Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Timothy D. Veenstra | -- | 2049 | 2023-10-30 13:25:43 | | | |
| 2 | Sirius Huang | Meta information modification | 2049 | 2023-10-31 02:30:44 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Van De Roovaart, H.J.; Nguyen, N.; Veenstra, T.D. Huntington’s Disease Drug Development. Encyclopedia. Available online: https://encyclopedia.pub/entry/50936 (accessed on 27 June 2026).
Van De Roovaart HJ, Nguyen N, Veenstra TD. Huntington’s Disease Drug Development. Encyclopedia. Available at: https://encyclopedia.pub/entry/50936. Accessed June 27, 2026.
Van De Roovaart, Hannah J., Nguyen Nguyen, Timothy D. Veenstra. "Huntington’s Disease Drug Development" Encyclopedia, https://encyclopedia.pub/entry/50936 (accessed June 27, 2026).
Van De Roovaart, H.J., Nguyen, N., & Veenstra, T.D. (2023, October 30). Huntington’s Disease Drug Development. In Encyclopedia. https://encyclopedia.pub/entry/50936
Van De Roovaart, Hannah J., et al. "Huntington’s Disease Drug Development." Encyclopedia. Web. 30 October, 2023.
Copy Citation
Huntington’s Disease (HD) is a severely debilitating neurodegenerative disorder in which sufferers exhibit different combinations of movement disorders, dementia, and behavioral or psychiatric abnormalities. The disorder is a result of a trinucleotide repeat expansion mutation that is inherited in an autosomal dominant manner. While there is still no treatment to alter the course of HD, there are medications that lessen abnormal movement and psychiatric symptoms.
Huntington’s disease
treatment
clinical trial
1. Introduction
Huntington’s Disease (HD) is a rare autosomal dominant neurodegenerative genetic disorder that has an average onset between 30 and 50 years of age [1]. The worldwide prevalence of HD is approximately 7 in 100,000 persons, with further evidence suggesting the worldwide prevalence may be increasing [2][3][4][5][6]. The regions of highest prevalence are North America and the United Kingdom, with Asia having the lowest [4]. This disease places a heavy cost burden on patients, caregivers, and the health system. A recent Huntington’s Disease Burden of Illness study comprising patients from five European countries and the United States estimated the annual direct medical, nonmedical, and indirect costs associated with HD at just over EUR 63,000 (i.e., about USD 68,000) per patient per annum [5]. Predictably, these costs escalate as patients progress from early- to late-stage HD. Hospital visits and long-term care account for the largest proportion of these expenses.
Caused by a mutation in the Huntington gene (HTT), HD patients have what is known as a “CAG repeat expansion,”. While most healthy people have between 10 and 30 repeats, individuals with HD have 40+ CAG repeats [7]. This trinucleotide repeat inserts additional glutamine residues (referred to as a polyQ domain) within the translated huntingtin protein (HTT). The average age of onset is between 30 and 50 years of age, with individuals possessing longer trinucleotide repeats generally exhibiting symptoms of HD at earlier ages [8]. In predictive models, the length of the trinucleotide repeats accounts for about 70% of the interpatient variability in the age of onset [9]. The highest concentrations of HTT are located within the brain and testes [10]. The main functions of HTT involve chemical signaling, cellular dynamics (i.e., the cytoskeleton, endocytosis, trafficking, and adhesion), metabolism, protein turnover, gene expression (transcription and RNA processing), and protection [11][12]. The aggregation of mutant HTT (mHTT) is the main pathophysiological signature of patients with HD [13]. These aggregates form inclusion bodies inside cells that lead to cell quiescence and neuronal degeneration [14][15]. The clinical manifestations of HD include chorea, dementia, and psychiatric symptoms that eventually lead to death between 15 and 20 years after symptoms are first detected [16][17].
While HD affects the entire brain, there is evidence of enhanced vulnerability within the striatum, which is part of the basal ganglia [18]. This brain region is involved in motor control, emotion, habit formation, and reward [19]. Neuronal degeneration within the basal ganglia is consistent with the symptoms observed in HD patients that include memory impairment, slurred speech, chorea, weight loss, and personality changes [20][21][22].
