truncation of the protein. At least 37 mutations have been reported that cause premature stop codons, occurring as early as in amino acid 33 (Y33*) or as late as in amino acid 976 (R976*). However, just three of them have been analyzed functionally. The most N-terminal one is W322*, which was described in Costa Rican patients diagnosed with Thomsen’s disease or Becker’s myotonia. Expression of this mutant in Xenopus oocytes showed a complete loss of function. It was very likely that the mutation led to a truncated, not functional protein, lacking 2/3 of the total sequence, or led to a nonsense-mediated mRNA decay [16].
Between W322 and Y686, there are nine stop codon mutants that have not been functionally described, but that probably also induce non-functional proteins. This is supported by the fact that the Y686* expressed in HEK293 cells did not generate ClC-1-related currents [36]. It would be interesting to functionally analyze stop codon-forming mutations beyond Y686*, as this could provide evidence of the location in which the protein starts being functional.
The last characterized mutant with an early stop codon is R894*, which lacks the C-terminal distal to the second CBS segment. When this truncated protein is expressed in human kidney cells (tsA201), it can produce functional channels, but with smaller current amplitudes than wild-type (WT) channels [37].
4.2. Membrane Localization
In addition to the channel’s biophysical function per se, protein folding efficiency, early posttranslational modification, and protein conformational stability are key aspects determining the trafficking, membrane localization, and, therefore, the physiological function of the channel. This is underlined by the fact that at least twelve different mutations affecting ClC-1 abundance at the plasma membrane have been associated with MC (Supplementary Table S2) [36][37][38][39][40][41][42][43][44][45]. It is not clear which ClC-1 segments are related to settling the fate of nascent ClC-1 protein and how they can affect channel membrane localization, but it has been suggested that the CBS segments are crucially involved in this [17].
Interestingly, the three mutations in the helical stretch L-M involve substitutions of hydrophobic for hydrophilic amino acid, and since this segment is located extracellularly (Figure 2), it is possible that the substitutions provoke structural changes in the helix, affecting protein interaction during trafficking or interactions with the extracellular matrix. The exact role of the helical stretch L-M and the C-terminal region in channel localization must be studied in the future; nevertheless, these studies should be taken with caution since, at least for the R894* mutant, a trafficking defect was reported in Xenopus oocytes, but it was normal in muscle cells [38][46].
4.3. Pore Properties
Each subunit of the ClC-1 channel forms its own Cl−-conducting pore, with a minimum diameter estimated at around 4.5 Å and an electropositive surface in its extracellular face [28][47]. Each Cl− pathway has three serial Cl− binding sites, named Sext, Scen, and Sint, for external (near extracellular compartment), central, and internal (near intracellular compartment), respectively. Interestingly, the pore pathway bifurcates on the intracellular side, one following the canonical Cl− pathway found in other ClC proteins and the other putatively being a secondary pathway directed toward the protomer–protomer boundary on the cytosolic surface [35].
It has been pointed out that the D, F, N, and R membrane helices contribute to the Cl− pathway, and among them, D, F, and N form the Cl− selectivity filter. Specifically, helices N (F484 and M485) and F (from G230 to G233, including the conserved Glu gate E232) are arranged to coordinate Cl− on Sext. The Cl− in Scen interacts with Y578 of helix R and with S189 of the C-D loop (just prior to the beginning of helix D). Finally, the Cl− in Sint seems to interact with amino acids around L577 and I581 of helix R [35].
In this regard, nine different mutations have been described to affect ion selectivity, i.e., directly modifying the pore pathway. Three of these mutations are located in helix D [48][49], changing negative or non-polar for positive or polar amino acids. Thus, similar mutations, like L198H located in helix D, could also affect ion selectivity.
4.4. Gating
In the ClC-1 channel, as in other ClC family members, two types of gates have been described: two independent protopore gates that control each subunit independently and one common gate for the two subunits [50]. Each of the gates has its own opening processes, and both are voltage-dependent [51]. The open probability of ClC-1 channels and their voltage dependence can be analyzed from macroscopic currents, but to allow Cl− movement, both ClC-1 channel gates must be opened. However, analysis can go further through protocols that allow the separation of independent and common gate open probabilities [51].
