 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ana Urbano | -- | 4047 | 2023-10-23 23:38:23 | | | |
| 2 | Peter Tang | Meta information modification | 4047 | 2023-10-24 03:16:28 | | |
Video Upload Options
Chromium (Cr) industries (metallurgical, chemical and refractory) employ several million workers worldwide. These workers may suffer from a variety of adverse health effects produced by airborne dusts, mists and fumes containing Cr in the hexavalent oxidation state, Cr(VI). Of major importance, occupational exposure to Cr(VI) compounds has been firmly associated with the development of lung cancer. Counterintuitively, Cr(VI) is largely unreactive towards most biomolecules, including nucleic acids and proteins. Yet, once inside cells, Cr(VI) undergoes reduction producing several species that react extensively with biomolecules. The diversity and chemical versatility of these species add great complexity to the study of the molecular mechanisms underlying Cr(VI) toxicity and carcinogenicity, which remain poorly understood. One such mechanism may involve the cellular stress response (also known as heat shock response), an intricate cellular system that combats proteotoxic stress, which is increasingly viewed as playing a critical role in carcinogenesis. Several studies, while not constituting a direct proof of a link between the cellular stress response and Cr(VI)-induced carcinogenesis, have shown the ability of Cr(VI) to modulate the expression of several components of this response under biologically relevant conditions.
1. Hexavalent Chromium: Applications, Chemical Properties and Biological Implications
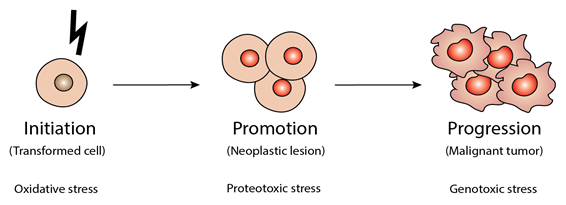
2. Cancer Initiation, Promotion and Progression: The Critical Importance of Oxidative, Proteotoxic and Genotoxic Stresses
3. Links between the Cellular Response to Stress and Carcinogenesis
3.1. Note on Nomenclature
3.2. The Cellular Stress Response: Basic Concepts
3.3. Cancer and the Cellular Stress Response
4. The Molecular Mechanisms of Hexavalent Chromium Carcinogenicity: A Brief State of the Art
Genetic mechanisms likely play a critical role Cr(VI) carcinogenesis, as supported by the observation of genetic lesions in both the lung cells of chromate workers and in cultured cells exposed to different Cr(VI) concentrations [78][79][80][81][82][83][84][85][86][87]. Thus, the initial observation, in test tube experiments, that Cr(VI) is mostly unreactive towards DNA (and most other biomolecules) puzzled researchers. However, it is now known that, following its rapid cellular uptake, Cr(VI) undergoes a multi-step reduction that generates a variety of species that react extensively with biomolecules, namely Cr(III), which is the final reduction species, and the unstable intermediates Cr(IV) and Cr(V) [88][89]. Under physiological conditions, ascorbate accounts for about 90% of Cr(VI) reduction, but non-protein thiols, such as glutathione and cysteine, also contribute significantly to its reduction [90]. Thus, Cr(VI) reduction generates additional reactive species, such as carbon-based radicals from ascorbate, thiyl radicals from glutathione and cysteine and, possibly, ROS. Among the Cr(III)-DNA complexes formed are Cr(III)-DNA adducts, DNA-protein crosslinks and DNA interstrand crosslinks [78][91].
By restraining the normal DNA replication and transcription processes, these adducts activate the various cellular DNA repair systems in a lesion-dependent manner. Mutations in key proteins involved in these DNA repair systems have been described both in Cr(VI)-induced lung cancer patients and in cultured cells exposed to Cr(VI) compounds, impairing their ability to remove chromium-DNA adducts [92]. Importantly, double-strand break (DSB) induction by the mismatch repair (MMR) system may drive genomic instability [3][93]. Hirose and co-workers reported a high incidence of microsatellite instability (MSI), a particular type of genomic instability that specifically affects the microsatellites, in lung cancers from chromate-exposed workers [3][94][95]. However, MSI was not be observed upon in vitro exposure of human lung epithelial cells to Cr(VI) [96].
Cr(VI) exposure can also generate DNA damage by indirect mechanisms. For instance, Cr(VI) exposure may lead to loss of thiol redox control through interference with antioxidant defense systems [97]. This and additional lines of evidence, namely the observation of 8-hydroxy-2’-deoxyguanosine formation in rat lungs following intratracheal administration of Cr(VI) [86], suggest that Cr(VI) exposure can damage DNA through the generation of oxidative stress [98][99][100]. Additionally, altered ROS levels affect gene expression [101].