2. Huntington’s Disease Drug Development
ClinicalTrials.gov was used to identify drugs that are currently in or have recently completed a phase III trial for an indication within HD. Only interventional phase 3 trials that were recruiting patients, were active but not recruiting patients, or had been completed were included. All the studies must have had an update posted within the past year.
Nine clinical trials were identified in accordance with the aforementioned criteria. There are eight different drug interventions included in the nine clinical trials. Four of these medications (dextromethorphan/quinidine, valbenazine, deutetrabenazine, and metformin) are FDA approved for alternative indications while four others (Cellavita HD, pridopidine, tominersen, and SAGE-718) have not yet been marketed and are seeking their first FDA approval.
The nine drugs described herein, along with some brief clinical trial information, are listed in Table 1.
Table 1. List of phase III clinical trials testing the effects of various drugs in the treatment of Huntington’s Disease.
| Drug | Clinical Trial Name | Identifier | Primary Outcome |
|---|---|---|---|
| Metformin | Testing METformin against cognitive decline in HD | NCT04826692 | Evaluate drug’s effect on scores obtained in different cognitive subtests that make up the Unified Huntington’s Disease Rating Scale (UHDRS) |
| Dextromethorphan/ quinidine |
Evaluating the Efficacy of Dextromethorphan/Quinidine in Treating Irritability in HD | NCT03854019 | Measure patient’s irritability using The Irritability Scale |
| Deutetrabenazine | Impact of Deutetrabenazine on Functional Speech and Gait Dynamics in Huntington Disease | NCT04713982 | Improvement on Sentence Intelligibility Test and Motor Speech Evaluation |
| Valbenazine | Efficacy, Safety, and Tolerability of Valbenazine for the Treatment of Chorea Associated with Huntington Disease (KINECT-HD) | NCT04102579 | Improvement in chorea symptoms and evaluation of treatment-emergent adverse events |
| An Open-Label Rollover Study for Continuing Valbenazine Administration for the Treatment of Chorea Associated With Huntington Disease | NCT04400331 | Evaluation of treatment-emergent adverse events (TEAEs) | |
| Cellavita HD | Clinical Extension Study for Safety and Efficacy Evaluation of the Cellavita HD Product in Huntington’s Patients (ADORE-EXT) | NCT04219241 | Evaluate treatment’s effectiveness as verified by comparing the total UHDRS |
| Pridopidine | Pridopidine’s Outcome on Function in Huntington Disease, PROOF-HD | NCT04556656 | Measure change from baseline in UHDRS-Total Functional Capacity score |
| SAGE-718 | A Study to Evaluate the Safety and Tolerability of SAGE-718 in Participants With Huntington’s Disease | NCT05655520 | Measure number and severity of TEAEs Measure number of participants that withdraw due to adverse events Measure change from baseline in clinical laboratory and electrocardiogram parameters Measure change in baseline in C-SSRS responses |
| RO7234292 (RG6042) | An Open-Label Extension Study to Evaluate Long-Term Safety and Tolerability of RO7234292 (RG6042) in Huntington’s Disease | NCT03842969 | Measure number and severity of TEAEs Measure number of participants with suicidal ideation/behavior and self-injurious behavior without suicidal intent based on C-SSRS scale Measure change from baseline in MoCA |
2.1. Metformin
Metformin is one of the oldest and most studied diabetic medications [23][24]. Besides type 2 diabetes (DM2), metformin may also offer cardiovascular protection and beneficial effects on obesity, musculoskeletal and reproductive diseases, cancer, and aging [25]. Not only is metformin able to penetrate nearly every bodily organ, it is also a well-known pleiotropic agent that modulates a plethora of metabolic pathways, extending its possible use beyond already FDA-approved indications [26]. The major molecular targets of metformin include complex I of the mitochondrial electron transport chain, the mechanistic target of rapamycin complex 1 (mTORC1), and adenosine monophosphate-activated protein kinase (AMPK) [27]. The interest in metformin as an HD treatment is in its ability to activate AMPK. Located throughout the body, this enzyme induces improved neuronal survival by inhibiting inflammation and promoting cell renewal processes [28]. Metformin has also been shown to induce autophagy, inhibit mHTT aggregation, and reduce the accumulation of this mutant protein within an HD mouse model [29]. While metformin activation of AMPK may offer some benefits, the optimal timing of AMPK activation appears to be an important variable in its effectiveness as an HD therapeutic. Data from in vivo/in vitro models and analyses of brain tissue have demonstrated that AMPK activation during late stages of HD could have negative effects [30]. Thus, AMPK may be an efficacious target in early-stage HD but may have contrasting effects during late-stage HD.
