 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Petra Hudler | -- | 6774 | 2023-10-31 11:29:37 | | | |
| 2 | Sirius Huang | Meta information modification | 6774 | 2023-11-06 02:17:45 | | |
Video Upload Options
Gastric cancer is characterised by high inter- and intratumour heterogeneity. The majority of patients are older than 65 years and the global burden of this disease is increasing due to the aging of the population. The disease is usually diagnosed at advanced stages, which is a consequence of nonspecific symptoms. A new field of mutational signatures has emerged in the past decade with advances in the genome sequencing technology. These distinct mutational patterns in the genome, caused by exogenous and endogenous mutational processes, can be associated with tumour aetiology and disease progression, and could provide novel perception on the treatment possibilities.
1. Introduction
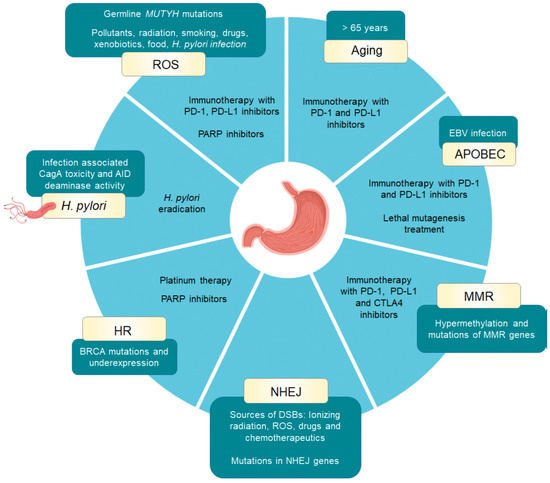
| Underlying Mechanism | Signature Type: Base Substitution Subtype 1 [References] | Molecular Consequences | Treatment Implications [References] |
Additional Biomarkers [References] |
|---|---|---|---|---|
| Aging | SBS1: C > T (NCG) [4][2][5][6] |
Deamination of 5-methylcytosine | immune checkpoint inhibition (PD-L1 and PD-1 inhibitors) [7][8][9] |
senescence score, EBV+, TMB [10] |
| APOBEC overactivity | SBS2: C > T (TCN) SBS13: C > A (TCA), C > G (TCA, TCC, TCT) [4][2][11][12][13][14] |
High mutational load in transcriptionally active genes | APOBEC3B inhibitors, immune checkpoint inhibition (PD-L1 and PD-1 inhibitors), lethal mutagenesis treatment [15][16][17][18] |
EBV+, TMB, L1-sequencing, APOBEC3B expression levels [2][12][17][19] |
| Homologous recombination repair (BRCA1/2 mutations) |
SBS3: C > A, C > G, C > T, T > A, T > C, T > G (NNN) 3 ID6: microhomology—deletion length: 5+ (microhomology length: 1, 2, 3, 4, 5+) [19][4][20] |
Higher mutational burden due to alternative error-prone DSBs repair by NHEJ | Pt-based therapy, PARP inhibitors [21][22][23][24] |
BRCA1/2 expression levels, ATM loss [25][26][27] |
| Mismatch repair | SBS20 (POLD1 mutations): C > A (CCC, CCT), C > T (ACA, GCA, GCC) SBS6, SBS14 (POLE mutations), SBS15, SBS21, SBS26, SBS44 2 DBS7: AC > NN (CA), CT > NN (TC), GC > NN (AT), TA > NN (AT), TT > NN (AA, AG, CA, GA) DBS10: CG > NN (TA), TT > NN (GG) ID1: 1 bp insertion (T, homopolymer length: 5+) ID2: 1 bp deletion (C, homopolymer length: 5, 6+) [2][28] |
Microsatellite instability, middle to high tumour mutational burden | Immune checkpoint inhibition (PD-L1, PD-1 and CTLA4 inhibitors) [29][30][31][32][33][34] |
MMR mutations, MSI status, PD-L1 status, EBV+, T-cell inflamed score, TMB status, POLD1 mutations, ARID1A mutations [35][36][37][38][39][40] |
| Nonhomologous end joining repair | ID6: microhomology—deletion length: 5+ (microhomology length: 1, 2, 3, 4, 5+) ID8: > 1 bp deletion at repeats—deletion length: 5+ (number of repeat units: 1); microhomology—deletion length: 5+ (microhomology length: 1, 2, 3) [2] |
Chromosomal instability | NHEJ inhibitors [41][42][43] |
CIN, KUs, DNA-PKcs, DNA ligase IV, XRCC4 expression levels [35][40] |
| Reactive oxygen species DNA damage | SBS18: C > A (ACA, CCA, GCA, GCT, TCA, TCC, TCT) [2][44] |
DNA damage | Immune checkpoint inhibition therapy (PD-L1 and PD-1 inhibitors) PARP inhibitors [45][46][46] |
MUTYH mutations and expression levels, CHEK1 mutations [44][47][48][49] |
| H. pylori infection | SBS3: C > A, C > G, C > T, T > A, T > C, T > G (NNN) 3 ID6: microhomology—deletion length: 5+ (microhomology length: 1, 2, 3, 4, 5+) [2][20][50] |
Inflammation, DNA damage | H. pylori eradication | AID expression levels [51] |
2. Aging
3. APOBEC Activity
Bobrovnitchaia et al. analysed 240 gastric cancer samples from the TCGA cohort and 112 samples from a different Brazilian validation cohort and showed that the expression of APOBEC genes was significantly higher in EBV+ tumours in comparison to EBV- tumour samples [22]. With the exception of APOBEC3A, all other members of the APOBEC3s were upregulated, with APOBEC3C being the most abundantly expressed. The authors also showed that APOBEC characteristic TpCpW mutation pattern was significantly enriched in EBV+ group and positively correlated with APOBEC3 expression, which further correlated with tumour purity, suggesting that APOBEC activity derives from tumour cells. APOBEC mutational load correlated with highly expressed genes, which were, as expected highly mutated. This is in concordance with the fact that the preferential editing substrate of APOBEC3s is ssDNA, which is available at transcriptionally active sites. The authors also observed enrichment of TpCpW mutations in the late-stage EBV+ tumours, which also carried higher proportion of mutated oncogenes in comparison to EBV- samples. Furthermore, in 40% of EBV+ patients with somatic mutations in PIK3CA, mutations were present in TpCpW motif, while this was not observed in EBV- patients.
Higher expression patterns of mRNA and protein levels of APOBEC3B were also found in gastric cancer tumour samples compared to paired normal tissue samples in a cohort of 236 patients [56]. APOBEC3B mRNA and protein levels were higher in tumour samples from Grade III stage in comparison to Grade I or Grade II stage and correlated to poor prognosis. High APOBEC3B expression was also associated with gender (female), tumour size (> 5.0 cm), histological grade (G3), and TNM staging (lower expression in TNM I). APOBEC3B down-regulation with shRNA resulted in enhanced cytotoxicity of PDCD2 (Programmed cell death protein 2) in gastric cancer cell line MKN28, probably due to the lower mutational load and non-interfered transcription of this tumour suppressor gene. APOBEC3B expression levels were associated with APOBEC mutational signatures, whereas APOBEC3C expression levels were associated with decreased APOBEC mutational signatures in gastric cancer [23].
Interestingly, analysis of cancer cell lines by sequencing single cells showed that at least 75% of investigated cancer cell lines (including gastric cancer) that previously encountered APOBEC mutagenesis persistently continued to generateSBS2 and SBS13 mutational signatures [25]. The authors suggested that APOBEC-associated mutagenesis in vivo appears to be episodic. Additionally, the procurement of APOBEC-associated mutational signatures continued in cell culture despite the absence of proposed initiators of APOBEC activity, immune system and exogenous viral infections [17][26]. Indeed, the presence of a virus is not necessarily required for initiating APOBEC mutagenesis [25]. This may occur also due to retrotransposition activities. There was significant correlation between the rates of the in vitro-acquired retrotransposition aberrations and burdens of SBS13 and, interestingly, SBS18 (associated to ROS) shown in cancer cell lines. However, the correlation was not significant when 2,353 primary cancers originating from different tissues were investigated. L1s are autonomous mobile elements, amounting to 17% of the human genome, and retrotranspose via an RNA intermediate through a “copy and paste” mechanism [27]. Somatic mobilization of retroelements can induce and accelerate insertional mutagenesis and genetic instability. Next-generation L1-resequencing (L1-seq) on paired tissue samples from seven patients with primary gastric cancer showed that somatic retrotransposition was present early in cancer development in gastro-intestinal epithelial cells [28]. L1-seq, APOBEC-mutational signature, and APOBEC-expression status in stomach tissues in combination with circulating biomarkers could therefore be valuable as biomarkers for early detection of gastric cancer.
Tumours characterized by APOBEC overactivity might be candidates for treatment by lethal mutagenesis [29]. Drugs, such as nucleoside analogues that increase the mutation load in tumour cells to toxic levels are already commonly used in addition to platinum-based chemotherapy to destroy tumour cells. On the other hand, inhibition of APOBEC enzymes may prevent cancer evolution. APOBEC3B specific inhibitors are promising, as this enzyme is non-essential in humans, whereas other members of the family are crucial for adaptive (AID) and innate (other APOBEC members) immune response [30]. Drug candidates for such “therapy by hypomutation” are being pursued. In recent years many studies of APOBEC3B crystal structures were published providing better understanding of APOBEC3B active site dynamics and setting the stage for design of selective small molecule inhibitors [31][32][33]. These may in the future in combination with other therapies be effective in preventing tumour recurrence and drug resistance.
Furthermore, a study by Wang and Jia et al. suggested that APOBEC3B and APOBEC-mutational signature might be a novel marker for predicting immunotherapy response [34]. They found a correlation between APOBEC3B expression and immune gene expression and known immunotherapy response biomarkers in patients with non-small cell lung cancer. APOBEC mutational signature was specifically enriched in patients with lasting clinical benefit after immunotherapy, suggesting that patients with APOBEC signatures might be candidates for checkpoint blockade immunotherapy with PD-1 and PD-L1 (discussed in detail in section 3.3.2). Similarly, Boichard and Pham et al. demonstrated that APOBEC related mutagenesis could be correlated with immunotherapy response in patients with various cancer types (gastric cancer excluded) [36].
All the factors, responsible for the expression of AID/APOBEC enzyme family are currently unknown. Nevertheless, altogether this data suggest that it might be worth analysing the presence of APOBEC mutational signature and the expression levels of APOBEC enzymes in gastric cancer biopsies as they can be a source of ongoing mutagenesis at transcriptionally active DNA sites or retrotranspozable elements providing a secondary driving force for subclonal expansions and intratumor heterogeneity propagation. This may manifest clinically as recurrence, metastasis, and drug resistance and could therefore have important prognostic and therapeutic implications. In a cohort analysed by Alexandrov et al. APOBEC signatures were present in < 50% of all gastric cancer samples indicating that this feature was not common for all gastric adenocarcinoma tumours and could therefore be useful for patient stratification in combination with other markers, such as PD-L1 positivity, TMB, and EBV infection status [2].