5. The Impact of Hexavalent Chromium on the Cellular Stress Response
Two studies have been conducted in the BEAS-2B cell line, established from normal human bronchial epithelium. Both studies used Cr(VI) concentrations that did not cause overt cytotoxicity. The first study aimed at identifying specific and sensitive biomarkers of toxic metal exposure [110]. One significant finding was the extreme specificity of the Cr(VI) effects: of the 1200 gene transcripts analyzed, only 44 had their expression altered after a 4 h Cr(VI) exposure. Of the 44 genes affected, 3 encoded HSP (HSP40, HSP60 and HSP90A) and were all down-regulated. The transcript levels of all other HSP analyzed (HSP27, HSP-70, HSP70.1, HSP-71) remained unchanged, giving further support to the perception that the impact of Cr(VI) is isoform-specific.
The second study employing BEAS-2B cells investigated the impact of Cr(VI) on the expression of the Hsp72 and Hsp90α isoforms at both the transcript and protein levels [111]. Importantly, this study unveiled decoupling of mRNA and protein levels for both Hsp72 and Hsp90α. After a 48 h incubation with Cr(VI), Hsp72 mRNA levels were decreased, whereas Hsp72 protein levels remained unchanged. For Hsp90α, mRNA levels were unaltered, whereas protein levels were decreased. This decoupling is likely multifactorial, potentially involving critical post-transcriptional regulators, such as RNA binding proteins and microRNAs [112][113]. Protein stability and turnover may also have to be considered [114][115]. Thus, in future studies, it will be important to conduct detailed time-courses of the effects of Cr(VI) on gene expression at both levels.
There are two other published cellular studies on the impact of Cr(VI) on HSP70 [116][117] and one on the impact of this carcinogen on HSP90 [117]. Altogether, these studies clearly show that this impact is dependent on both the cellular model employed and on the experimental design.
Another study, conducted in rat lung epithelial cells, showed the impact of Cr(VI) on additional HSP isoforms, namely Hsp10 and Hsp105, whose protein levels were increased after a 24 h incubation, which was shown to produce significant cytotoxicity [118]. Two other studies, one employing HaCaT [119] cells and the other employing human primary skin fibroblasts [120], unveiled the ability of Cr(VI) to alter the phosphorylation state of HSP27. Of note, aberrant phosphorylation of HSP27 has been associated with cancer [121]. In HaCaT cells, HSP27 expression was upregulated by Cr(VI) at both transcript and protein levels, but the phosphorylation of this HSP was decreased [119]. On the contrary, levels of phosphorylated HSP27 were found to be increased in Cr(VI)-exposed in human primary skin fibroblasts [120]. This apparent contradiction might be explained by differences in cell model, Cr(VI) concentration and duration of exposure.
In the only two in vivo studies conducted to date, one employing Institute of Cancer Research (ICR) mice [122] and the other one Sprague-Dawley rats [103], Cr(VI) administration induced HSP expression. In ICR mice, Cr(VI) intraperitoneal injection increased liver HSP27 and HSP70 protein levels. In Sprague-Dawley rats, Cr(VI) intratracheal instillation increased HSP70 mRNA levels in the lungs, but not in the liver. HSP60, Grp75 and Grp94 mRNA levels, on the other hand, were unaffected in both lung and liver. In fact, none of the 216 genes assessed had their liver mRNA levels altered, whereas changes in lung mRNA levels were observed for 52 genes. The observed lack of effect in the liver was ascribed to the upstream reduction and consequent detoxification of Cr(VI), firstly in the lung, then in the blood of the general circulation and finally in the liver itself.
While the results obtained in the studies published thus far do not constitute a direct proof of a link between the cellular stress response and Cr(VI)-induced carcinogenesis, they do show the ability of this carcinogen to modulate the expression of several components of this response under conditions of biological relevance. It has also become clear that the observed effects are dependent on tissue, cell type, Cr(VI) concentration, duration of exposure and HSP isoform. Future studies must address the issue of biological relevance and should also include adequate time courses, as HSP transcript and protein levels change over time during the recovery period.References
- Cotton, F.A. Advanced Inorganic Chemistry, 6th ed.; Wiley: New York, NY, USA, 1999; p. 1355.
- Urbano, A.M.; Ferreira, L.M.R.; Alpoim, M.C. Molecular and cellular mechanisms of hexavalent chromium-induced lung cancer: An updated perspective. Curr. Drug Metab. 2012, 13, 284–305.