2.2. Dextromethorphan/Quinidine
Dextromethorphan/quinidine (DM/Q) is a fixed-dose combination therapy that was approved by the FDA in 2010 [31]. It is marketed by Avanir Pharmaceuticals under the brand name Nuedexta. Dextromethorphan is a noncompetitive N-methyl-D-aspartate (NMDA) receptor antagonist and a sigma-1-receptor (S1R) agonist [32]. Quinidine prolongs the plasma levels of dextromethorphan by inhibiting the CYP2D6 enzyme. There is evidence that NMDA receptor excitotoxicity is implicated in the pathogenesis of HD. Enhanced NMDA receptor signaling is detected at ages prior to motor dysfunction and neuronal loss. Specifically, agents such as quinolinic acid (a selective NMDA receptor agonist) can produce striatal degeneration. There are studies in HD mice treated with NMDA receptor antagonists that showed the reversal of nuclear signaling and motor learning deficits. Pridopidine, an S1R agonist, has previously been shown to have neuroprotective effects in cellular and animal models of HD (as well as Alzheimer's Disease) presumably by increasing mitochondrial activity. This increased activity led to improvements in motor coordination and a delay in symptom onset in mouse HD models.
2.3. Deutetrabenazine
As a reversible inhibitor of vesicular monoamine transporter 2 (VMAT2), deutetrabenazine decreases the reuptake and stores monoamines (i.e., dopamine, serotonin, histamine, and norepinephrine) within presynaptic vesicles [33]. VMAT2 is located on the synaptic vesicles of presynaptic neurons [34]. Responsible for the packaging and release of monoamines into the synapse, drugs targeting this transporter have shown efficacy in treating symptoms that manifest from the imbalance of monoamines [35].
Deutetrabenazine is a deuterated form of tetrabenazine, meaning that there is a substitution with deuterium for hydrogen throughout the molecule, giving it a plasma half-life (t½) almost two-fold greater than tetrabenazine [36]. This longer t½ decreases plasma fluctuations, resulting in fewer adverse effects such as somnolence, insomnia, depression, and Parkinsonism, which are associated with peak drug concentration.
2.4. Valbenazine
Hyperkinetic movements including those representative of chorea have been associated with high dopamine states. Thus, the current standard of care aims to decrease dopamine activity through the off-label use of antipsychotics that antagonize postsynaptic receptors or the on-label use of the existing FDA-approved VMAT2 inhibitors deutetrabenazine and tetrabenazine [37]. As a VMAT2 inhibitor, valbenazine works similarly to deutetrabenazine and tetrabenazine to treat chorea associated with HD. Valbenazine differs from deutetrabenazine and tetrabenazine in its affinity for VMAT2. Deutetrabenazine and tetrabenazine are broken down into four different stereoisomers, all of which have shown varying affinity for VMAT2. Valbenazine is broken down into only one stereoisomer, [+]-α-dihydrotetrabenazine, which has the strongest affinity for VMAT2 of all the stereoisomers [37]. In addition to the affinity observed with respect to VMAT2 receptors, valbenazine’s formulation allows for easier titration and daily dosing. Deutetrabenazine and tetrabenazine require multiple episodes of daily dosing, with tetrabenazine specifically requiring a slower, more complicated uptitration schedule.