4. DNA Integrity Machinery
4.1. Homologous Recombination DNA Repair
4.2. DNA Mismatch Repair Deficiency
| SBS | Base Substitution Subtype 1 | Associated ID | Transcriptional (T) and Replicational (R) Strand Asymmetry in Stomach Cancer |
|---|---|---|---|
| SBS6 | C > T (ACA, ACG, CCG, GCN) |
ID1, ID2 | T:/ R:/ |
| SBS14 | C > A (ACT, CCT, GCT, TCT) |
ID1, ID2 | T:/ R:/ |
| SBS15 | C > A (CCA) C > T (ACG, GCN) |
ID1, ID2 | T:/ R:/ |
| SBS20 2 | C > A (CCC, CCT) C > T (ACA, GCA, GCC) |
ID1, ID2 | T: no significance R: lagging, C > A |
| SBS21 | T > C (GTN, TTA, TTC, TTT) |
ID1, ID2 | T: no significance R: lagging strand, T > C |
| SBS26 | T > C (ATA, ATC, CTA, CTG, CTT, GTA, GTG, GTT, TTT) |
ID1, ID2 | T: untranscribed strand, T > C R: lagging strand, T > C |
| SBS44 | C > A (CCT) C > T (ACA, GCN) |
ID1, ID2 | T: no significance R: lagging strand, C > A; leading strand, C > T and T > A |
MLH1 Promoter Hypermethylation and MSI Phenotype
POLD1/POLE Mutations and MSI Phenotype
Mutational Status of ARID1A and MSI Phenotype
4.3. Double Strand Break Repair by Nonhomologous End Joining
5. Reactive Oxygen Species
6. Helicobacter Pylori Infection
7. Mutational Signatures of Unknown Origin
8. Conclusion
In the future, it will be important to understand signature penetrance and integrate genomic, epigenomic, transcriptomic and molecular markers for a more accurate cancer subtyping with defined hallmarks. These integrated signature profiles could offer predictive diagnostic and prognostic values in order to develop necessary frameworks to design successful patient-tailored treatment strategies. Understanding the genomic background of gastric cancer would together with molecular and clinical data help with patient stratification and selection of the most appropriate targeted therapy.
References
- Ludmil B. Alexandrov; Serena Nik-Zainal; David C. Wedge; Peter J. Campbell; Michael R. Stratton; Deciphering Signatures of Mutational Processes Operative in Human Cancer. Cell Rep. 2013, 3, 246-259.
- Ludmil B. Alexandrov; Jaegil Kim; Nicholas J. Haradhvala; Mi Ni Huang; Alvin Wei Tian Ng; Yang Wu; Arnoud Boot; Kyle R. Covington; Dmitry A. Gordenin; Erik N. Bergstrom; et al.S. M. Ashiqul IslamNuria Lopez-BigasLeszek J. KlimczakSandro MorganellaRadhakrishnan SabarinathanDavid A. WheelerVille MustonenKin ChanAkihiro FujimotoGad GetzIñigo MartincorenaHidewaki NakagawaPaz PolakSteven A. RobertsSteven G. RozenNatalie SainiTatsuhiro ShibataYuichi ShiraishiMichael R. StrattonBin Tean TehIgnacio Vázquez-GarcíaFouad YousifWillie YuLauri A. AaltonenFederico AbascalAdam AbeshouseHiroyuki AburataniDavid J. AdamsNishant AgrawalKeun Soo AhnSung-Min AhnHiroshi AikataRehan AkbaniKadir C. AkdemirHikmat Al-AhmadieSultan T. Al-SedairyFatima Al-ShahrourMalik AlawiMonique AlbertKenneth AldapeAdrian AllyKathryn AlsopEva G. AlvarezFernanda AmarySamirkumar B. AminBrice AminouOle AmmerpohlMatthew J. AndersonYeng AngDavide AntonelloPavana AnurSamuel AparicioElizabeth L. AppelbaumYasuhito AraiAxel AretzKoji ArihiroShun-Ichi AriizumiJoshua ArmeniaLaurent ArnouldSylvia AsaYassen AssenovGurnit AtwalSietse AukemaJ. Todd AumanMiriam R. R. AurePhilip AwadallaMarta AymerichGary D. BaderAdrian Baez-OrtegaMatthew H. BaileyPeter J. BaileyMiruna BalasundaramSaianand BaluPratiti BandopadhayayRosamonde E. BanksStefano BarbiAndrew P. BarbourJonathan BarenboimJill Barnholtz-SloanHugh BarrElisabet BarreraJohn BartlettJavier BartolomeClaudio BassiOliver F. BatheDaniel BaumhoerPrashant BaviStephen B. BaylinWojciech BazantDuncan BeardsmoreTimothy A. BeckSam BehjatiAndreas BehrenBeifang NiuCindy BellSergi BeltranChristopher BenzAndrew BerchuckAnke K. BergmannBenjamin P. BermanDaniel M. BerneyStephan H. BernhartRameen BeroukhimMario BerriosSamantha BersaniJohanna BertlMiguel BetancourtVinayak BhandariShriram G. BhosleAndrew V. BiankinMatthias BiegDarell BignerHans BinderEwan BirneyMichael BirrerNidhan K. BiswasBodil BjerkehagenTom BodenheimerLori BoiceGiada BonizzatoJohann S. De BonoMoiz S. BootwallaAke BorgArndt BorkhardtKeith A. BoroevichIvan BorozanChristoph BorstMarcus BosenbergMattia BosioJacqueline BoultwoodGuillaume BourquePaul C. BoutrosG. Steven BovaDavid T. BowenReanne BowlbyDavid D. L. BowtellSandrine BoyaultRich BoyceJeffrey BoydAlvis BrazmaPaul BrennanDaniel S. BrewerArie B. BrinkmanRobert G. BristowRussell R. BroaddusJane E. BrockMalcolm BrockAnnegien BroeksAngela N. BrooksDenise BrooksBenedikt BrorsSøren BrunakTimothy J. C. BruxnerAlicia L. BruzosAlex BuchananIvo BuchhalterChristiane BuchholzSusan BullmanHazel BurkeBirgit BurkhardtKathleen H. BurnsJohn BusanovichCarlos D. BustamanteAdam P. ButlerAtul J. ButteNiall J. ByrneAnne-Lise Børresen-DaleSamantha J. Caesar-JohnsonAndy CafferkeyDeclan CahillClaudia CalabreseCarlos CaldasFabien CalvoNiedzica CamachoPeter J. CampbellElias CampoCinzia CantùShaolong CaoThomas E. CareyJoana Carlevaro-FitaRebecca CarlsenIvana CataldoMario CazzolaJonathan CebonRobert CerfolioDianne E. ChadwickDimple ChakravartyDon ChalmersCalvin Wing Yiu ChanMichelle Chan-Seng-YueVishal S. ChandanDavid K. ChangStephen J. ChanockLorraine A. ChantrillAurélien ChateignerNilanjan ChatterjeeKazuaki ChayamaHsiao-Wei ChenJieming ChenKen ChenYiwen ChenZhaohong ChenAndrew D. CherniackJeremy ChienYoke-Eng ChiewSuet-Feung ChinJuok ChoSunghoon ChoJung Kyoon ChoiWan ChoiChristine ChomienneZechen ChongSu Pin ChooAngela ChouAngelika N. ChristElizabeth L. ChristieEric ChuahCarrie CibulskisKristian CibulskisSara CingarliniPeter ClaphamAlexander ClaviezSean ClearyNicole CloonanMarek CmeroColin C. CollinsAshton A. ConnorSusanna L. CookeColin S. CooperLeslie CopeVincenzo CorboMatthew G. CordesStephen M. CordnerIsidro Cortés-CirianoPrue A. CowinBrian CraftDavid CraftChad J. CreightonYupeng CunErin CurleyIoana CutcutacheKarolina CzajkaBogdan CzerniakRebecca A. DaggLudmila DanilovaMaria Vittoria DaviNatalie R. DavidsonHelen DaviesIan J. DavisBrandi N. Davis-DusenberyKevin J. DawsonFrancisco M. De La VegaRicardo De Paoli-IseppiTimothy DefreitasAngelo P. Dei TosOlivier DelaneauJohn A. DemchokJonas DemeulemeesterGerman M. DemidovDeniz DemircioğluNening M. DennisRobert E. DenrocheStefan C. DentroNikita DesaiVikram DeshpandeAmit G. DeshwarChristine DesmedtJordi Deu-PonsNoreen DhallaNeesha C. DhaniPriyanka DhingraRajiv DhirAnthony DiBiaseKlev DiamantiLi DingShuai DingHuy Q. DinhLuc DirixHarshaVardhan DoddapaneniNilgun DonmezMichelle T. DowRonny DrapkinOliver DrechselRuben M. DrewsSerge SergeTim DudderidgeAna Dueso-BarrosoAndrew J. DunfordMichael DunnLewis Jonathan DursiFraser R. DuthieKen Dutton-RegesterJenna EaglesDouglas F. EastonStuart EdmondsPaul A. EdwardsSandra E. EdwardsRosalind A. EelesAnna EhingerJuergen EilsRoland EilsAdel El-NaggarMatthew EldridgeKyle EllrottSerap ErkekGeorgia EscaramisShadrielle M. G. EspirituXavier EstivillDariush EtemadmoghadamJorunn E. EyfjordBishoy M. FaltasDaiming FanYu FanWilliam C. FaquinClaudiu FarcasMatteo FassanAquila FatimaFrancesco FaveroNodirjon FayzullaevIna FelauSian FeredayMartin L. FergusonVincent FerrettiLars FeuerbachMatthew A. FieldJ. Lynn FinkGaetano FinocchiaroCyril FisherMatthew W. FittallAnna FitzgeraldRebecca C. FitzgeraldAdrienne M. FlanaganNeil E. FleshnerPaul FlicekJohn A. FoekensKwun M. FongNuno A. FonsecaChristopher S. FosterNatalie S. FoxMichael FraserScott FrazerMilana Frenkel-MorgensternWilliam FriedmanJoan FrigolaCatrina C. FronickMasashi FujitaMasashi FukayamaLucinda A. FultonRobert S. FultonMayuko FurutaP. Andrew FutrealAnja FüllgrabeStacey B. GabrielSteven GallingerCarlo Gambacorti-PasseriniJianjiong GaoShengjie GaoLevi GarrawayØystein GarredErik GarrisonDale W. GarsedNils GehlenborgJosep L. L. GelpiJoshy GeorgeDaniela S. GerhardClarissa GerhauserJeffrey E. GershenwaldMark GersteinMoritz