- Urbano, A.M.; Rodrigues, C.F.D.; Alpoim, M.C. Hexavalent chromium exposure, genomic instability and lung cancer. Gene Mol. Biol. 2008, 12B, 219–238.
- Anderson, R.A. Chromium as an essential nutrient for humans. Regul. Toxicol. Pharm. 1997, 26, S35–S41.
- Jeejeebhoy, K.N. The role of chromium in nutrition and therapeutics and as a potential toxin. Nutr. Rev. 1999, 57, 329–335.
- EFSA. Scientific opinion on dietary reference values for chromium. Efsa J. 2014, 12, 25.
- IARC. Chromium, nickel and welding. Iarc Monogr. Eval. Carcinog. Risks Hum. 1990, 49, 1–648.
- IARC. Arsenic, metals, fibres and dusts. Iarc Monogr. Eval. Carcinog. Risks Hum. 2012, 100, 1–465.
- NTP. Report on Carcinogens, 13th ed. Research Triangle Park, NC, USA, 2015.
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767.
- Pitot, H.C. The molecular biology of carcinogenesis. Cancer 1993, 72, 962–970.
- Ferreira, L.M. Cancer metabolism: The Warburg effect today. Exp. Mol. Pathol. 2010, 89, 372–380.
- Abreu, P.L.; Urbano, A.M. Targeting the Warburg effect for cancer therapy: A long and winding road. Front. Clin. Drug Res.—Anti-Cancer Agents 2016, 3, 271–324.
- Acharya, A.; Das, I.; Chandhok, D.; Saha, T. Redox regulation in cancer: A double-edged sword with therapeutic potential. Oxid. Med. Cell Longev. 2010, 3, 23–34.
- Schar, P. Spontaneous DNA damage, genome instability, and cancer—When DNA replication escapes control. Cell 2001, 104, 329–332.
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic Biol. Med. 2017, 104, 144–164.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Hartl, F.U.; Hayer-Hartl, M. Converging concepts of protein folding in vitro and in vivo. Nat. Struct. Mol. Biol. 2009, 16, 574–581.
- Vandewynckel, Y.P.; Laukens, D.; Geerts, A.; Bogaerts, E.; Paridaens, A.; Verhelst, X.; Janssens, S.; Heindryckx, F.; Van Vlierberghe, H. The paradox of the unfolded protein response in cancer. Anticancer Res. 2013, 33, 4683–4694.
- Bussard, K.M.; Mutkus, L.; Stumpf, K.; Gomez-Manzano, C.; Marini, F.C. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016, 18, 84.
- Thommen, D.S.; Schumacher, T.N. T cell dysfunction in cancer. Cancer Cell 2018, 33, 547–562.
- Kerbel, R.S. Tumor angiogenesis. N. Engl. J. Med. 2008, 358, 2039–2049.
- Pflaum, J.; Schlosser, S.; Muller, M. p53 family and cellular stress responses in cancer. Front. Oncol. 2014, 4, 285.
- Herr, I.; Debatin, K.M. Cellular stress response and apoptosis in cancer therapy. Blood 2001, 98, 2603–2614.
- Neri, D.; Supuran, C.T. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discov. 2011, 10, 767–777.
- Bertout, J.A.; Patel, S.A.; Simon, M.C. The impact of O2 availability on human cancer. Nat. Rev. Cancer 2008, 8, 967–975.
- Bolhaqueiro, A.C.F.; Ponsioen, B.; Bakker, B.; Klaasen, S.J.; Kucukkose, E.; van Jaarsveld, R.H.; Vivie, J.; Verlaan-Klink, I.; Hami, N.; Spierings, D.C.J.; et al. Ongoing chromosomal instability and karyotype evolution in human colorectal cancer organoids. Nat. Genet. 2019, 51, 824–834.
- Kost, G.C.; Patierno, S.R.; Wise, S.S.; Holmes, A.L.; Wise, J.P., Sr.; Ceryak, S. Protein tyrosine phosphatase (PTP) inhibition enhances chromosomal stability after genotoxic stress: Decreased chromosomal instability (CIN) at the expense of enhanced genomic instability (GIN)? Mutat. Res. 2012, 735, 51–55.
- Velegzhaninov, I.O.; Ievlev, V.A.; Pylina, Y.I.; Shadrin, D.M.; Vakhrusheva, O.M. Programming of cell resistance to genotoxic and oxidative stress. Biomedicines 2018, 6.