2.5. Cellavita HD
Cellavita HD represents a novel therapy within the treatment landscape for HD. Cellavita HD is a stem cell therapy that may help restore lost brain cells [38]. Cellavita HD involves dental-pulp-derived mesenchymal stem cells that have been associated with both multifaceted differential effects as well as immunomodulatory functions [39]. Capable of crossing the blood–brain barrier, Cellavita HD promotes the proliferation of neuronal stem cells. Available evidence supporting the use of dental pulp stem cells in neurodegenerative disorders comes from their ability to regulate different molecular mediators including those within the anti-inflammatory, neurogenic, antiapoptotic, angiogenic, and osteogenic classes [40].
2.6. Pridopidine
In HD, there is progressive neuronal loss in the striatum. This process includes the dysregulation of endoplasmic reticulum (ER) calcium, altered mitochondrial function, reduced autophagy, increased ER stress, and reduced brain-derived neurotrophic factor, all ultimately leading to neuronal death [41]. Pridopidine was originally identified as a low-affinity dopamine D2 receptor ligand that modulated dopamine-dependent behaviors [42]. In the R6/2 mouse model of HD, pridopidine had been shown to improve motor performance and offer neuroprotective effects [43]. Recent evidence, however, has shown that pridopidine’s affinity for S1R is ~100- and ~30-fold higher than that for the D2 and D3 receptors, respectively [42]. These results suggest that the positive effects of pridopidine are modulated through its interaction with S1R. Studies have shown that pridopidine activation of S1R reduces cell stress and inflammation, while increasing cellular energy production and the clearance of toxic proteins. Since S1R is also involved in synapse plasticity, pridopidine can enhance neuronal connectivity. Prilenia Pharma currently holds orphan drug designation for the treatment of HD in both the US and EU using pridopidine, while simultaneously receiving fast-track designation from the FDA [44].
2.7. SAGE-718
After receiving orphan drug status and fast-track designation for the treatment of HD by the European Medicine Agency, SAGE-718 has gained traction in both its clinical development and regulatory review process. As a first-in-class NMDA receptor modulator, SAGE-718 was designed to improve cognitive function in disorders associated with NMDA dysfunction such as AD and HD [45]. Modulation of the NMDA receptor enhances long-term effects at neuronal synapses, which is an essential process in learning and memory.
2.8. Tominersen (RO7234292 or RG6042)
Tominersen is also known as RO7234292 or RG6042. Ionis Pharmaceuticals designed tominersen and licensed the treatment to Roche, who is currently developing and marketing it as a potential HD treatment. Tominersen is an antisense RNA that binds to mRNA transcribed from the mutant HTT gene found in HD patients. This interaction creates an HTT mRNA/tominersen hybrid, which is degraded by the cell and is therefore not translated into the mHTT protein (Figure 1). The net result is a reduced production of mHTT.
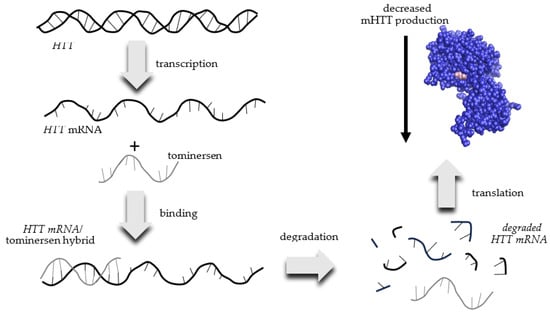
Figure 1. Mechanism of action of tominersen for the decreased production of mutant huntingtin (mHTT) protein in patients with Huntington’s Disease.
References
- Anil, M.; Mason, S.L.; Barker, R.A. The clinical features and progression of late-onset versus younger-onset in an adult cohort of Huntington’s Disease patients. J. Huntington’s Dis. 2020, 9, 275–282.