GerstungMohammed GhoriRonald GhosseinNasra H. GiamaRichard A. GibbsBob GibsonAnthony J. GillPelvender GillDilip D. GiriDominik GlodzikVincent J. GnanapragasamMaria Elisabeth GoeblerMary J. GoldmanCarmen GomezSantiago GonzalezAbel Gonzalez-PerezJames GossageKunihito GotohRamaswamy GovindanDorthe GrabauJanet S. GrahamRobert C. GrantAnthony R. GreenEric GreenLiliana GregerNicola GrehanSonia GrimaldiSean M. GrimmondRobert L. GrossmanAdam GrundhoffGunes GundemQianyun GuoManaswi GuptaShailja GuptaIvo G. GutMarta GutJonathan GökeGavin HaAndrea HaakeDavid HaanSiegfried HaasKerstin HaaseJames E. HaberNina HabermannFaraz HachSyed HaiderNatsuko HamaFreddie C. HamdyAnne HamiltonMark P. HamiltonLeng HanGeorge B. HannaMartin HansmannOlivier HarismendyIvon HarliwongArif O. HarmanciEoghan HarringtonTakanori HasegawaDavid HausslerSteve HawkinsShinya HayamiShuto HayashiD. Neil HayesStephen J. HayesNicholas K. HaywardSteven HazellYao HeAllison P. HeathSimon C. HeathDavid HedleyApurva M. HegdeDavid I. HeimanMichael C. HeinoldZachary HeinsLawrence E. HeislerEva Hellstrom-LindbergMohamed HelmySeong Gu HeoAustin J. HepperlaJosé María Heredia-GenestarCarl HerrmannPeter HerseyJulian M. HessHolmfridur HilmarsdottirJonathan HintonSatoshi HiranoNobuyoshi HiraokaKatherine A. HoadleyAsger HobolthErmin HodzicJessica I. HoellSteve HoffmannOliver HofmannAndrea HolbrookAliaksei Z. HolikMichael A. HollingsworthOliver HolmesRobert A. HoltChen HongEun Pyo HongJongwhi H. HongGerrit K. HooijerHenrik HornshøjFumie HosodaYong HouVolker HovestadtWilliam HowatAlan P. HoyleRalph H. HrubanJianhong HuTaobo HuXing HuaKuan-Lin HuangMei HuangVincent HuangYi HuangWolfgang HuberThomas J. HudsonMichael HummelJillian A. HungDavid HuntsmanTed R. HuppJason HuseMatthew R. HuskaBarbara HutterCarolyn M. HutterDaniel HübschmannChristine A. Iacobuzio-DonahueCharles David ImbuschMarcin ImielinskiSeiya ImotoWilliam B. IsaacsKeren IsaevShumpei IshikawaMurat IskarMichael IttmannSinisa IvkovicJose M. G. IzarzugazaJocelyne JacquemierValerie JakrotNigel B. JamiesonGun Ho JangSe Jin JangJoy C. JayaseelanReyka JayasingheStuart R. JefferysKarine JegalianJennifer L. JenningsSeung-Hyup JeonLara JermanYuan JiWei JiaoPeter A. JohanssonAmber L. JohnsJeremy JohnsRory JohnsonTodd A. JohnsonClemency JollyYann JolyJon G. JonassonCorbin D. JonesDavid T. W. JonesNic JonesSteven J. M. JonesJos JonkersYoung Seok JuHartmut JuhlJongsun JungMalene JuulRandi Istrup JuulSissel JuulNatalie JägerRolf KabbeAndre KahlesAbdullah KahramanVera B. KaiserHojabr KakavandSangeetha KalimuthuChristof von KalleKoo Jeong KangKatalin KarasziBeth KarlanRosa KarlićDennis KarschKatayoon KasaianKarin S. KassahnHitoshi KataiMamoru KatoHiroto KatohYoshiiku KawakamiJonathan D. KayStephen H. KazakoffMarat D. KazanovMaria KeaysElectron KebebewRichard F. KeffordManolis KellisJames G. KenchCatherine J. KennedyJules N. A. KerssemakersDavid KhooVincent KhooNarong KhuntikeoEkta KhuranaHelena KilpinenHark Kyun KimHyung-Yong KimHyunghwan KimJihoon KimJong K. KimYoungwook KimTari A. KingWolfram KlapperKortine KleinheinzStian KnappskogMichael KnebaBartha M. KnoppersYoungil KohJan KomorowskiDaisuke KomuraMitsuhiro KomuraGu KongMarcel KoolJan O. KorbelViktoriya KorchinaAndrey KorshunovMichael KoscherRoelof KosterZsofia Kote-JaraiAntonios KouresMilena KovacevicBarbara KremeyerHelene KretzmerMarkus KreuzSavitri KrishnamurthyDieter KubeKiran KumarPardeep KumarSushant KumarYogesh KumarRitika KundraKirsten KüblerRalf KüppersJesper LagergrenPhillip H. LaiPeter W. LairdSunil R. LakhaniChristopher M. LalansinghEmilie LalondeFabien C. LamazeAdam LambertEric LanderPablo LandgrafLuca LandoniAnita LangerødAndrés LanzósDenis LarsimontErik LarssonMark LathropLoretta M. S. LauChris LawerenzRita T. LawlorMichael S. LawrenceAlexander J. LazarAna Mijalkovic LazicXuan LeDarlene LeeDonghoon LeeEunjung Alice LeeHee Jin LeeJake June-Koo LeeJeong-Yeon LeeJuhee LeeMing Ta Michael LeeHenry Lee-SixKjong-Van LehmannHans LehrachDido LenzeConrad R. LeonardDaniel A. LeongamornlertIgnaty LeshchinerLouis LetourneauIvica LetunicDouglas A. LevineLora LewisTim LeyChang LiConstance H. LiHaiyan Irene LiJun LiLin LiShantao LiSiliang LiXiaobo LiXiaotong LiXinyue LiYilong LiHan LiangSheng-Ben LiangPeter LichterPei LinZiao LinW. M. LinehanOle Christian LingjærdeDongbing LiuEric Minwei LiuFei-Fei Fei LiuFenglin LiuJia LiuXingmin LiuJulie LivingstoneDimitri LivitzNaomi LivniLucas LochovskyMarkus LoefflerGeorgina V. LongArmando Lopez-GuillermoShaoke LouDavid N. LouisLaurence B. LovatYiling LuYong-Jie LuYouyong LuClaudio LuchiniIlinca LunguXuemei LuoHayley J. LuxtonAndy G. LynchLisa LypeCristina LópezCarlos López-OtínEric Z. MaYussanne MaGaetan MacGroganShona MacRaeGeoff MacintyreTobias MadsenKazuhiro MaejimaAndrea MafficiniDennis T. MaglinteArindam MaitraPartha P. MajumderLuca MalcovatiSalem MalikicGiuseppe MalleoGraham J. MannLuisa Mantovani-LöfflerKathleen MarchalGiovanni MarchegianiElaine R. MardisAdam A. MargolinMaximillian G. MarinFlorian MarkowetzJulia MarkowskiJeffrey MarksTomas Marques-BonetMarco A. MarraLuke MarsdenJohn W. M. MartensSancha MartinJose I. Martin-SuberoAlexander Martinez-FundichelyYosef E. MaruvkaR. Jay MashlCharlie E. MassieThomas J. MatthewLucy MatthewsErik MayerSimon MayesMichael MayoFaridah MbabaaliKaren McCuneUltan McDermottPatrick D. McGillivrayMichael D. McLellanJohn D. McPhersonTreasa A. McPhersonSamuel R. MeierAlice MengShaowu MengAndrew MenziesNeil D. MerrettSue MersonMatthew MeyersonWilliam MeyersonPiotr A. MieczkowskiGeorge L. MihaiescuSanja MijalkovicTom MikkelsenMichele MilellaLinda MileshkinChristopher A. MillerDavid K. MillerJessica K. MillerGordon B. MillsAna MilovanovicSarah MinnerMarco MiottoGisela Mir ArnauLisa MirabelloChris MitchellThomas J. MitchellSatoru MiyanoNaoki MiyoshiShinichi MizunoFruzsina Molnár-GáborMalcolm J. MooreRichard A. MooreQuaid D. MorrisCarl MorrisonLisle E. MoseCatherine D. MoserFerran MuiñosLoris MularoniAndrew J. MungallKaren MungallElizabeth A. MusgroveDavid MutchFrancesc MuyasDonna M. MuznyAlfonso MuñozJerome MyersOla MyklebostPeter MöllerGenta NagaeAdnan M. NagrialHardeep K. Nahal-BoseHitoshi NakagamaHiromi NakamuraToru NakamuraKaoru NakanoTannistha NandiJyoti NangaliaMia NasticArcadi NavarroFabio C. P. NavarroDavid E. NealGerd NettekovenFelicity NewellSteven J. NewhouseYulia NewtonAnthony NgJonathan NicholsonDavid NicolYongzhan NieG. Petur NielsenMorten Muhlig NielsenSerena Nik-ZainalMichael S. NobleKatia NonesPaul A. NorthcottFaiyaz NottaBrian D. O’connorPeter O’donnellMaria O’donovanSarah O’mearaBrian Patrick O’neillJ. Robert O’neillDavid OcanaAngelica OchoaLayla OesperChristopher OgdenHideki OhdanKazuhiro OhiLucila Ohno-MachadoKarin A. OienAkinyemi I. OjesinaHidenori OjimaTakuji OkusakaLarsson OmbergChoon Kiat OngStephan OssowskiGerman OttB. F. Francis OuelletteChristine P’ngMarta PaczkowskaSalvatore PaiellaChawalit PairojkulMarina PajicQiang Pan-HammarströmElli PapaemmanuilIrene PapatheodorouNagarajan ParamasivamJi Wan ParkJoong-Won ParkKeunchil ParkKiejung ParkPeter J. ParkJoel S. ParkerSimon L. ParsonsHarvey PassDanielle PasternackAlessandro PastoreAnn-Marie PatchIris PauportéAntonio PeaJohn V. PearsonChandra Sekhar PedamalluJakob Skou PedersenPaolo PederzoliMartin PeiferNathan A. PennellCharles M. PerouMarc D. PerryGloria M. PetersenMyron PetoNicholas PetrelliRobert PetryszakStefan M. PfisterMark PhillipsOriol PichHilda A. PickettTodd D. PihlNischalan PillaySarah PinderMark PineseAndreia V. PinhoEsa PitkänenXavier PivotElena Piñeiro-YáñezLaura PlankoChristoph PlassTirso PonsIrinel PopescuOlga PotapovaAparna PrasadShaun R. PrestonManuel PrinzAntonia L. PritchardStephenie D. ProkopecElena ProvenzanoXose S. PuenteSonia PuigMontserrat PuiggròsSergio Pulido-TamayoGulietta M. PupoColin