As mentioned before, ClC-1 is a channel with an open probability of about 20–40% at muscle resting potential. The open probability is increased by depolarization and decreased by hyperpolarization, even though it never closes completely (Figure 3A,B) [1][28][51]. In voltage-dependent cation channels, a transmembrane segment containing positively charged Arg and Lys residues plays the role of a voltage-sensing domain, which is independent of the pore domain and the ion movement through the pore pathway [52]. No such domain is present in ClC channels; in contrast, the gating process of the two ClC-1 protopores is strongly coupled to the anion movements in the pore [53][54].
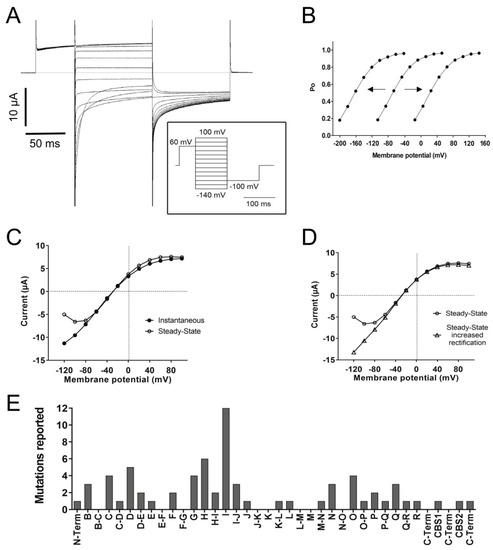
Figure 3. Functional characteristics of ClC-1. (A) The left panel shows a representative voltage-clamp recording of ClC-1 currents elicited by the stimulation protocol shown in the insert. Note the increase in currents with depolarization and the deactivation upon hyperpolarization. (B) The apparent open probability (Po) is estimated by measuring the initial current at the constant “tail” voltage of −100 mV [I(V)] as a function of the test potential V (varying between −140 and +100 mV, insert in 3A) and calculated by the normalization Po = I(V)/Imax, where Imax is the maximal tail current. Note that the expected Po with ~40% probability around muscle resting potential (−85 mV) in the central line (WT condition). Hypothetical “shifts” of Po to the left (i.e., to more negative voltages) or to the right (i.e., to more positive voltages) are also shown. (C) Current–voltage (I-V) relationship of the “instantaneous currents” measured at the beginning of the pulse step and the steady-state current measured towards the end of the step. (D) I-V relationship of the steady-state currents and an example of mutant current with increased inward rectification. (E) Frequency histogram of reported mutations inducing a positive shift in the voltage-dependence of open probability according to the different helices and loops.
At least 74 mutations have been reported to affect the open probability with positive shifts, negative shifts (Figure 3B), or even inversions in the voltage dependence. The most common effect (thus far) reported is a positive shift (at least 69 mutations). A positive shift in the voltage dependence of the open probability means that the channel responsiveness to voltage changes decreased; therefore, stronger depolarizations are needed to increase the open probability. A frequency histogram of these mutants pointed to three main positions in the protein related to this behavior (Figure 3E): the first around helix I, the second towards helix D, and the third and smaller one around helices N, O, P, and Q.
The fast gate has been related to the conserved gating glutamate (E232) in helix F, where the mutation G230E has been shown to abolish currents or trigger slower and less complete deactivation by hyperpolarization and even a change in Erev, suggesting affected anion selectivity. The identity of the slow gate remains elusive, but since the slow gate is a common gate for both protopores, it has been suggested that the helices that form the interface between monomers (H, I, P, and Q) could be part of the slow gate, in addition to the CBS and C-terminal region as modulators through interactions with transmembrane structures [47].
The kinetics of activation and/or deactivation of the fast and slow gates can also be affected by mutations. Fast gate activation by depolarization is likely most important during the period of the muscle’s action potential [53]. A characteristic feature of ClC-1 is that it deactivates within about 50 ms with at least two exponential components when the voltage is stepped to values more negative than the normal muscle resting potential (Figure 3A) [55]. The faster time constant of deactivation is primarily associated with almost complete fast gate closure, while the slower component mostly reflects (incomplete) slow gate closure [51].