- Ross, C.A.; Aylward, E.H.; Wild, E.J.; Langbehn, D.R.; Long, J.D.; Warner, J.H.; Scahill, R.I.; Leavitt, B.R.; Stout, J.C.; Paulsen, J.S.; et al. Huntington disease: Natural history, biomarkers and prospects for therapeutics. Nat. Rev. Neurol. 2014, 10, 204–216.
- McColgan, P.; Tabrizi, S.J. Huntington’s disease: A clinical review. Eur. J. Neurol. 2018, 25, 24–34.
- Rawlins, M.D.; Wexler, N.S.; Wexler, A.R.; Tabrizi, S.J.; Douglas, I.; Evans, S.J.W.; Smeeth, L. The prevalence of Huntington’s disease. Neuroepidemiology 2016, 46, 144–153.
- Rodríguez-Santana, I.; Mestre, T.; Squitieri, F.; Willock, R.; Arnesen, A.; Clarke, A.; D’Alessio, B.; Fisher, A.; Fuller, R.; Hamilton, J.L.; et al. Economic burden of Huntington disease in Europe and the USA: Results from the Huntington’s Disease Burden of Illness study. Eur. J. Neurol. 2023, 30, 1109–1117.
- Shaw, E.; Mayer, M.; Ekwaru, P.; McMullen, S.; Graves, E.; Wu, J.W.; Budd, N.; Maturi, B.; Cowling, T.; Mestre, T.A. Epidemiology and economic burden of Huntington’s disease: A Canadian provincial public health system perspective. J. Med. Econ. 2022, 1, 212–219.
- Myers, R.H. Huntington’s disease genetics. NeuroRx 2004, 1, 255–262.
- van der Zwaan, K.F.; Mentink, M.D.C.; Jacobs, M.; Roos, R.A.C.; de Bot, S.T. Huntington’s disease influences employment before and during clinical manifestation: A systematic review. Park. Relat. Disord. 2022, 96, 100–108.
- Wright, G.E.B.; Black, H.F.; Collins, J.A.; Gall-Duncan, T.; Caron, N.S.; Pearson, C.E.; Hayden, M.R. Interrupting sequence variants and age of onset in Huntington’s disease: Clinical implications and emerging therapies. Lancet Neurol. 2020, 19, 930–939.
- Li, S.H.; Schilling, G.; Young, W.S., 3rd; Li, X.J.; Margolis, R.L.; Stine, O.C.; Wagster, M.V.; Abbott, M.H.; Franz, M.L.; Ranen, N.G.; et al. Huntington’s disease gene (IT15) is widely expressed in human and rat tissues. Neuron 1993, 11, 985–993.
- Schulte, J.; Littleton, J.T. The biological function of the Huntingtin protein and its relevance to Huntington’s Disease pathology. Curr. Trends Neurol. 2011, 5, 65–78.
- Saudou, F.; Humbert, S. The biology of Huntingtin. Neuron 2016, 89, 910–926.
- Hackam, A.S.; Hodgson, J.G.; Singaraja, R.; Zhang, T.; Gan, L.; Gutekunst, C.A.; Hersch, S.M.; Hayden, M.R. Evidence for both the nucleus and cytoplasm as subcellular sites of pathogenesis in Huntington’s disease in cell culture and in transgenic mice expressing mutant huntingtin. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1999, 354, 1047–1055.
- Davies, S.W.; Turmaine, M.; Cozens, B.A.; DiFiglia, M.; Sharp, A.H.; Ross, C.A.; Scherzinger, E.; Wanker, E.E.; Mangiarini, L.; Bates, G.P. Formation of Neuronal Intranuclear Inclusions Underlies the Neurological Dysfunction in Mice Transgenic for the HD Mutation. Cell 1997, 90, 537–548.
- Reiner, A.; Dragatsis, I.; Dietrich, P. Genetics and Neuropathology of Huntington’s Disease. Int. Rev. Neurobiol. 2011, 98, 325–372.
- Almqvist, E.W.; Bloch, M.; Brinkman, R.; Craufurd, D.; Hayden, M.R. A worldwide assessment of the frequency of suicide, suicide attempts, or psychiatric hospitalization after predictive testing for Huntington disease. Am. J. Hum. Genet. 1999, 64, 1293–1304.