A. PurdieMichael C. QuinnRaquel RabionetJanet S. RaderBernhard RadlwimmerPetar RadovicBenjamin RaederKeiran M. RaineManasa RamakrishnaKamna RamakrishnanSuresh RamalingamBenjamin J. RaphaelW. Kimryn RathmellTobias RauschGuido ReifenbergerJüri ReimandJorge Reis-FilhoVictor ReuterIker Reyes-SalazarMatthew A. ReynaSheila M. ReynoldsEsther RheinbayYasser RiazalhosseiniAndrea L. RichardsonJulia RichterMatthew RingelMarkus RingnérYasushi RinoKarsten RippeJeffrey RoachLewis R. RobertsNicola D. RobertsA. Gordon RobertsonJavier Bartolomé RodriguezBernardo Rodriguez-MartinF. Germán Rodríguez-GonzálezMichael H. A. RoehrlMarius RohdeHirofumi RokutanGilles RomieuIlse RoomanTom RoquesDaniel RosebrockMara RosenbergPhilip C. RosenstielAndreas RosenwaldEdward W. RoweRomina RoyoYulia RubanovaMark A. RubinCarlota Rubio-PerezVasilisa A. RudnevaBorislav C. RusevAndrea RuzzenenteGunnar RätschVeronica Y. SabelnykovaSara SadeghiS. Cenk SahinalpMihoko Saito-AdachiGordon SaksenaAdriana SalcedoRoberto SalgadoLeonidas SalichosRichard SallariCharles SallerRoberto SalviaMichelle SamJaswinder S. SamraFrancisco Sanchez-VegaChris SanderGrant SandersRajiv SarinIman SarrafiAya Sasaki-OkuTorill SauerGuido SauterRobyn P. M. SawMaria ScardoniChristopher J. ScarlettAldo ScarpaGhislaine SceloDirk SchadendorfJacqueline E. ScheinMarkus B. SchilhabelMatthias SchlesnerThorsten SchlommHeather K. SchmidtSarah-Jane SchrammStefan SchreiberNikolaus SchultzSteven E. SchumacherRoland F. SchwarzRichard A. ScolyerDavid ScottRalph ScullyRaja SeethalaAyellet V. SegreIris SelanderColin A. SempleYasin SenbabaogluSubhajit SenguptaElisabetta SereniStefano SerraDennis C. SgroiMark ShackletonNimish C. ShahSagedeh ShahabiCatherine A. ShangPing ShangOfer ShapiraTroy SheltonCiyue ShenHui ShenRebecca ShepherdRuian ShiYan ShiYu-Jia ShiahJuliann ShihEigo ShimizuKiyo ShimizuSeung Jun ShinTal ShmayaIlya ShmulevichSolomon I. ShorserCharles ShortRaunak ShresthaSuyash S. ShringarpureCraig ShriverShimin ShuaiNikos SidiropoulosReiner SiebertAnieta M. SieuwertsLina SieverlingSabina SignorettiKatarzyna O. SikoraMichele SimboloRonald SimonJanae V. SimonsJared T. SimpsonPeter T. SimpsonSamuel SingerNasa Sinnott-ArmstrongPayal SipahimalaniTara J. SkellyMarcel SmidJaclyn SmithKaren Smith-McCuneNicholas D. SocciHeidi J. SofiaMatthew G. SolowayLei SongAnil K. SoodSharmila SothiChristos SotiriouCameron M. SoulettePaul N. SpanPaul T. SpellmanNicola SperandioAndrew J. SpillaneOliver SpiroJonathan SpringJohan StaafPeter F. StadlerPeter StaibStefan G. StarkLucy StebbingsÓlafur Andri StefánssonOliver StegleLincoln D. SteinAlasdair StenhouseChip StewartStephan StilgenbauerMiranda D. StobbeJonathan R. StretchAdam J. StruckJoshua M. StuartHenk G. StunnenbergHong SuXiaoping SuRen X. SunStephanie SungaleeHana SusakAkihiro SuzukiFred SweepMonika SzczepanowskiHolger SültmannTakashi YugawaAngela TamDavid TamboreroBenita Kiat Tee TanDonghui TanPatrick TanHiroko TanakaHirokazu TaniguchiTomas J. TanskanenMaxime TarabichiRoy TarnuzzerPatrick TarpeyMorgan L. TaschukKenji TatsunoSimon TavaréDarrin F. TaylorAmaro Taylor-WeinerJon W. TeagueVarsha TembeJavier TemesKevin ThaiSarah P. ThayerNina ThiessenGilles ThomasSarah ThomasAlan ThompsonAlastair M. ThompsonJohn F. F. ThompsonR. Houston ThompsonHeather ThorneLeigh B. ThorneAdrian ThorogoodGrace TiaoNebojsa TijanicLee E. TimmsRoberto TiraboscoMarta TojoStefania TommasiChristopher W. ToonUmut H. ToprakDavid TorrentsGiampaolo TortoraJörg TostYasushi TotokiDavid TownendNadia TraficanteIsabelle TreilleuxJean-Rémi TrottaLorenz H. P. TrümperMing TsaoTatsuhiko TsunodaJose M. C. TubioOlga TuckerRichard TurkingtonDaniel J. TurnerAndrew TuttMasaki UenoNaoto T. UenoChristopher UmbrichtHusen M. UmerTimothy J. UnderwoodLara UrbanTomoko UrushidateTetsuo UshikuLiis Uusküla-ReimandAlfonso ValenciaDavid J. Van Den BergSteven Van LaerePeter Van LooErwin G. Van MeirGert G. Van Den EyndenTheodorus Van der KwastNaveen VasudevMiguel VazquezRavikiran VedururuUmadevi VeluvoluShankar VembuLieven P. C. VerbekePeter VermeulenClare VerrillAlain ViariDavid VicenteCaterina VicentiniK. VijayRaghavanJuris ViksnaRicardo E. VilainIzar VillasanteAnne Vincent-SalomonTapio VisakorpiDouglas VoetParesh VyasNick M. WaddellNicola WaddellClaes WadeliusLina WadiRabea WagenerJeremiah A. WalaJiayin WangLinghua WangQi WangWenyi WangYumeng WangZhining WangPaul M. WaringHans-Jörg WarnatzJonathan WarrellAnne Y. WarrenSebastian M. WaszakDavid C. WedgeDieter WeichenhanPaul WeinbergerJohn N. WeinsteinJoachim WeischenfeldtDaniel J. WeisenbergerIan WelchMichael C. WendlJohannes WernerJustin P. WhalleyHayley C. WhitakerDennis WigleMatthew D. WilkersonAshley WilliamsJames S. WilmottGavin W. WilsonJulie M. WilsonRichard K. WilsonBoris WinterhoffJeffrey A. WintersingerMaciej WiznerowiczStephan WolfBernice H. WongTina WongWinghing WongYoungchoon WooScott WoodBradly G. WoutersAdam J. WrightDerek W. WrightMark H. WrightChin-Lee WuDai-Ying WuGuanming WuJianmin WuKui WuZhenggang WuLiu XiTian XiaQian XiangXiao XiaoRui XingHeng XiongQinying XuYanxun XuHong XueShinichi YachidaSergei YakneenRui YamaguchiTakafumi N. YamaguchiMasakazu YamamotoShogo YamamotoHiroki YamaueFan YangHuanming YangJean Y. YangLixing YangShanlin YangTsun-Po YangYang YangXiaotong YaoMarie-Laure YaspoLucy YatesChristina YauChen YeKai YeVenkata D. YellapantulaChristopher J. YoonSung-Soo YoonJun YuKaixian YuYingyan YuKe YuanYuan YuanDenis YuenChristina K. YungOlga ZaikovaJorge ZamoraMarc ZapatkaJean C. ZenklusenThorsten ZenzNikolajs ZepsCheng-Zhong ZhangFan ZhangHailei ZhangHongwei ZhangHongxin ZhangJiashan ZhangJing ZhangJunjun ZhangXuanping ZhangYan ZhangZemin ZhangZhongming ZhaoLiangtao ZhengXiuqing ZhengWanding ZhouYong ZhouBin ZhuHongtu ZhuJingchun ZhuShida ZhuLihua ZouXueqing ZouAnna DefazioNicholas van AsCarolien H. M. van DeurzenMarc J. van de VijverL. Van’t VeerChristian von MeringPCAWG Mutational Signatures Working GroupPCAWG Consortium The repertoire of mutational signatures in human cancer. Nat. 2020, 578, 94-101.
- John G Tate; Sally Bamford; Harry C Jubb; Zbyslaw Sondka; David M Beare; Nidhi Bindal; Harry Boutselakis; Charlotte G Cole; Celestino Creatore; Elisabeth Dawson; et al.Peter FishBhavana HarshaCharlie HathawaySteve C JupeChai Yin KokKate NobleLaura PontingChristopher C RamshawClaire E RyeHelen E SpeedyRay StefancsikSam L ThompsonShicai WangSari WardPeter J CampbellSimon A Forbes COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2018, 47, D941-D947.
- Ludmil B. Alexandrov; Australian Pancreatic Cancer Genome Initiative; Serena Nik-Zainal; David C. Wedge; Samuel A. J. R. Aparicio; Sam Behjati; Andrew V. Biankin; Graham R. Bignell; Niccolò Bolli; Ake Borg; et al.Anne-Lise Børresen-DaleSandrine BoyaultBirgit BurkhardtAdam P. ButlerCarlos CaldasHelen R. DaviesChristine DesmedtRoland EilsJórunn Erla EyfjördJohn A. FoekensMel GreavesFumie HosodaBarbara HutterTomislav IlicicSandrine ImbeaudMarcin ImielinskiNatalie JägerDavid T. W. JonesStian KnappskogMarcel KoolSunil R. LakhaniCarlos López-OtínSancha MartinNikhil C. MunshiHiromi NakamuraPaul A. NorthcottMarina PajicElli PapaemmanuilAngelo ParadisoJohn V. PearsonXose S. PuenteKeiran RaineManasa RamakrishnaAndrea L. RichardsonJulia RichterPhilip RosenstielMatthias SchlesnerTon N. SchumacherPaul N. SpanJon W. TeagueYasushi TotokiAndrew N. J. TuttRafael Valdés-MasMarit M. van BuurenLaura van ’T VeerAnne Vincent-SalomonNicola WaddellLucy R. YatesJessica Zucman-RossiP. Andrew FutrealUltan McDermottPeter LichterMatthew MeyersonSean M. GrimmondReiner SiebertElías CampoTatsuhiro ShibataStefan M. PfisterPeter J. CampbellMichael R. StrattonICGC Breast Cancer ConsortiumICGC MMML-Seq ConsortiumICGC PedBrain Signatures of mutational processes in human cancer. Nat. 2013, 500, 415-421.