- Kampinga, H.H.; Hageman, J.; Vos, M.J.; Kubota, H.; Tanguay, R.M.; Bruford, E.A.; Cheetham, M.E.; Chen, B.; Hightower, L.E. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 2009, 14, 105–111.
- Csermely, P.; Schnaider, T.; Soti, C.; Prohaszka, Z.; Nardai, G. The 90-kDa molecular chaperone family: Structure, function, and clinical applications. A comprehensive review. Pharmacol. Ther. 1998, 79, 129–168.
- Makhnevych, T.; Houry, W.A. The role of Hsp90 in protein complex assembly. Biochim. Biophys. Acta 2012, 1823, 674–682.
- Lindquist, S. The heat-shock response. Annu. Rev. Biochem. 1986, 55, 1151–1191.
- Vihervaara, A.; Sistonen, L. HSF1 at a glance. J. Cell Sci. 2014, 127, 261–266.
- Morimoto, R.I. Regulation of the heat shock transcriptional response: Cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 1998, 12, 3788–3796.
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell Mol. Life Sci. 2016, 73, 79–94.
- Diaz-Villanueva, J.F.; Diaz-Molina, R.; Garcia-Gonzalez, V. Protein folding and mechanisms of proteostasis. Int. J. Mol. Sci. 2015, 16, 17193–17230.
- Hetz, C.; Chevet, E.; Oakes, S.A. Proteostasis control by the unfolded protein response. Nat. Cell Biol. 2015, 17, 829–838.
- Gardner, B.M.; Pincus, D.; Gotthardt, K.; Gallagher, C.M.; Walter, P. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb. Perspect. Biol. 2013, 5, a013169.
- Cubillos-Ruiz, J.R.; Bettigole, S.E.; Glimcher, L.H. Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell 2017, 168, 692–706.
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J., Jr.; Lee, K.P.; Boise, L.H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107, 4907–4916.
- Ni, M.; Lee, A.S. ER chaperones in mammalian development and human diseases. Febs Lett. 2007, 581, 3641–3651.
- Galluzzi, L.; Yamazaki, T.; Kroemer, G. Linking cellular stress responses to systemic homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 731–745.
- Fulda, S.; Gorman, A.M.; Hori, O.; Samali, A. Cellular stress responses: Cell survival and cell death. Int. J. Cell Biol. 2010, 2010, 214074.
- Tsai, Y.C.; Weissman, A.M. The Unfolded Protein Response, degradation from endoplasmic reticulum and cancer. Genes Cancer 2010, 1, 764–778.
- Carreras-Sureda, A.; Pihan, P.; Hetz, C. The Unfolded Protein Response: At the intersection between endoplasmic reticulum function and mitochondrial bioenergetics. Front. Oncol 2017, 7, 55.
- Rieusset, J. The role of endoplasmic reticulum-mitochondria contact sites in the control of glucose homeostasis: An update. Cell Death Dis. 2018, 9, 388.
- Altieri, D.C.; Stein, G.S.; Lian, J.B.; Languino, L.R. TRAP-1, the mitochondrial Hsp90. Biochim. Biophys. Acta 2012, 1823, 767–773.
- Takemoto, K.; Miyata, S.; Takamura, H.; Katayama, T.; Tohyama, M. Mitochondrial TRAP1 regulates the unfolded protein response in the endoplasmic reticulum. Neurochem. Int. 2011, 58, 880–887.
- Li, J.; Lee, B.; Lee, A.S. Endoplasmic reticulum stress-induced apoptosis: Multiple pathways and activation of p53-up-regulated modulator of apoptosis (PUMA) and NOXA by p53. J. Biol. Chem. 2006, 281, 7260–7270.
- Hori, O.; Ichinoda, F.; Tamatani, T.; Yamaguchi, A.; Sato, N.; Ozawa, K.; Kitao, Y.; Miyazaki, M.; Harding, H.P.; Ron, D.; et al. Transmission of cell stress from endoplasmic reticulum to mitochondria: Enhanced expression of Lon protease. J. Cell Biol. 2002, 157, 1151–1160.
- Pinti, M.; Gibellini, L.; Nasi, M.; De Biasi, S.; Bortolotti, C.A.; Iannone, A.; Cossarizza, A. Emerging role of Lon protease as a master regulator of mitochondrial functions. Biochim. Biophys. Acta 2016, 1857, 1300–1306.
- Schneider, K.; Bertolotti, A. Surviving protein quality control catastrophes - from cells to organisms. J. Cell Sci. 2015, 128, 3861–3869.