4.5. Channel Rectification
Additionally, nine mutations, three of them without a reported positive shift in open probability in helices D, G, and Q, have been reported to prevent saturation at positive potentials, hence increasing outward rectification. This suggests a role of these helices in the modulation of saturation, a possibility that should be tested in future studies.
Another characteristic feature of ClC-1 is that, after maximal activation, the instantaneous current–voltage relationship exhibits stronger inward rectification than steady-state currents (Figure 3C). As mentioned before, at least 23 mutations have been reported to completely abolish or decrease the deactivation of the fast and/or the slow gate, an effect that can be related to increased inward rectification (Figure 3D). However, an increase in inward rectification was reported for just six mutations on helices B, N, and O and the H-I loop.
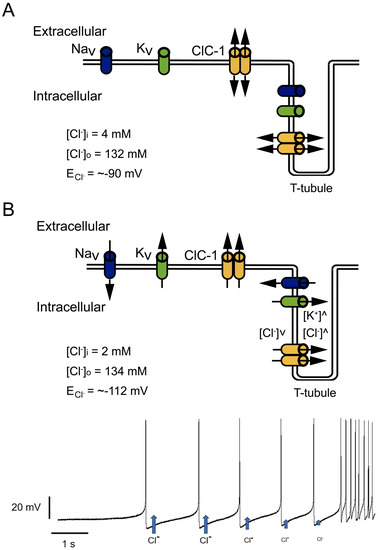
Special attention should be paid to the relationship between increased inward rectification and decreased outward current amplitudes. Mutations such as D136G showed loss of deactivation with hyperpolarization and the absence of outward currents at voltages positive to the Cl− reversal potential [56]. This behavior can allow the Cl− efflux but prevents Cl− influx (Figure 4A). At resting conditions, this behavior should not affect Em since, as mentioned before, Em is principally set by the resting K+ conductance. However, in a muscle under repetitive stimulation, a mutant channel with this behavior could cause Cl− depletion and changes in the equilibrium potential of this ion to values much more negative than muscle resting membrane potential (Figure 4A,B), affecting Cl− electromotive force and hampering the stabilizing function of the channel [56].
Figure 4. Effect of increased inward but decreased outward currents in Cl− currents. (A) At physiological conditions, Cl− inside and outside concentrations allow an equilibrium potential (ECl−) close to muscle resting potential, where Cl− influx and outflux play a stabilizing role. (B) Mutant channels with pronounced inward rectification allow Cl− outflux during trains of action potentials without influx during action potential repolarization. In this condition, repetitive activity can cause intracellular Cl− depletion and T-tubule Cl− accumulation, progressively decreasing Cl− outflux during repetitive firing (blue arrows getting smaller after each action potential in bottom panel), and shifting ECl− to more negative potentials.
4.6. ClC-1 Modulation
As pointed out before, intracellular pH affects ClC-1 gating, triggering faster deactivation at more alkaline pH and channel inhibition by intracellular acidification when studied in the presence of ATP at physiological concentrations [28][57][58]. ATP acts synergistically with acidic pH to inhibit ClC-1 by shifting the voltage dependence of common gating to more positive potentials [57]. Both protons and ATP stabilize the closed state of the common gate and together induce an alteration of the voltage dependence of the closing rate [57].
In this regard, ATP binding site is between the two CBS segments [21], and the structural analysis performed by Wang et al. [47] showed a more rigid CBS segment at lower pH in the presence of nucleotides, but more flexibility at higher pH in the absence of nucleotides.
4.7. Apparently WT-like Channels
Several CLCN1 variants with strong genetic evidence of causal implication in myotonia have been reported to behave very similarly to WT channels in in vitro electrophysiological studies. Normally, such variants might be considered non-disease-causing variants, i.e., benign polymorphisms. However, sometimes, the current–voltage relationships and the voltage dependence are the only analyses performed. Deeper analysis must be performed to test, for example, activation and deactivation kinetics, membrane localization, rectification, or ion selectivity.