- Francis Blokzijl; Joep de Ligt; Myrthe Jager; Valentina Sasselli; Sophie Roerink; Nobuo Sasaki; Meritxell Huch; Sander Boymans; Ewart Kuijk; Pjotr Prins; et al.Isaac J. NijmanInigo MartincorenaMichal MokryCaroline L. WiegerinckSabine MiddendorpToshiro SatoGerald SchwankEdward E. S. NieuwenhuisMonique M. A. VerstegenLuc J. W. van der LaanJeroen de JongeJan N. M. IjzermansRobert G. VriesMarc van de WeteringMichael R. StrattonHans CleversEdwin CuppenRuben van Boxtel Tissue-specific mutation accumulation in human adult stem cells during life. Nat. 2016, 538, 260-264.
- Ludmil B Alexandrov; Philip H Jones; David C Wedge; Julian E Sale; Peter J Campbell; Serena Nik-Zainal; Michael R Stratton; Clock-like mutational processes in human somatic cells. Nat. Genet. 2015, 47, 1402-1407.
- Seung Tae Kim; Razvan Cristescu; Adam J. Bass; Kyoung-Mee Kim; Justin I. Odegaard; Kyung Kim; Xiao Qiao Liu; Xinwei Sher; Hun Jung; Mijin Lee; et al.Sujin LeeSe Hoon ParkJoon Oh ParkYoung Suk ParkHo Yeong LimHyuk LeeMingew ChoiAmirAli TalasazPeter Soonmo KangJonathan ChengAndrey LobodaJeeyun LeeWon Ki Kang Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat. Med. 2018, 24, 1449-1458.
- Anshuman Panda; Janice M Mehnert; Kim M Hirshfield; Greg Riedlinger; Sherri Damare; Tracie Saunders; Michael Kane; Levi Sokol; Mark N Stein; Elizabeth Poplin; et al.Lorna Rodriguez-RodriguezAnn W SilkJoseph AisnerNancy ChanJyoti MalhotraMelissa FrankelHoward L KaufmanSiraj AliJeffrey S RossEileen P WhiteGyan BhanotShridar Ganesan Immune Activation and Benefit From Avelumab in EBV-Positive Gastric Cancer. JNCI J. Natl. Cancer Inst. 2017, 110, 316-320.
- Dung T. Le; Jennifer N. Durham; Kellie N. Smith; Hao Wang; Bjarne R. Bartlett; Laveet K. Aulakh; Steve Lu; Holly Kemberling; Cara Wilt; Brandon S. Luber; et al.Fay WongNilofer S. AzadAgnieszka A. RuckiDan LaheruRoss DonehowerAtif ZaheerGeorge A. FisherTodd S. CrocenziJames J. LeeTim F. GretenAustin G. DuffyKristen K. CiomborAleksandra D. EyringBao H. LamAndrew JoeS. Peter KangMatthias HoldhoffLudmila DanilovaLeslie CopeChristian MeyerShibin ZhouRichard M. GoldbergDeborah K. ArmstrongKatherine M. BeverAmanda N. FaderJanis TaubeFranck HousseauDavid SpetzlerNianqing XiaoDrew M. PardollNickolas PapadopoulosKenneth W. KinzlerJames R. EshlemanBert VogelsteinRobert A. AndersLuis A. Diaz Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Sci. 2017, 357, 409-413.
- Lulin Zhou; Zubiao Niu; Yuqi Wang; You Zheng; Yichao Zhu; Chenxi Wang; Xiaoyan Gao; Lihua Gao; Wen Zhang; Kaitai Zhang; et al.Gerry MelinoHongyan HuangXiaoning WangQiang Sun Senescence as a dictator of patient outcomes and therapeutic efficacies in human gastric cancer. Cell Death Discov. 2022, 8, 1-14.
- Thomas M. Reid; Lawrence A. Loeb; Effect of DNA-repair enzymes on mutagenesis by oxygen free radicals. Mutat. Res. Mol. Mech. Mutagen. 1993, 289, 181-186.
- Irina Bobrovnitchaia; Renan Valieris; Rodrigo D. Drummond; Joao P. Lima; Helano C. Freitas; Thais F. Bartelli; Maria G. de Amorim; Diana N. Nunes; Emmanuel Dias‐Neto; Israel T. da Silva; et al. APOBEC‐mediated DNA alterations: A possible new mechanism of carcinogenesis in EBV‐positive gastric cancer. Int. J. Cancer 2019, 146, 181-191.
- Zhishan Chen; Wanqing Wen; Jiandong Bao; Krystle L. Kuhs; Qiuyin Cai; Jirong Long; Xiao-Ou Shu; Wei Zheng; Xingyi Guo; Integrative genomic analyses of APOBEC-mutational signature, expression and germline deletion of APOBEC3 genes, and immunogenicity in multiple cancer types. BMC Med Genom. 2019, 12, 1-13.
- Mia Petljak; Ludmil B. Alexandrov; Jonathan S. Brammeld; Stacey Price; David C. Wedge; Sebastian Grossmann; Kevin J. Dawson; Young Seok Ju; Francesco Iorio; Jose M.C. Tubio; et al.Ching Chiek KohIlias Georgakopoulos-SoaresBernardo Rodríguez–MartínBurçak OtluSarah O’mearaAdam P. ButlerAndrew MenziesShriram G. BhosleKeiran RaineDavid R. JonesJon W. TeagueKathryn BealCalli LatimerLaura O’neillJorge ZamoraElizabeth AndersonNikita PatelMark MaddisonBee Ling NgJennifer GrahamMathew J. GarnettUltan McDermottSerena Nik-ZainalPeter J. CampbellMichael R. Stratton Characterizing Mutational Signatures in Human Cancer Cell Lines Reveals Episodic APOBEC Mutagenesis. Cell 2019, 176, 1282-1294.e20.
- Edward J. Fox; Lawrence A. Loeb; Lethal Mutagenesis: Targeting the Mutator Phenotype in Cancer. Semin. Cancer Biol. 2010, 20, 353-359.
- Jeffrey M Kidd; Tera L Newman; Eray Tuzun; Rajinder Kaul; Evan E Eichler; Population Stratification of a Common APOBEC Gene Deletion Polymorphism. PLOS Genet. 2007, 3, e63.
- Shixiang Wang; Mingming Jia; Zaoke He; Xue-Song Liu; APOBEC3B and APOBEC mutational signature as potential predictive markers for immunotherapy response in non-small cell lung cancer. Oncogene 2018, 37, 3924-3936.
- Amélie Boichard; Timothy V. Pham; Huwate Yeerna; Aaron Goodman; Pablo Tamayo; Scott Lippman; Garrett M. Frampton; Igor F. Tsigelny; Razelle Kurzrock; APOBEC-related mutagenesis and neo-peptide hydrophobicity: implications for response to immunotherapy. OncoImmunology 2018, 8, 1550341.
- Serena Nik-Zainal; Ludmil B. Alexandrov; David C. Wedge; Peter Van Loo; Christopher D. Greenman; Keiran Raine; David Jones; Jonathan Hinton; John Marshall; Lucy A. Stebbings; et al.Andrew MenziesSancha MartinKenric LeungLina ChenCatherine LeroyManasa RamakrishnaRichard RanceKing Wai LauLaura J. MudieIgnacio VarelaDavid J. McBrideGraham R. BignellSusanna L. CookeAdam ShlienJohn GambleIan WhitmoreMark MaddisonPatrick S. TarpeyHelen R. DaviesElli PapaemmanuilPhilip J. StephensStuart McLarenAdam P. ButlerJon W. TeagueGöran JönssonJudy E. GarberDaniel SilverPenelope MironAquila FatimaSandrine BoyaultAnita LangerødAndrew TuttJohn W.M. MartensSamuel A.J.R. AparicioÅke BorgAnne Vincent SalomonGilles ThomasAnne-Lise Børresen-DaleAndrea L. RichardsonMichael S. NeubergerP. Andrew FutrealPeter J. CampbellMichael R. Stratton Mutational Processes Molding the Genomes of 21 Breast Cancers. Cell 2012, 149, 979-993.
- Ludmil B. Alexandrov; Serena Nik-Zainal; Hoi Cheong Siu; Suet Yi Leung; Michael R Stratton; A mutational signature in gastric cancer suggests therapeutic strategies. Nat. Commun. 2015, 6, 8683.
- J. A. Ledermann; PARP inhibitors in ovarian cancer. Ann. Oncol. 2016, 27, i40-i44.
- Man Yee T. Keung; Yanyuan Wu; Jaydutt V. Vadgama; PARP Inhibitors as a Therapeutic Agent for Homologous Recombination Deficiency in Breast Cancers. J. Clin. Med. 2019, 8, 435.
- M. Catherine Pietanza; Saiama N. Waqar; Lee M. Krug; Afshin Dowlati; Christine L. Hann; Alberto Chiappori; Taofeek K. Owonikoko; Kaitlin M. Woo; Robert J. Cardnell; Junya Fujimoto; et al.Lihong LongLixia DiaoJing WangYevgeniva BensmanBrenda HurtadoPatricia de GrootErik P. SulmanIgnacio I. WistubaAlice ChenMartin FleisherJohn V. HeymachMark G. KrisCharles M. RudinLauren Averett Byers Randomized, Double-Blind, Phase II Study of Temozolomide in Combination With Either Veliparib or Placebo in Patients With Relapsed-Sensitive or Refractory Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 2386-2394.
- Waddell N., Pajic M., Patch A.M., Chang D.K., Kassahn K.S., Bailey P., Johns A.L., Miller D., Nones K., Quek K., et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501.
- Yung-Jue Bang; Rui-Hua Xu; Keisho Chin; Keun-Wook Lee; Se Hoon Park; Sun Young Rha; Lin Shen; Shukui Qin; Nong Xu; Seock-Ah Im; et al.Gershon LockerPhil RoweXiaojin ShiDarren HodgsonYu-Zhen LiuNarikazu Boku Olaparib in combination with paclitaxel in patients with advanced gastric cancer who have progressed following first-line therapy (GOLD): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1637-1651.
- Wenjiao Chen; Jian Wang; Xiao Li; Jingying Li; Li Zhou; Tianzhu Qiu; Meiling Zhang; Ping Liu; Prognostic significance of BRCA1 expression in gastric cancer. Med Oncol. 2013, 30, 423.
- Jin Won Kim; Hyun Jin Cho; Miso Kim; Kyung-Hun Lee; Min A. Kim; Sae-Won Han; Do-Youn Oh; Hyuk-Joon Lee; Seock-Ah Im; Tae-You Kim; et al.Han-Kwang YangWoo Ho KimYung-Jue Bang Differing effects of adjuvant chemotherapy according to BRCA1 nuclear expression in gastric cancer. Cancer Chemother. Pharmacol. 2013, 71, 1435-1443.
- Jaqueline Ramalho Buttura; Monize Nakamoto Provisor Santos; Renan Valieris; Rodrigo Duarte Drummond; Alexandre Defelicibus; João Paulo Lima; Vinicius Fernando Calsavara; Helano Carioca Freitas; Vladmir C. Cordeiro de Lima; Thais Fernanda Bartelli; et al.Marc WiednerRafael RosalesKenneth John GollobJoanna LoizouEmmanuel Dias-NetoDiana Noronha NunesIsrael Tojal da Silva Mutational Signatures Driven by Epigenetic Determinants Enable the Stratification of Patients with Gastric Cancer for Therapeutic Intervention. Cancers 2021, 13, 490.