- Petrella, L.I.; Castelhano, J.M.; Ribeiro, M.; Sereno, J.V.; Gonçalves, S.I.; Laço, M.N.; Hayden, M.R.; Rego, A.C.; Castelo-Branco, M. A whole brain longitudinal study in the YAC128 mouse model of Huntington’s disease shows distinct trajectories of neurochemical, structural connectivity and volumetric changes. Hum. Mol. Genet. 2018, 27, 2125–2137.
- Blumenstock, S.; Dudanova, I. Cortical and striatal circuits in Huntington’s disease. Front. Neurosci. 2020, 14, 82–99.
- Rocha, G.S.; Freire, M.A.M.; Britto, A.M.; Paiva, K.M.; Oliveira, R.F.; Fonseca, I.A.T.; Araújo, D.P.; Oliveira, L.C.; Guzen, F.P.; Morais, P.L.A.G.; et al. Basal ganglia for beginners: The basic concepts you need to know and their role in movement control. Front. Syst. Neurosci. 2023, 17, 1242929–1242936.
- Zielonka, D.; Piotrowska, I.; Marcinkowski, J.T.; Mielcarek, M. Skeletal muscle pathology in Huntington’s disease. Front. Physiol. 2014, 5, 380–384.
- Bonomo, R.; Elia, A.E.; Bonomo, G.; Romito, L.M.; Mariotti, C.; Devigili, G.; Cilia, R.; Giossi, R.; Eleopra, R. Deep brain stimulation in Huntington’s disease: A literature review. Neurol. Sci. 2021, 42, 4447–4457.
- Bonelli, R.M.; Cummings, J.L. Frontal-subcortical dementias. Neurologist 2008, 14, 100–107.
- Blonde, L. Management of type 2 diabetes: Update on new pharmacological options. Manag. Care 2000, 9, 11–17.
- Kajbaf, F.; De Broe, M.E.; Lalau, J.D. Therapeutic concentrations of metformin: A systematic review. Clin. Pharmacokinet. 2016, 55, 439–459.
- Song, Y.; Wu, Z.; Zhao, P. The function of metformin in aging-related musculoskeletal disorders. Front. Pharmacol. 2022, 13, 865524–865537.
- Trujillo-Del Río, C.; Tortajada-Pérez, J.; Gómez-Escribano, A.P.; Casterá, F.; Peiró, C.; Millán, J.M.; Herrero, M.J.; Vázquez-Manrique, R.P. Metformin to treat Huntington disease: A pleiotropic drug against a multi-system disorder. Mech. Ageing Dev. 2022, 204, 111670–111682.
- Faria, J.; Negalha, G.; Azevedo, A.; Martel, F. Metformin and breast cancer: Molecular targets. J. Mammary Gland Biol. Neoplasia 2019, 24, 111–123.
- Ronnett, G.V.; Ramamurthy, S.; Kleman, A.M.; Landree, L.E.; Aja, S. AMPK in the brain: Its roles in energy balance and neuroprotection. J. Neurochem. 2009, 109 (Suppl. S1), 17–23.
- Sanchis, A.; García-Gimeno, M.A.; Cañada-Martínez, A.J.; Sequedo, M.D.; Millán, J.M.; Sanz, P.; Vázquez-Manrique, R.P. Metformin treatment reduces motor and neuropsychiatric phenotypes in the zQ175 mouse model of Huntington disease. Exp. Mol. Med. 2019, 51, 1–16.
- Hervás, D.; Fornés-Ferrer, V.; Gómez-Escribano, A.P.; Sequedo, M.D.; Peiró, C.; Millán, J.M.; Vázquez-Manrique, R.P. Metformin intake associates with better cognitive function in patients with Huntington’s disease. PLoS ONE 2017, 12, e0179283.
- FDA Center for Drug Evaluation and Research Medical Review Application Number: 021879Orig1s000. 30 April 2010. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2010/021879Orig1s000MedR.pdf (accessed on 5 September 2023).