- Lin, Y.F.; Haynes, C.M. Metabolism and the UPR(mt). Mol. Cell 2016, 61, 677–682.
- Melber, A.; Haynes, C.M. UPR(mt) regulation and output: A stress response mediated by mitochondrial-nuclear communication. Cell Res. 2018, 28, 281–295.
- Fiorese, C.J.; Haynes, C.M. Integrating the UPR(mt) into the mitochondrial maintenance network. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 304–313.
- Fiorese, C.J.; Schulz, A.M.; Lin, Y.F.; Rosin, N.; Pellegrino, M.W.; Haynes, C.M. The transcription factor ATF5 mediates a mammalian mitochondrial UPR. Curr. Biol. 2016, 26, 2037–2043.
- Xia, M.; Zhang, Y.; Jin, K.; Lu, Z.; Zeng, Z.; Xiong, W. Communication between mitochondria and other organelles: A brand-new perspective on mitochondria in cancer. Cell Biosci. 2019, 9, 27.
- Hsu, C.C.; Tseng, L.M.; Lee, H.C. Role of mitochondrial dysfunction in cancer progression. Exp. Biol. Med. (Maywood) 2016, 241, 1281–1295.
- Ciocca, D.R.; Arrigo, A.P.; Calderwood, S.K. Heat shock proteins and heat shock factor 1 in carcinogenesis and tumor development: An update. Arch. Toxicol. 2013, 87, 19–48.
- Whitesell, L.; Lindquist, S. Inhibiting the transcription factor HSF1 as an anticancer strategy. Expert Opin. Targets 2009, 13, 469–478.
- Dai, C.; Whitesell, L.; Rogers, A.B.; Lindquist, S. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell 2007, 130, 1005–1018.
- Saretzki, G.; Armstrong, L.; Leake, A.; Lako, M.; von Zglinicki, T. Stress defense in murine embryonic stem cells is superior to that of various differentiated murine cells. Stem Cells 2004, 22, 962–971.
- Oesterreich, S.; Weng, C.N.; Qiu, M.; Hilsenbeck, S.G.; Osborne, C.K.; Fuqua, S.A. The small heat shock protein hsp27 is correlated with growth and drug resistance in human breast cancer cell lines. Cancer Res. 1993, 53, 4443–4448.
- Nahleh, Z.; Tfayli, A.; Najm, A.; El Sayed, A.; Nahle, Z. Heat shock proteins in cancer: Targeting the ‘chaperones’. Future Med. Chem. 2012, 4, 927–935.
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772.
- Trepel, J.; Mollapour, M.; Giaccone, G.; Neckers, L. Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 2010, 10, 537–549.
- Barrott, J.J.; Haystead, T.A. Hsp90, an unlikely ally in the war on cancer. Febs J. 2013, 280, 1381–1396.
- Abreu, P.L.; Ferreira, L.M.R.; Cunha-Oliveira, T.; Alpoim, M.C.; Urbano, A.M. HSP90: A key player in metal-induced carcinogenesis? In Heat Shock Protein 90 in Human Diseases and Disorders; Asea, A.A., Kaur, P., Eds.; Springer International Publishing: New York, NY, USA, 2019.
- Ziemiecki, A.; Catelli, M.G.; Joab, I.; Moncharmont, B. Association of the heat shock protein hsp90 with steroid hormone receptors and tyrosine kinase oncogene products. Biochem. Biophys. Res. Commun. 1986, 138, 1298–1307.
- Holt, S.E.; Aisner, D.L.; Baur, J.; Tesmer, V.M.; Dy, M.; Ouellette, M.; Trager, J.B.; Morin, G.B.; Toft, D.O.; Shay, J.W.; et al. Functional requirement of p23 and Hsp90 in telomerase complexes. Genes Dev. 1999, 13, 817–826.
- Basso, A.D.; Solit, D.B.; Chiosis, G.; Giri, B.; Tsichlis, P.; Rosen, N. Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. J. Biol. Chem. 2002, 277, 39858–39866.
- Isaacs, J.S.; Jung, Y.J.; Mimnaugh, E.G.; Martinez, A.; Cuttitta, F.; Neckers, L.M. Hsp90 regulates a von Hippel Lindau-independent hypoxia-inducible factor-1 alpha-degradative pathway. J. Biol. Chem. 2002, 277, 29936–29944.
- Ferreira, L.M.; Hebrant, A.; Dumont, J.E. Metabolic reprogramming of the tumor. Oncogene 2012, 31, 3999–4011.