- Kazuhiro Togasaki; Yasutaka Sukawa; Takanori Kanai; Hiromasa Takaishi; Clinical efficacy of immune checkpoint inhibitors in the treatment of unresectable advanced or recurrent gastric cancer: an evidence-based review of therapies. OncoTargets Ther. 2018, ume 11, 8239-8250.
- Cong Chen; Fan Zhang; Ning Zhou; Yan-Mei Gu; Ya-Ting Zhang; Yi-Di He; Ling Wang; Lu-Xi Yang; Yang Zhao; Yu-Min Li; et al. Efficacy and safety of immune checkpoint inhibitors in advanced gastric or gastroesophageal junction cancer: a systematic review and meta-analysis. OncoImmunology 2019, 8, e1581547.
- Mingyu Zhu; Haiyan Cui; Lu Zhang; Kuo Zhao; Xiaochen Jia; Hao Jin; Assessment of POLE and POLD1 mutations as prognosis and immunotherapy biomarkers for stomach adenocarcinoma. Transl. Cancer Res. 2022, 11, 193-205.
- Xi Jiao; Xin Wei; Shuang Li; Chang Liu; Huan Chen; Jifang Gong; Jian Li; Xiaotian Zhang; Xicheng Wang; Zhi Peng; et al.Changsong QiZhenghang WangYujiao WangYanni WangNa ZhuoHenghui ZhangZhihao LuLin Shen A genomic mutation signature predicts the clinical outcomes of immunotherapy and characterizes immunophenotypes in gastrointestinal cancer. npj Precis. Oncol. 2021, 5, 1-9.
- Keigo Chida; Akihito Kawazoe; Masahito Kawazu; Toshihiro Suzuki; Yoshiaki Nakamura; Tetsuya Nakatsura; Takeshi Kuwata; Toshihide Ueno; Yasutoshi Kuboki; Daisuke Kotani; et al.Takashi KojimaHiroya TaniguchiHiroyuki ManoMasafumi IkedaKohei ShitaraItaru EndoTakayuki Yoshino A Low Tumor Mutational Burden and PTEN Mutations Are Predictors of a Negative Response to PD-1 Blockade in MSI-H/dMMR Gastrointestinal Tumors. Clin. Cancer Res. 2021, 27, 3714-3724.
- Chang Gon Kim; Moonki Hong; Hei-Cheul Jeung; Garden Lee; Hyun Cheol Chung; Sun Young Rha; Hyo Song Kim; Choong-Kun Lee; Ji Hyun Lee; Yejeong Han; et al.Jee Hung KimSeo Young LeeHyunki KimSu-Jin ShinSong-Ee BaekMinkyu Jung Hyperprogressive disease during PD-1 blockade in patients with advanced gastric cancer. Eur. J. Cancer 2022, 172, 387-399.
- The Cancer Genome Atlas Research Network; Comprehensive molecular characterization of gastric adenocarcinoma. Nat. 2014, 513, 202-209.
- Feng Wang; Qi Zhao; Ying-Nan Wang; Ying Jin; Ming-Ming He; Ze-Xian Liu; Rui-Hua Xu; Evaluation of POLE and POLD1 Mutations as Biomarkers for Immunotherapy Outcomes Across Multiple Cancer Types. JAMA Oncol. 2019, 5, 1504-1506.
- Yun Gu; Puran Zhang; Jieti Wang; Chao Lin; Hao Liu; He Li; Hongyong He; Ruochen Li; Heng Zhang; Weijuan Zhang; et al. Somatic ARID1A mutation stratifies patients with gastric cancer to PD-1 blockade and adjuvant chemotherapy. Cancer Immunol. Immunother. 2022, 72, 1199-1208.
- Minyu Wang; Yu‐Kuan Huang; Joseph Ch Kong; Yu Sun; Daniela G Tantalo; Han Xian Aw Yeang; Le Ying; Feng Yan; Dakang Xu; Heloise Halse; et al.Natasha Di CostanzoIan R GordonCatherine MitchellLaura K MackayRita A BusuttilPaul J NeesonAlex Boussioutas High‐dimensional analyses reveal a distinct role of T‐cell subsets in the immune microenvironment of gastric cancer. Clin. Transl. Immunol. 2020, 9, e1127.
- Elisabetta Puliga; Simona Corso; Filippo Pietrantonio; Silvia Giordano; Microsatellite instability in Gastric Cancer: Between lights and shadows. Cancer Treat. Rev. 2021, 95, 102175.
- Wei Li; Chuan Xie; Zhen Yang; Jiang Chen; Nong-Hua Lu; Abnormal DNA-PKcs and Ku 70/80 expression may promote malignant pathological processes in gastric carcinoma. World J. Gastroenterol. 2013, 19, 6894-6901.
- Meghana Manjunath; Bibha Choudhary; Sathees C. Raghavan; SCR7, a potent cancer therapeutic agent and a biochemical inhibitor of nonhomologous DNA end‐joining. Cancer Rep. 2021, 4, e1341.
- Yufeng Wang; Yasuhiro Kuramitsu; Byron Baron; Takao Kitagawa; Kazuhiro Tokuda; Junko Akada; Shin-Ichiro Maehara; Yoshihiko Maehara; Kazuyuki Nakamura; PI3K inhibitor LY294002, as opposed to wortmannin, enhances AKT phosphorylation in gemcitabine-resistant pancreatic cancer cells. Int. J. Oncol. 2016, 50, 606-612.
- M.T. Niazi; G. Mok; M. Heravi; L. Lee; T. Vuong; R. Aloyz; L. Panasci; T. Muanza; Effects of dna-Dependent Protein Kinase Inhibition by NU7026 on dna Repair and Cell Survival in Irradiated Gastric Cancer Cell Line N87. Curr. Oncol. 2014, 21, 91-96.
- Seung-Gi Jin; Yingying Meng; Jennifer Johnson; Piroska E. Szabó; Gerd P. Pfeifer; Concordance of hydrogen peroxide–induced 8-oxo-guanine patterns with two cancer mutation signatures of upper GI tract tumors. Sci. Adv. 2022, 8, eabn3815.
- Nikita M. Volkov; Grigoriy A. Yanus; Alexandr O. Ivantsov; Fedor V. Moiseenko; Olga G. Matorina; Ilya V. Bizin; Vladimir M. Moiseyenko; Evgeny N. Imyanitov; Efficacy of immune checkpoint blockade in MUTYH-associated hereditary colorectal cancer. Investig. New Drugs 2019, 38, 894-898.
- Zachary R McCaw; Michael A Liu; Lee-Jen Wei; Olaparib in Metastatic Castration-Resistant Prostate Cancer. New Engl. J. Med. 2021, 384, 1174-1176.
- G Kantersmoler; Jan Björk; Kaisa Fritzell; Yvonne Engwall; Birgitta Hallberg; Göran Karlsson; Henrik Grönberg; Per Karlsson; Arne Wallgren; Jan Wahlström; et al.Gunilla Kanter–SmolerRolf HultcrantzMargareta Nordling Novel Findings in Swedish Patients With MYH-Associated Polyposis: Mutation Detection and Clinical Characterization. Clin. Gastroenterol. Hepatol. 2006, 4, 499-506.
- Stefanie Vogt; Natalie Jones; Daria Christian; Christoph Engel; Maartje Nielsen; Astrid Kaufmann; Verena Steinke; Hans F. Vasen; Peter Propping; Julian R. Sampson; et al.Frederik J. HesStefan Aretz Expanded Extracolonic Tumor Spectrum in MUTYH-Associated Polyposis. Gastroenterology 2009, 137, 1976-1985.e10.
- Yuanying Zhang; Xiaorong Liu; Yimei Fan; Jianhua Ding; Ailing Xu; Xuefu Zhou; Xu Hu; Ming Zhu; Xiaomei Zhang; Suping Li; et al.Jianzhong WuHaixia CaoJintian LiYaping Wang Germline mutations and polymorphic variants inMMR,E-cadherin andMYH genes associated with familial gastric cancer in Jiangsu of China. Int. J. Cancer 2006, 119, 2592-2596.
- Satoshi Imai; Takuya Ooki; Naoko Murata-Kamiya; Daisuke Komura; Kamrunnesa Tahmina; Weida Wu; Atsushi Takahashi-Kanemitsu; Christopher Takaya Knight; Akiko Kunita; Nobumi Suzuki; et al.Adriana A. Del ValleMayo TsuboiMasahiro HataYoku HayakawaNaomi OhnishiKoji UedaMasashi FukayamaTetsuo UshikuShumpei IshikawaMasanori Hatakeyama Helicobacter pylori CagA elicits BRCAness to induce genome instability that may underlie bacterial gastric carcinogenesis. Cell Host Microbe 2021, 29, 941-958.e10.
- Takahiro Shimizu; Hiroyuki Marusawa; Yoko Endo; Tsutomu Chiba; Inflammation-mediated genomic instability: roles of activation-induced cytidine deaminase in carcinogenesis. Cancer Sci. 2012, 103, 1201-1206.
- G. P. Pfeifer. Mutagenesis at Methylated CpG Sequences; Springer Science and Business Media LLC: Dordrecht, GX, Netherlands, 2006; pp. 259-281.
- David N Cooper; Matthew Mort; Peter D Stenson; Edward V Ball; Nadia A Chuzhanova; Methylation-mediated deamination of 5-methylcytosine appears to give rise to mutations causing human inherited disease in CpNpG trinucleotides, as well as in CpG dinucleotides. Hum. Genom. 2010, 4, 406-410.
- Akira Sassa; Yuki Kanemaru; Nagisa Kamoshita; Masamitsu Honma; Manabu Yasui; Mutagenic consequences of cytosine alterations site-specifically embedded in the human genome. Genes Environ. 2016, 38, 1-6.
- Brittan M., Wright N.A. Stem cell in gastrointestinal structure and neoplastic development. Gut 2004, 53, 899–910.
- Demaria M., O’Leary M.N., Chang J., Shao L., Liu S., Alimirah F., Koenig K., Le C., Mitin N., Deal A.M., et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176.
- Valdimara C. Vieira; Marcelo A. Soares; The Role of Cytidine Deaminases on Innate Immune Responses against Human Viral Infections. BioMed Res. Int. 2013, 2013, 1-18.
- Silvestro G Conticello; The AID/APOBEC family of nucleic acid mutators. Genome Biol. 2008, 9, 229-229.
- Steven A. Roberts; Joan Sterling; Cole Thompson; Shawn Harris; Deepak Mav; Ruchir Shah; Leszek J. Klimczak; Gregory V. Kryukov; Ewa Malc; Piotr A. Mieczkowski; et al.Michael A. ResnickDmitry A. Gordenin Clustered Mutations in Yeast and in Human Cancers Can Arise from Damaged Long Single-Strand DNA Regions. Mol. Cell 2012, 46, 424-435.