- Price, D.D.; Mao, J.; Frenk, H.; Mayer, D.J. The N-methyl-D-asparate receptor antagonist dextromethorphan selectively reduces temporal summation of second pain in man. Pain 1994, 59, 165–174.
- Scorr, L.M.; Factor, S.A. VMAT2 inhibitors for the treatment of tardive dyskinesia. J. Neurol. Sci. 2018, 389, 43–47.
- Yaffe, D.; Forrest, L.R.; Schuldiner, S. The ins and outs of vesicular monoamine transporters. J. Gen. Physiol. 2018, 150, 671–682.
- Harriott, N.D.; Williams, J.P.; Smith, E.B.; Bozigian, H.P.; Grigoriadis, D.E. VMAT2 inhibitors and the path to Ingrezza (valbenazine). Prog. Med. Chem. 2018, 57, 87–111.
- Dean, M.; Sung, V.W. Review of deutetrabenazine: A novel treatment for chorea associated with Huntington’s disease. Drug Des. Dev. Ther. 2018, 12, 313–319.
- Furr Stimming, E.; Claassen, D.O.; Kayson, E.; Goldstein, J.; Mehanna, R.; Zhang, H.; Liang, G.S.; Haubenberger, D.; Huntington Study Group KINECT-HD Collaborators. Safety and efficacy of valbenazine for the treatment of chorea associated with Huntington’s disease (KINECT-HD): A phase 3, randomized, double-blind, placebo-controlled trial. Lancet Neurol. 2023, 22, 494–504.
- Ferguson, M.W.; Kennedy, C.J.; Palpagama, T.H.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. Current and possible future therapeutic options for Huntington’s Disease. J. Cent. Nerv. Syst. Dis. 2022, 14, 11795735221092517–11795735221092553.
- Song, W.P.; Jin, L.Y.; Zhu, M.D.; Wang, H.; Xia, D.S. Clinical trials using dental stem cells: 2022 update. World J. Stem Cells 2023, 15, 31–51.
- Ueda, T.; Inden, M.; Ito, T.; Kurita, H.; Hozumi, I. Characteristics and therapeutic potential of dental pulp stem cells on neurodegenerative diseases. Front. Neurosci. 2020, 14, 407–413.
- Waters, S.; Tedroff, J.; Ponten, H.; Klamer, D.; Sonesson, C.; Waters, N. Pridopidine: Overview of pharmacology and rationale for its use in Huntington’s Disease. J. Huntington’s Dis. 2018, 7, 1–16.
- Grachev, I.D.; Meyer, P.M.; Becker, G.A.; Bronzel, M.; Marsteller, D.; Pastino, G.; Voges, O.; Rabinovich, L.; Knebel, H.; Zientek, F.; et al. Sigma-1 and dopamine D2/D3 receptor occupancy of pridopidine in healthy volunteers and patients with Huntington disease: A fluspidine and fallypride PET study. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 1103–1115.
- Squitieri, F.; Di Pardo, A.; Favellato, M.; Amico, E.; Maglione, V.; Frati, L. Pridopidine, a dopamine stabilizer, improves motor performance and shows neuroprotective effects in Huntington disease R6/2 mouse model. J. Cell. Mol. Med. 2015, 19, 2540–2548.
- What is Pridopidine? Available online: https://www.Prilenia.com (accessed on 17 September 2023).
- Hill, M.D.; Blanco, M.J.; Salituro, F.G.; Bai, Z.; Beckley, J.T.; Ackley, M.A.; Dai, J.; Doherty, J.J.; Harrison, B.L.; Hoffmann, E.C.; et al. SAGE-718: A first-in-class N-methyl-d-aspartate receptor positive allosteric modulator for the potential treatment of cognitive impairment. J. Med. Chem. 2022, 65, 9063–9075.
More
Information
Subjects:
Neurosciences
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
648
Revisions:
2 times
(View History)
Update Date:
31 Oct 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No