- Eustace, B.K.; Sakurai, T.; Stewart, J.K.; Yimlamai, D.; Unger, C.; Zehetmeier, C.; Lain, B.; Torella, C.; Henning, S.W.; Beste, G.; et al. Functional proteomic screens reveal an essential extracellular role for hsp90 alpha in cancer cell invasiveness. Nat. Cell Biol. 2004, 6, 507–514.
- Pockley, A.G.; Multhoff, G. Cell stress proteins in extracellular fluids: Friend or foe? Novartis Found. Symp. 2008, 291, 86–95.
- Sidera, K.; Patsavoudi, E. HSP90 inhibitors: Current development and potential in cancer therapy. Recent Pat. Anticancer Drug Discov. 2014, 9, 1–20.
- O’Brien, T.J.; Ceryak, S.; Patierno, S.R. Complexities of chromium carcinogenesis: Role of cellular response, repair and recovery mechanisms. Mutat. Res. 2003, 533, 3–36.
- Ishikawa, Y.; Nakagawa, K.; Satoh, Y.; Kitagawa, T.; Sugano, H.; Hirano, T.; Tsuchiya, E. Hot spots of chromium accumulation at bifurcations of chromate workers bronchi. Cancer Res. 1994, 54, 2342–2346.
- Kondo, K.; Takahashi, Y.; Ishikawa, S.; Uchihara, H.; Hirose, Y.; Yoshizawa, K.; Tsuyuguchi, M.; Takizawa, H.; Miyoshi, T.; Sakiyama, S.; et al. Microscopic analysis of chromium accumulation in the bronchi and lung of chromate workers. Cancer 2003, 98, 2420–2429.
- Ishikawa, Y.; Nakagawa, K.; Satoh, Y.; Kitagawa, T.; Sugano, H.; Hirano, T.; Tsuchiya, E. Characteristics of chromate workers cancers, chromium lung deposition and precancerous bronchial lesions—An autopsy study. Br. J. Cancer 1994, 70, 160–166.
- Arakawa, H.; Weng, M.W.; Chen, W.C.; Tang, M.S. Chromium (VI) induces both bulky DNA adducts and oxidative DNA damage at adenines and guanines in the p53 gene of human lung cells. Carcinogenesis 2012, 33, 1993–2000.
- Wise, S.S.; Holmes, A.L.; Qin, Q.; Xie, H.; Katsifis, S.P.; Thompson, W.D.; Wise, J.P. Comparative genotoxicity and cytotoxicity of four hexavalent chromium compounds in human bronchial cells. Chem. Res. Toxicol. 2010, 23, 365–372.
- Thompson, C.M.; Fedorov, Y.; Brown, D.D.; Suh, M.; Proctor, D.M.; Kuriakose, L.; Haws, L.C.; Harris, M.A. Assessment of Cr(VI)-induced cytotoxicity and genotoxicity using high content analysis. PLoS ONE 2012, 7.
- Reynolds, M.; Armknecht, S.; Johnston, T.; Zhitkovich, A. Undetectable role of oxidative DNA damage in cell cycle, cytotoxic and clastogenic effects of Cr(VI) in human lung cells with restored ascorbate levels. Mutagenesis 2012, 27, 437–443.
- Izzotti, A.; Bagnasco, M.; Camoirano, A.; Orlando, M.; De Flora, S. DNA fragmentation, DNA-protein crosslinks, P-32 postlabeled nucleotidic modifications, and 8-hydroxy-2’-deoxyguanosine in the lung but not in the liver of rats receiving intratracheal instillations of chromium(VI). Chemoprevention by oral N-acetylcysteine. Mutat. Res. 1998, 400, 233–244.
- Figgitt, M.; Newson, R.; Leslie, I.J.; Fisher, J.; Ingham, E.; Case, C.P. The genotoxicity of physiological concentrations of chromium (Cr(III) and Cr(VI)) and cobalt (Co(II)): An in vitro study. Mutat. Res. 2010, 688, 53–61.
- Reynolds, M.; Zhitkovich, A. Cellular vitamin C increases chromate toxicity via a death program requiring mismatch repair but not p53. Carcinogenesis 2007, 28, 1613–1620.
- Wetterhahn, K.E.; Hamilton, J.W.; Aiyar, J.; Borges, K.M.; Floyd, R. Mechanisms of Chromium(VI) carcinogenesis—Reactive intermediates and effect on gene-expression. Biol. Trace Elem. Res. 1989, 21, 405–411.
- Standeven, A.M.; Wetterhahn, K.E. Ascorbate is the principal reductant of chromium(VI) in rat lung ultrafiltrates and cytosols, and mediates chromium—DNA-binding invitro. Carcinogenesis 1992, 13, 1319–1324.