5. Possible Implications for Channel Pharmacological Modulation
So far, no commercial drugs directly target ClC-1. However, taking into consideration the structure–function relationships revealed by naturally occurring mutations and the role of ClC-1 in MC, it is feasible to suggest some channel segments as targets of future drugs developed to modulate the effect of specific CLCN1 mutations found in myotonic patients.
For example, in cases of dominant gate-shifting mutations, such as S132C, S189C, I290M, V299L, or F306L, among others, the development of gating corrector molecules might be useful. In particular, molecules might be developed to target the segments related to gating shift mutations, like helix I, helix D, and around helices N, O, P, and Q (Figure 3E).
The screening of small-molecule correctors that can be useful in the development of drugs targeting specific defects in channels has been conducted for the cystic fibrosis transmembrane conductance regulator (CFTR), an epithelial Cl− channel. It has been possible to identify molecules that correct open channel probability, stabilize the protein fold, facilitate translocation to the cellular membrane, and/or act at several levels [59][60].
References
- Cannon, S.C. Channelopathies of skeletal muscle excitability. Compr. Physiol. 2015, 5, 761–790.
- Morales, F.; Pusch, M. An Up-to-Date Overview of the Complexity of Genotype-Phenotype Relationships in Myotonic Channelopathies. Front. Neurol. 2020, 10, 1404.
- Heatwole, C.R.; Moxley, R.T., 3rd. The nondystrophic myotonias. Neurotherapeutics 2007, 4, 238–251.
- Platt, D.; Griggs, R. Skeletal muscle channelopathies: New insights into the periodic paralyses and nondystrophic myotonias. Curr. Opin. Neurol. 2009, 22, 524–531.
- Heatwole, C.R.; Statland, J.M.; Logigian, E.L. The diagnosis and treatment of myotonic disorders. Muscle Nerve 2013, 47, 632–648.
- Soltanzadeh, P. Myotonic Dystrophies: A Genetic Overview. Genes 2022, 13, 367.
- Koch, M.C.; Steinmeyer, K.; Lorenz, C.; Ricker, K.; Wolf, F.; Otto, M.; Zoll, B.; Lehmann-Horn, F.; Grzeschik, K.-H.; Jentsch, T.J. The skeletal muscle chloride channel in dominant and recessive human myotonia. Science 1992, 257, 797–800.
- Matthews, E.; Fialho, D.; Tan, S.V.; Venance, S.L.; Cannon, S.C.; Sternberg, D.; Fontaine, B.; Amato, A.A.; Barohn, R.J.; Griggs, R.C.; et al. The non-dystrophic myotonias: Molecular pathogenesis, diagnosis and treatment. Brain 2010, 133, 9–22.
- Becker, P.E. Myotonia Congenita and Syndromes Associated with Myotonia; Thieme: Sttutgart, Germany, 1977.
- Thomsen, J. Tonische Krampfe in willkurlich beweglichen Muskeln in Folge von ererbterpsychischer Disposition (Ataxia muscularis?). Arch. Psychiatr. Nervenkr. 1876, 6, 702–718.
- Sun, C.; Tranebjaerg, L.; Torbergsen, T.; Holmgren, G.; Van Ghelue, M. Spectrum of CLCN1 mutations in patients with myotonia congenita in Northern Scandinavia. Eur. J. Hum. Genet. 2001, 9, 903–909.
- Jurkat-Rott, K.; Lerche, H.; Lehmann-Horn, F. Skeletal muscle channelopathies. J. Neurol. 2002, 249, 1493–1502.
- Koch, M.C.; Ricker, K.; Otto, M.; Wolf, F.; Zoll, B.; Lorenz, C.; Jentsch, T.J. Evidence for genetic homogeneity in autosomal recessive generalized myotonia (Becker). J. Med. Genet. 1993, 30, 914–917.
- Ricker, K.; Hertel, G.; Langscheid, K.; Stodieck, G. Myotonia not aggravated by cooling. Force and relaxation of the adductor pollicis in normal subjects and in myotonia as compared to paramyotonia. J. Neurol. 1977, 216, 9–20.