- Thomas Helleday; Saeed Eshtad; Serena Nik-Zainal; Mechanisms underlying mutational signatures in human cancers. Nat. Rev. Genet. 2014, 15, 585-598.
- Steven A. Roberts; Dmitry A. Gordenin; Hypermutation in human cancer genomes: footprints and mechanisms. Nat. Rev. Cancer 2014, 14, 786-800.
- Razvan Cristescu; Jeeyun Lee; Michael Nebozhyn; Kyoung-Mee Kim; Jason C Ting; Swee Seong Wong; Jiangang Liu; Yong Gang Yue; Jian Wang; Kun Yu; et al.Xiang S YeIn-Gu DoShawn LiuLara GongJake FuJason Gang JinMin Gew ChoiTae Sung SohnJoon Ho LeeJae Moon BaeSeung Tae KimSe Hoon ParkInsuk SohnSin-Ho JungPatrick TanRonghua ChenJames HardwickWon Ki KangMark AyersDai HongyueChristoph ReinhardAndrey LobodaSung KimAmit Aggarwal Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat. Med. 2015, 21, 449-456.
- Rohini Roy; Jarin Chun; Simon N. Powell; BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat. Rev. Cancer 2011, 12, 68-78.
- Jackson S.P. Sensing and repairing DNA double-strand breaks. Carcinogenesis 2002, 23, 687–696.
- Tom Walsh; Silvia Casadei; Kathryn Hale Coats; Elizabeth Swisher; Sunday M. Stray; Jake Higgins; Kevin C. Roach; Jessica Mandell; Ming K. Lee; Sona Ciernikova; et al.Lenka ForetovaPavel SoucekMary-Claire King Spectrum of Mutations in BRCA1, BRCA2, CHEK2, and TP53 in Families at High Risk of Breast Cancer. JAMA 2006, 295, 1379-1388.
- Nik-Zainal S., Davies H., Staaf J., Ramakrishna M., Glodzik D., Zou X., Martincorena I., Alexandrov L.B., Martin S., Wedge D.C. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54.
- Philip Gotwals; Scott Cameron; Daniela Cipolletta; Viviana Cremasco; Adam Crystal; Becker Hewes; Britta Mueller; Sonia Quaratino; Catherine Sabatos-Peyton; Lilli Petruzzelli; et al.Jeffrey A. EngelmanGlenn Dranoff Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat. Rev. Cancer 2017, 17, 286-301.
- Hanxiao Li; P. Anton van der Merwe; Shivan Sivakumar; Biomarkers of response to PD-1 pathway blockade. Br. J. Cancer 2022, 126, 1663-1675.
- Yanan Cheng; Dechao Bu; Qiaoling Zhang; Rebecca Sun; Stephen Lyle; Gang Zhao; Li Dong; Hui Li; Yi Zhao; Jinpu Yu; et al.Xishan Hao Genomic and transcriptomic profiling indicates the prognosis significance of mutational signature for TMB-high subtype in Chinese patients with gastric cancer. J. Adv. Res. 2023, 51, 121-134.
- Elizabeth D Thompson; Marianna Zahurak; Adrian Murphy; Toby Cornish; Nathan Cuka; Eihab Abdelfatah; Stephen Yang; Mark Duncan; Nita Ahuja; Janis M Taube; et al.Robert A AndersRonan J Kelly Patterns of PD-L1 expression and CD8 T cell infiltration in gastric adenocarcinomas and associated immune stroma. Gut 2016, 66, 794-801.
- Emmanuelle Nicolas; Erica A. Golemis; Sanjeevani Arora; POLD1: Central mediator of DNA replication and repair, and implication in cancer and other pathologies. Gene 2016, 590, 128-141.
- Scott A. Lujan; Jessica S. Williams; Thomas A. Kunkel; DNA Polymerases Divide the Labor of Genome Replication. Trends Cell Biol. 2016, 26, 640-654.
- William Douglass Wright; Shanaya Shital Shah; Wolf-Dietrich Heyer; Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10524-10535.
- Charles S. Fuchs; Toshihiko Doi; Raymond W. Jang; Kei Muro; Taroh Satoh; Manuela Machado; Weijing Sun; Shadia I. Jalal; Manish A. Shah; Jean-Phillipe Metges; et al.Marcelo GarridoTalia GolanMario MandalaZev A. WainbergDaniel V. CatenacciAtsushi OhtsuKohei ShitaraRavit GevaJonathan BleekerAndrew H. KoGeoffrey KuPhilip PhilipPeter C. EnzingerYung-Jue BangDiane LevitanJiangdian WangMinori RosalesRita P. DalalHarry H. Yoon Safety and Efficacy of Pembrolizumab Monotherapy in Patients With Previously Treated Advanced Gastric and Gastroesophageal Junction Cancer. JAMA Oncol. 2018, 4, e180013-e180013.
- Yelena Y. Janjigian; Francisco Sanchez-Vega; Philip Jonsson; Walid K. Chatila; Jaclyn F. Hechtman; Geoffrey Y. Ku; Jamie C. Riches; Yaelle Tuvy; Ritika Kundra; Nancy Bouvier; et al.Efsevia VakianiJianjiong GaoZachary J. HeinsBenjamin E. GrossDavid P. KelsenLiying ZhangVivian E. StrongMark SchattnerHans GerdesDaniel G. CoitManjit BainsZsofia K. StadlerValerie W. RuschDavid R. JonesDaniela MolenaJinru ShiaMark E. RobsonMarinela CapanuSumit MiddhaAhmet ZehirDavid M. HymanMaurizio ScaltritiMarc LadanyiNeal RosenDavid H. IlsonMichael F. BergerLaura TangBarry S. TaylorDavid B. SolitNikolaus Schultz Genetic Predictors of Response to Systemic Therapy in Esophagogastric Cancer. Cancer Discov. 2018, 8, 49-58.
- Secrier M., Li X., de Silva N., Eldridge M.D., Contino G., Bornschein J., MacRae S., Grehan N., O’Donovan M., Miremadi A., et al. Mutational signatures in esophageal adenocarcinoma define etiologically distinct subgroups with therapeutic relevance.. Nat. Genet. 2016, 48, 1131–1141.
- Aaron M. Goodman; Shumei Kato; Lyudmila Bazhenova; Sandip P. Patel; Garrett M. Frampton; Vincent Miller; Philip J. Stephens; Gregory A. Daniels; Razelle Kurzrock; Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598-2608.
- Robert M. Samstein; Chung-Han Lee; Alexander N. Shoushtari; Matthew D. Hellmann; Ronglai Shen; Yelena Y. Janjigian; David A. Barron; Ahmet Zehir; Emmet J. Jordan; Antonio Omuro; et al.Thomas J. KaleySviatoslav M. KendallRobert J. MotzerA. Ari HakimiMartin H. VossPaul RussoJonathan RosenbergGopa IyerBernard H. BochnerDean F. BajorinHikmat A. Al-AhmadieJamie E. ChaftCharles M. RudinGregory J. RielyShrujal BaxiAlan L. HoRichard J. WongDavid G. PfisterJedd D. WolchokChristopher A. BarkerPhilip H. GutinCameron W. BrennanViviane TabarIngo K. MellinghoffLisa M. DeAngelisCharlotte E. AriyanNancy LeeWilliam D. TapMrinal M. GounderSandra P. D’angeloLeonard SaltzZsofia K. StadlerHoward I. ScherJose BaselgaPedram RazaviChristopher A. KlebanoffRona YaegerNeil H. SegalGeoffrey Y. KuRonald P. DeMatteoMarc LadanyiNaiyer A. RizviMichael F. BergerNadeem RiazDavid B. SolitTimothy A. ChanLuc G. T. Morris Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202-206.
- Janice M. Mehnert; Anshuman Panda; Hua Zhong; Kim Hirshfield; Sherri Damare; Katherine Lane; Levi Sokol; Mark N. Stein; Lorna Rodriguez-Rodriquez; Howard L. Kaufman; et al.Siraj AliJeffrey S. RossDean C. PavlickGyan BhanotEileen P. WhiteRobert S. DiPaolaAnn LovellJonathan ChengShridar Ganesan Immune activation and response to pembrolizumab in POLE-mutant endometrial cancer. J. Clin. Investig. 2016, 126, 2334-2340.
- Naiyer A. Rizvi; Matthew D. Hellmann; Alexandra Snyder; Pia Kvistborg; Vladimir Makarov; Jonathan J. Havel; William Lee; Jianda Yuan; Phillip Wong; Teresa S. Ho; et al.Martin L. MillerNatasha RekhtmanAndre L. MoreiraFawzia IbrahimCameron BruggemanBillel GasmiRoberta ZappasodiYuka MaedaChris SanderEdward B. GaronTaha MerghoubJedd D. WolchokTon N. SchumacherTimothy A. Chan Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Sci. 2015, 348, 124-128.
- Michael J. Overman; Sara Lonardi; Ka Yeung Mark Wong; Heinz-Josef Lenz; Fabio Gelsomino; Massimo Aglietta; Michael A. Morse; Eric Van Cutsem; Ray McDermott; Andrew Hill; et al.Michael B. SawyerAlain HendliszBart NeynsMagali SvrcekRebecca A. MossJean-Marie LedeineZ. Alexander CaoShital KambleScott KopetzThierry André Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair–Deficient/Microsatellite Instability–High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773-779.
- Zhiyong Mao; Michael Bozzella; Andrei Seluanov; Vera Gorbunova; DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 2008, 7, 2902-2906.
- Michael R. Lieber; The Mechanism of Double-Strand DNA Break Repair by the Nonhomologous DNA End-Joining Pathway. Annu. Rev. Biochem. 2010, 79, 181-211.
- Jan H. J. Hoeijmakers; Genome maintenance mechanisms for preventing cancer. Nat. 2001, 411, 366-374.
- Sarah L. Thompson; Samuel F. Bakhoum; Duane A. Compton; Mechanisms of Chromosomal Instability. Curr. Biol. 2010, 20, R285-R295.
- Brock J. Sishc; Anthony J. Davis; The Role of the Core Non-Homologous End Joining Factors in Carcinogenesis and Cancer. Cancers 2017, 9, 81.
- Utkarsh Jain; Kirti Saxena; Nidhi Chauhan; Helicobacter pylori induced reactive oxygen Species: A new and developing platform for detection. Helicobacter 2021, 26, e12796.
- James A. Swenberg; Kun Lu; Benjamin C. Moeller; Lina Gao; Patricia B. Upton; Jun Nakamura; Thomas B. Starr; Endogenous versus Exogenous DNA Adducts: Their Role in Carcinogenesis, Epidemiology, and Risk Assessment. Toxicol. Sci. 2010, 120, S130-S145.