- Nickens, K.P.; Patierno, S.R.; Ceryak, S. Chromium genotoxicity: A double-edged sword. Chem. Biol. Interact. 2010, 188, 276–288.
- O’Brien, T.J.; Brooks, B.R.; Patierno, S.R. Nucleotide excision repair functions in the removal of chromium-induced DNA damage in mammalian cells. Mol. Cell Biochem. 2005, 279, 85–95.
- Xie, H.; Wise, S.S.; Holmes, A.L.; Xu, B.; Wakeman, T.P.; Pelsue, S.C.; Singh, N.P.; Wise, J.P., Sr. Carcinogenic lead chromate induces DNA double-strand breaks in human lung cells. Mutat. Res. 2005, 586, 160–172.
- Hirose, T.; Kondo, K.; Takahashi, Y.; Ishikura, H.; Fujino, H.; Tsuyuguchi, M.; Hashimoto, M.; Yokose, T.; Mukai, K.; Kodama, T.; et al. Frequent microsatellite instability in lung cancer from chromate-exposed workers. Mol. Carcinog. 2002, 33, 172–180.
- Takahashi, Y.; Kondo, K.; Hirose, T.; Nakagawa, H.; Tsuyuguchi, M.; Hashimoto, M.; Sano, T.; Ochiai, A.; Monden, Y. Microsatellite instability and protein expression of the DNA mismatch repair gene, hMLH1, of lung cancer in chromate-exposed workers. Mol. Carcinog. 2005, 42, 150–158.
- Rodrigues, C.F.; Urbano, A.M.; Matoso, E.; Carreira, I.; Almeida, A.; Santos, P.; Botelho, F.; Carvalho, L.; Alves, M.; Monteiro, C.; et al. Human bronchial epithelial cells malignantly transformed by hexavalent chromium exhibit an aneuploid phenotype but no microsatellite instability. Mutat. Res. 2009, 670, 42–52.
- Abreu, P.L.; Ferreira, L.M.R.; Alpoim, M.C.; Urbano, A.M. Impact of hexavalent chromium on mammalian cell bioenergetics: Phenotypic changes, molecular basis and potential relevance to chromate-induced lung cancer. Biometals 2014, 27, 409–443.
- Tully, D.B.; Collins, B.J.; Overstreet, J.D.; Smith, C.S.; Dinse, G.E.; Mumtaz, M.M.; Chapin, R.E. Effects of arsenic, cadmium, chromium, and lead on gene expression regulated by a battery of 13 different promoters in recombinant HepG2 cells. Toxicol. Appl. Pharm. 2000, 168, 79–90.
- Dubrovskaya, V.A.; Wetterhahn, K.E. Effects of Cr(VI) on the expression of the oxidative stress genes in human lung cells. Carcinogenesis 1998, 19, 1401–1407.
- Ye, J.P.; Zhang, X.Y.; Young, H.A.; Mao, Y.; Shi, X.L. Chromium(VI)-induced nuclear factor-kappa-B activation in intact-cells via free-radical reactions. Carcinogenesis 1995, 16, 2401–2405.
- Dalton, T.P.; Shertzer, H.G.; Puga, A. Regulation of gene expression by reactive oxygen. Annu. Rev. Pharm. Toxicol. 1999, 39, 67–101.
- Holmes, A.L.; Wise, S.S.; Wise, J.P., Sr. Carcinogenicity of hexavalent chromium. Indian J. Med. Res. 2008, 128, 353–372.
- Izzotti, A.; Cartiglia, C.; Balansky, R.; D’Agostini, F.; Longobardi, M.; De Flora, S. Selective induction of gene expression in rat lung by hexavalent chromium. Mol. Carcinog. 2002, 35, 75–84.
- Ye, J.P.; Shi, X.L. Gene expression profile in response to chromium-induced cell stress in A549 cells. Mol. Cell Biochem. 2001, 222, 189–197.
- Delmas, F.; Schaak, S.; Gaubin, Y.; Croute, F.; Arrabit, C.; Murat, J.C. Hsp72 mRNA production in cultured human cells submitted to nonlethal aggression by heat, ethanol, or propanol. Application to the detection of low concentrations of chromium(VI) (potassium dichromate). Cell Biol. Toxicol. 1998, 14, 39–46.
- Delmas, F.; Trocheris, V.; Murat, J.C. Expression of stress proteins in cultured HT29 human cell-line: A model for studying environmental aggression. Int. J. Biochem. Cell Biol. 1995, 27, 385–391.