- Altamura, C.; Desaphy, J.F.; Conte, D.; De Luca, A.; Imbrici, P. Skeletal muscle ClC-1 chloride channels in health and diseases. Pflug. Arch. 2020, 472, 961–975.
- Brenes, O.; Barbieri, R.; Vásquez, M.; Vindas-Smith, R.; Roig, J.; Romero, A.; Morales, F. Functional and Structural Characterization of ClC-1 and Nav1.4 Channels Resulting from CLCN1 and SCN4A Mutations Identified Alone and Coexisting in Myotonic Patients. Cells 2021, 10, 374.
- Estevez, R.; Jentsch, T.J. CLC chloride channels: Correlating structure with function. Curr. Opin. Struct. Biol. 2002, 12, 531–539.
- Imbrici, P.; Altamura, C.; Camerino, G.M.; Mangiatordi, G.F.; Conte, E.; Maggi, L.; Camerino, D.C. Multidisciplinary study of a new ClC-1 mutation causing myotonia congenita: A paradigm to understand and treat ion channelopathies. FASEB J. 2016, 30, 3285–3295.
- Imbrici, P.; Altamura, C.; Pessia, M.; Mantegazza, R.; Desaphy, J.F.; Camerino, D.C. ClC-1 chloride channels: State-of-the-art research and future challenges. Front. Cell. Neurosci. 2015, 9, 156.
- Seong, J.Y.; Ha, K.; Hong, C.; Myeong, J.; Lim, H.-H.; Yang, D.; So, I. Helix O modulates voltage dependency of ClC-1. Pflug. Arch. 2017, 469, 183–193.
- Tseng, P.Y.; Yu, W.P.; Liu, H.Y.; Zhang, X.D.; Zou, X.; Chen, T.Y. Binding of ATP to the CBS domains in the C-terminal region of ClC-1. J. Gen. Physiol. 2011, 137, 357–368.
- Hille, B. Ion Channels of Excitable Membranes, 3rd ed.; Sinauer: Sunderland, MA, USA, 2001.
- Kandel, E.R.; Koester, J.D.; Mack, S.H.; Siegelbaum, S. Principles of Natural Science, 6th ed.; McGraw Hill: New York, NY, USA, 2021.
- Afzali, A.M.; Ruck, T.; Herrmann, A.M.; Iking, J.; Sommer, C.; Kleinschnitz, C.; Meuth, S.G. The potassium channels TASK2 and TREK1 regulate functional differentiation of murine skeletal muscle cells. Am. J. Physiol. Cell Physiol. 2016, 311, C583–C595.
- Stefani, E.; Chiarandini, D.J. Ionic channels in skeletal muscle. Annu. Rev. Physiol. 1982, 44, 357–372.
- Stolting, G.; Fischer, M.; Fahlke, C. CLC channel function and dysfunction in health and disease. Front. Physiol. 2014, 5, 378.
- Zifarelli, G.; Pusch, M. CLC chloride channels and transporters: A biophysical and physiological perspective. Rev. Physiol. Biochem. Pharmacol. 2007, 158, 23–76.
- Jentsch, T.J.; Pusch, M. CLC Chloride Channels and Transporters: Structure, Function, Physiology, and Disease. Physiol. Rev. 2018, 98, 1493–1590.
- Ruff, R.L.; Lennon, V.A. How myasthenia gravis alters the safety factor for neuromuscular transmission. J. Neuroimmunol. 2008, 201–202, 13–20.
- Plomp, J.J. Neuromuscular junction physiology and pthophysiology. In Myasthenia Gravis and Related Disorders; Kaminski, H.L., Kusner, L.L., Eds.; Springer Nature: Berlin/Heidelberg, Germany, 2018.
- Adrian, R.H.; Bryant, S.H. On the repetitive discharge in myotonic muscle fibres. J. Physiol. 1974, 240, 505–515.
- Hodgkin, A.L.; Horowicz, P. The effect of sudden changes in ionic concentrations on the membrane potential of single muscle fibres. J. Physiol. 1960, 153, 370–385.