- Filomena Mazzei; Alessandra Viel; Margherita Bignami; Role of MUTYH in human cancer. Mutat. Res. Mol. Mech. Mutagen. 2013, 743-744, 33-43.
- Nada Al-Tassan; Nikolas H. Chmiel; Julie Maynard; Nick Fleming; Alison L. Livingston; Geraint T. Williams; Angela K. Hodges; D. Rhodri Davies; Sheila S. David; Julian R. Sampson; et al.Jeremy P. Cheadle Inherited variants of MYH associated with somatic G:C→T:A mutations in colorectal tumors. Nat. Genet. 2002, 30, 227-232.
- Alessandra Viel; Alessandro Bruselles; Ettore Meccia; Mara Fornasarig; Michele Quaia; Vincenzo Canzonieri; Eleonora Policicchio; Emanuele Damiano Urso; Marco Agostini; Maurizio Genuardi; et al.Emanuela Lucci-CordiscoTiziana VenesioAline MartayanMaria Grazia DiodoroLupe Sanchez-MeteVittoria StiglianoFilomena MazzeiFrancesca GrassoAlessandro GiulianiMarta BaiocchiRoberta MaestroGiuseppe GianniniMarco TartagliaLudmil B. AlexandrovMargherita Bignami A Specific Mutational Signature Associated with DNA 8-Oxoguanine Persistence in MUTYH-defective Colorectal Cancer. EBioMedicine 2017, 20, 39-49.
- Maartje Nielsen; Liza N. van Steenbergen; Natalie Jones; Stefanie Vogt; Hans F. A. Vasen; Hans Morreau; Stefan Aretz; Julian R. Sampson; Olaf M. Dekkers; Maryska L. G. Janssen-Heijnen; et al.Frederik J. Hes Survival of MUTYH-Associated Polyposis Patients With Colorectal Cancer and Matched Control Colorectal Cancer Patients. JNCI J. Natl. Cancer Inst. 2010, 102, 1724-1730.
- Mouw K.W., Goldberg M.S., Konstantinopoulos P.A., D’Andrea A.D. DNA Damage and Repair Biomarkers of Immunotherapy Response.. Cancer Discov. 2017, 7, 675–693.
- Soo-Hyun Hahm; Jong-Hwa Park; Sung-Il Ko; You-Ri Lee; In-Sik Chung; Ji-Hyung Chung; Lin-Woo Kang; Ye-Sun Han; Knock-down of human MutY homolog (hMYH) decreases phosphorylation of checkpoint kinase 1 (Chk1) induced by hydroxyurea and UV treatment. BMB Rep. 2011, 44, 352-357.
- Huang M., Miao Z.H., Zhu H., Cai Y.J., Lu W., Ding J. Chk1 and Chk2 are differentially involved in homologous recombination repair and cell cycle arrest in response to DNA double-strand breaks induced by camptothecins. Mol. Cancer Ther. 2008, 7, 1440–1449.
- Carla Oliveira; Hugo Pinheiro; Joana Figueiredo; Raquel Seruca; Fátima Carneiro; Familial gastric cancer: genetic susceptibility, pathology, and implications for management. Lancet Oncol. 2015, 16, e60-e70.
- Peek R.M., Jr., Crabtree J.E. Helicobacter infection and gastric neoplasia. J. Pathol. 2006, 208, 233–248.
- Masanori Hatakeyama; Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat. Rev. Cancer 2004, 4, 688-694.
- Naoko Murata‐Kamiya; Masanori Hatakeyama; Helicobacter pylori ‐induced DNA double‐stranded break in the development of gastric cancer. Cancer Sci. 2022, 113, 1909-1918.
- Masanori Hatakeyama; Helicobacter pylori CagA and Gastric Cancer: A Paradigm for Hit-and-Run Carcinogenesis. Cell Host Microbe 2014, 15, 306-316.
- Andrea K. Byrum; Alessandro Vindigni; Nima Mosammaparast; Defining and Modulating ‘BRCAness’. Trends Cell Biol. 2019, 29, 740-751.
- Yunlong Hu; Mingzhou Guo; Synthetic lethality strategies: Beyond BRCA1/2 mutations in pancreatic cancer. Cancer Sci. 2020, 111, 3111-3121.
- Takahiro Shimizu; Hiroyuki Marusawa; Yuko Matsumoto; Tadashi Inuzuka; Atsuyuki Ikeda; Yosuke Fujii; Sachiko Minamiguchi; Shin’ichi Miyamoto; Tadayuki Kou; Yoshiharu Sakai; et al.Jean E. CrabtreeTsutomu Chiba Accumulation of Somatic Mutations in TP53 in Gastric Epithelium With Helicobacter pylori Infection. Gastroenterology 2014, 147, 407-417.e3.
- Caleb F. Davis; Christopher J. Ricketts; Min Wang; Andrew D. Cherniack; Hui Shen; Christian Buhay; Hyojin Kang; Sang Cheol Kim; Catherine C. Fahey; Kathryn E. Hacker; et al.Gyan BhanotDmitry A. GordeninAndy ChuPreethi H. GunaratneMichael BiehlSahil SethBenny A. KaipparettuChristopher A. BristowLawrence A. DonehowerEric M. WallenAngela B. SmithSatish K. TickooPheroze TamboliVictor ReuterLaura S. SchmidtJames J. HsiehToni K. ChoueiriA. Ari HakimiLynda ChinMatthew MeyersonRaju KucherlapatiWoong-Yang ParkA. Gordon RobertsonPeter W. LairdElizabeth P. HenskeDavid J. KwiatkowskiPeter J. ParkMargaret MorganBrian ShuchDonna MuznyDavid A. WheelerW. Marston LinehanRichard A. GibbsW. Kimryn RathmellChad J. CreightonSabina SignorettiMichael SeilerHsu ChaoMike DahdouliLiu XiNipun KakkarJeffrey G. ReidBrittany DownsJennifer DrummondDonna MortonHarsha DoddapaneniLora LewisAdam EnglishQingchang MengChristie KovarQiaoyan WangWalker HaleAlicia HawesDivya KalraKimberly WalkerBradley A. MurrayCarrie SougnezGordon SaksenaScott L. CarterSteven E. SchumacherBarbara TabakTravis I. ZackGad GetzRameen BeroukhimStacey B. GabrielAdrian AllyMiruna BalasundaramInanc BirolDenise BrooksYaron S.N. ButterfieldEric ChuahAmanda ClarkeNoreen DhallaRanabir GuinRobert A. HoltKatayoon KasaianDarlene LeeHaiyan I. LiEmilia LimYussanne MaMichael MayoRichard A. MooreAndrew J. MungallJacqueline E. ScheinPayal SipahimalaniAngela TamNina ThiessenTina WongSteven J.M. JonesMarco A. MarraJ. Todd AumanDonghui TanShaowu MengCorbin D. JonesKatherine A. HoadleyPiotr A. MieczkowskiLisle E. MoseStuart R. JefferysJeffrey RoachUmadevi VeluvoluMatthew D. WilkersonScot WaringElizabeth BudaJunyuan WuTom BodenheimerAlan P. HoyleJanae V. SimonsMathew G. SolowaySaianand BaluJoel S. ParkerD. Neil HayesCharles M. PerouDaniel J. WeisenbergerMoiz S. BootwallaTimothy TrichePhillip H. LaiDavid J. Van Den BergStephen B. BaylinFengju ChenCristian CoarfaMichael S. NobleDaniel DiCaraHailei ZhangJuok ChoDavid I. HeimanNils GehlenborgDoug VoetPei LinScott FrazerPetar StojanovYingchun LiuLihua ZouJaegil KimMichael S. LawrenceAlexei ProtopopovXingzhi SongJianhua ZhangAngeliki PantaziAngela HadjipanayisEunjung LeeLovelace J. LuquetteSemin LeeMichael ParfenovNetty SantosoJonathan SeidmanAndrew W. XuJunehawk LeeSteven A. RobertsLeszek J. KlimczakDavid FargoMartin LangYoon-La ChoiJune-Koo LeeWenyi WangYu FanJaeil AhnRehan AkbaniJohn N. WeinsteinDavid HausslerSinger MaAmie RadenbaughJingchun ZhulTara M. LichtenbergErik ZmudaAaron D. BlackBenjamin HanfNilsa C. RamirezLisa WiseJay BowenKristen M. LeraasTracy M. HallJulie M. Gastier-FosterWilliam G. KaelinLeigh ThorneLori BoiceMei HuangCathy VockeJames PetersonRobert WorrellMaria MerinoBogdan A. CzerniakKenneth D. AldapeChristopher G. WoodPaul T. SpellmanMichael B. AtkinsJohn ChevilleR. Houston ThompsonMark A. JensenTodd PihlYunhu WanBrenda AyalaJulien BaboudSudhakar VelagaJessica WaltonJia LiuSudha ChudamaniYe WuMargi ShethKenna R. Mills ShawJohn A. DemchokTanja DavidsenLiming YangZhining WangRoy W. TarnuzzerJiashan ZhangGreg EleyIna FelauJean Claude ZenklusenCarolyn M. HutterMark S. GuyerBradley A. OzenbergerHeidi J. Sofia The Somatic Genomic Landscape of Chromophobe Renal Cell Carcinoma. Cancer Cell 2014, 26, 319-330.
- Haiyan Chu; Dongying Gu; Ming Xu; Zhi Xu; Yonglin Gong; Weida Gong; Yongfei Tang; Jianwei Zhou; N. Tong; Zhengdong Zhang; et al.Jinfei ChenMeilin Wang A genetic variant in ERCC2 is associated with gastric cancer prognosis in a Chinese population. Mutagen. 2013, 28, 441-446.
- Xue H., Lu Y., Lin B., Chen J., Tang F., Huang G. The effect of XPD/ERCC2 polymorphisms on gastric cancer risk among different ethnicities: A systematic review and meta-analysis.. PLoS ONE 2012, 7, e43431.
- Jackson S.P. Sensing and repairing DNA double-strand breaks. Carcinogenesis 2002, 23, 687–696.
- Helleday T., Eshtad S., Nik-Zainal S. Mechanisms underlying mutational signatures in human cancers. Nat. Rev. Genet. 2014, 15, 585–598.
- Jackson S.P. Sensing and repairing DNA double-strand breaks. Carcinogenesis 2002, 23, 687–696.
- Helleday T., Eshtad S., Nik-Zainal S. Mechanisms underlying mutational signatures in human cancers. Nat. Rev. Genet. 2014, 15, 585–598.
- Jackson S.P. Sensing and repairing DNA double-strand breaks. Carcinogenesis 2002, 23, 687–696.
- Xue H., Lu Y., Lin B., Chen J., Tang F., Huang G. The effect of XPD/ERCC2 polymorphisms on gastric cancer risk among different ethnicities: A systematic review and meta-analysis.. PLoS ONE 2012, 7, e43431.