- Ge, H.; Li, Z.; Jiang, L.; Li, Q.; Geng, C.; Yao, X.; Shi, X.; Liu, Y.; Cao, J. Cr (VI) induces crosstalk between apoptosis and autophagy through endoplasmic reticulum stress in A549 cells. Chem. Biol. Interact. 2019, 298, 35–42.
- Liang, Q.; Zhang, Y.; Huang, M.; Xiao, Y.; Xiao, F. Role of mitochondrial damage in Cr(VI)induced endoplasmic reticulum stress in L02 hepatocytes. Mol. Med. Rep. 2019, 19, 1256–1265.
- Xiao, F.; Li, Y.; Dai, L.; Deng, Y.; Zou, Y.; Li, P.; Yang, Y.; Zhong, C. Hexavalent chromium targets mitochondrial respiratory chain complex I to induce reactive oxygen species-dependent caspase-3 activation in L-02 hepatocytes. Int. J. Mol. Med. 2012, 30, 629–635.
- Andrew, A.S.; Warren, A.J.; Barchowsky, A.; Temple, K.A.; Klei, L.; Soucy, N.V.; O’Hara, K.A.; Hamilton, J.W. Genomic and proteomic profiling of responses to toxic metals in human lung cells. Environ. Health Perspect. 2003, 111, 825–835.
- Abreu, P.L.; Cunha-Oliveira, T.; Ferreira, L.M.R.; Urbano, A.M. Hexavalent chromium, a lung carcinogen, confers resistance to thermal stress and interferes with heat shock protein expression in human bronchial epithelial cells. Biometals 2018, 31, 477–487.
- Glisovic, T.; Bachorik, J.L.; Yong, J.; Dreyfuss, G. RNA-binding proteins and post-transcriptional gene regulation. Febs Lett. 2008, 582, 1977–1986.
- Janga, S.C.; Vallabhaneni, S. MicroRNAs as post-transcriptional machines and their interplay with cellular networks. Adv. Exp. Med. Biol. 2011, 722, 59–74.
- Doherty, M.K.; Hammond, D.E.; Clague, M.J.; Gaskell, S.J.; Beynon, R.J. Turnover of the human proteome: Determination of protein intracellular stability by dynamic SILAC. J. Proteome Res. 2009, 8, 104–112.
- Sadoul, K.; Boyault, C.; Pabion, M.; Khochbin, S. Regulation of protein turnover by acetyltransferases and deacetylases. Biochimie 2008, 90, 306–312.
- Majumder, S.; Ghoshal, K.; Summers, D.; Bai, S.; Datta, J.; Jacob, S.T. Chromium(VI) down-regulates heavy metal-induced metallothionein gene transcription by modifying transactivation potential of the key transcription factor, metal-responsive transcription factor 1. J. Biol. Chem. 2003, 278, 26216–26226.
- Banu, S.K.; Stanley, J.A.; Lee, J.; Stephen, S.D.; Arosh, J.A.; Hoyer, P.B.; Burghardt, R.C. Hexavalent chromium-induced apoptosis of granulosa cells involves selective sub-cellular translocation of Bcl-2 members, ERK1/2 and p53. Toxicol. Appl. Pharm. 2011, 251, 253–266.
- Lei, T.; He, Q.Y.; Cai, Z.; Zhou, Y.; Wang, Y.L.; Si, L.S.; Chiu, J.F. Proteomic analysis of chromium cytotoxicity in cultured rat lung epithelial cells. Proteomics 2008, 8, 2420–2429.
- Zhang, Q.; Zhang, L.; Xiao, X.; Su, Z.; Zou, P.; Hu, H.; Huang, Y.; He, Q.Y. Heavy metals chromium and neodymium reduced phosphorylation level of heat shock protein 27 in human keratinocytes. Toxicol. Vitr. 2010, 24, 1098–1104.
- Rudolf, E.; Cervinka, M. Nickel modifies the cytotoxicity of hexavalent chromium in human dermal fibroblasts. Toxicol. Lett. 2010, 197, 143–150.
- Katsogiannou, M.; Andrieu, C.; Rocchi, P. Heat shock protein 27 phosphorylation state is associated with cancer progression. Front. Genet. 2014, 5, 346.
- Lee, J.; Lim, K.T. Inhibitory effect of SJSZ glycoprotein (38 kDa) on expression of heat shock protein 27 and 70 in chromium (VI)-treated hepatocytes. Mol. Cell Biochem. 2012, 359, 45–57.