- Gurnett, C.A.; Kahl, S.D.; Anderson, R.D.; Campbell, K.P. Absence of the skeletal muscle sarcolemma chloride channel ClC-1 in myotonic mice. J. Biol. Chem. 1995, 270, 9035–9038.
- Papponen, H.; Kaisto, T.; Myllyla, V.V.; Myllyla, R.; Metsikko, K. Regulated sarcolemmal localization of the muscle-specific ClC-1 chloride channel. Exp. Neurol. 2005, 191, 163–173.
- Park, E.; MacKinnon, R. Structure of the ClC-1 chloride channel from Homo sapiens. Elife 2018, 7, e36629.
- Desaphy, J.-F.; Gramegna, G.; Altamura, C.; Dinardo, M.M.; Imbrici, P.; George, A.L.; Modoni, A.; LoMonaco, M.; Camerino, D.C. Functional characterization of ClC-1 mutations from patients affected by recessive myotonia congenita presenting with different clinical phenotypes. Exp. Neurol. 2013, 248, 530–540.
- Meyer-Kleine, C.; Steinmeyer, K.; Ricker, K.; Jentsch, T.J.; Koch, M.C. Spectrum of mutations in the major human skeletal muscle chloride channel gene (CLCN1) leading to myotonia. Am. J. Hum. Genet. 1995, 57, 1325–1334.
- Papponen, H.; Nissinen, M.; Kaisto, T.; Myllyla, V.V.; Myllyla, R.; Metsikko, K. F413C and A531V but not R894X myotonia congenita mutations cause defective endoplasmic reticulum export of the muscle-specific chloride channel ClC-1. Muscle Nerve 2008, 37, 317–325.
- Hebeisen, S.; Fahlke, C. Carboxy-terminal truncations modify the outer pore vestibule of muscle chloride channels. Biophys J. 2005, 89, 1710–1720.
- Altamura, C.; Ivanova, E.A.; Imbrici, P.; Conte, E.; Camerino, G.M.; Dadali, E.L.; Desaphy, J.F. Pathomechanisms of a CLCN1 Mutation Found in a Russian Family Suffering From Becker’s Myotonia. Front. Neurol. 2020, 11, 1019.
- Gaitan-Penas, H.; Armand-Ugon, M.; Macaya, A.; Estevez, R. CLCN1 Myotonia congenita mutation with a variable pattern of inheritance suggests a novel mechanism of dominant myotonia. Muscle Nerve 2018, 58, 157–160.
- Ronstedt, K.; Sternberg, D.; Detro-Dassen, S.; Gramkow, T.; Begemann, B.; Becher, T.; Kilian, P.; Grieschat, M.; Machtens, J.-P.; Schmalzing, G.; et al. Impaired surface membrane insertion of homo- and heterodimeric human muscle chloride channels carrying amino-terminal myotonia-causing mutations. Sci. Rep. 2015, 5, 15382.
- Simpson, B.J.; Height, T.A.; Rychkov, G.Y.; Nowak, K.J.; Laing, N.G.; Hughes, B.P.; Bretag, A.H. Characterization of three myotonia-associated mutations of the CLCN1 chloride channel gene via heterologous expression. Hum. Mutat. 2004, 24, 185.
- Vindas-Smith, R.; Fiore, M.; Vásquez, M.; Cuenca, P.; del Valle, G.; Lagostena, L.; Gaitán-Peñas, H.; Estevez, R.; Pusch, M.; Morales, F. Identification and Functional Characterization of CLCN1 Mutations Found in Nondystrophic Myotonia Patients. Hum. Mutat. 2016, 37, 74–83.
- Zhang, J.; Sanguinetti, M.C.; Kwiecinski, H.; Ptacek, L.J. Mechanism of inverted activation of ClC-1 channels caused by a novel myotonia congenita mutation. J. Biol. Chem. 2000, 275, 2999–3005.
- Macías, M.J.; Teijido, O.; Zifarelli, G.; Martin, P.; Ramirez-Espain, X.; Zorzano, A.; Palacín, M.; Pusch, M.; Estévez, R. Myotonia-related mutations in the distal C-terminus of ClC-1 and ClC-0 chloride channels affect the structure of a poly-proline helix. Biochem. J. 2007, 403, 79–87.
- Wang, K.; Preisler, S.S.; Zhang, L.; Cui, Y.; Missel, J.W.; Grønberg, C.; Gotfryd, K.; Lindahl, E.; Andersson, M.; Calloe, K.; et al. Structure of the human ClC-1 chloride channel. PLoS Biol. 2019, 17, e3000218.
- Grunnet, M.; Jespersen, T.; Colding-Jørgensen, E.; Schwartz, M.; Klaerke, D.A.; Vissing, J.; Olesen, S.-P.; Dunø, M. Characterization of two new dominant ClC-1 channel mutations associated with myotonia. Muscle Nerve 2003, 28, 722–732.
- Ulzi, G.; Lecchi, M.; Sansone, V.; Redaelli, E.; Corti, E.; Saccomanno, D.; Pagliarani, S.; Corti, S.; Magri, F.; Raimondi, M.; et al. Myotonia congenita: Novel mutations in CLCN1 gene and functional characterizations in Italian patients. J. Neurol. Sci. 2012, 318, 65–71.
- Miller, C. Open-state substructure of single chloride channels from Torpedo electroplax. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1982, 299, 401–411.
- Accardi, A.; Pusch, M. Fast and slow gating relaxations in the muscle chloride channel ClC-1. J. Gen. Physiol. 2000, 116, 433–444.
- Pan, X.; Li, Z.; Zhou, Q.; Shen, H.; Wu, K.; Huang, X.; Yan, N. Structure of the human voltage-gated sodium channel Na(v)1.4 in complex with beta1. Science 2018, 362, eaau2486.
- Pedersen, T.H.; Riisager, A.; de Paoli, F.V.; Chen, T.Y.; Nielsen, O.B. Role of physiological ClC-1 Cl- ion channel regulation for the excitability and function of working skeletal muscle. J. Gen. Physiol. 2016, 147, 291–308.
- Pusch, M.; Ludewig, U.; Rehfeldt, A.; Jentsch, T.J. Gating of the voltage-dependent chloride channel CIC-0 by the permeant anion. Nature 1995, 373, 527–531.
- Pusch, M.; Steinmeyer, K.; Jentsch, T.J. Low single channel conductance of the major skeletal muscle chloride channel, ClC-1. Biophys. J. 1994, 66, 149–152.
- Fahlke, C.; Rudel, R.; Mitrovic, N.; Zhou, M.; George, A.L., Jr. An aspartic acid residue important for voltage-dependent gating of human muscle chloride channels. Neuron 1995, 15, 463–472.
- Bennetts, B.; Parker, M.W.; Cromer, B.A. Inhibition of skeletal muscle ClC-1 chloride channels by low intracellular pH and ATP. J. Biol. Chem. 2007, 282, 32780–32791.
- Tseng, P.Y.; Bennetts, B.; Chen, T.Y. Cytoplasmic ATP inhibition of ClC-1 is enhanced by low pH. J. Gen. Physiol. 2007, 130, 217–221.
- Yang, H.; Shelat, A.A.; Guy, R.K.; Gopinath, V.S.; Ma, T.; Du, K.; Verkman, A.S. Nanomolar affinity small molecule correctors of defective Delta F508-CFTR chloride channel gating. J. Biol. Chem. 2003, 278, 35079–35085.
- Liu, J.; Bihler, H.; Farinha, C.M.; Awatade, N.T.; Romão, A.M.; Mercadante, D.; Cheng, Y.; Musisi, I.; Jantarajit, W.; Wang, Y.; et al. Partial rescue of F508del-cystic fibrosis transmembrane conductance regulator channel gating with modest improvement of protein processing, but not stability, by a dual-acting small molecule. Br. J. Pharmacol. 2018, 175, 1017–1038.
More
Information
Subjects:
Biophysics; Physiology; Genetics & Heredity
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
887
Revisions:
2 times
(View History)
Update Date:
02 Nov 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No