Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Patrick Wagner | -- | 2183 | 2023-10-19 15:33:06 | | | |
| 2 | Camila Xu | Meta information modification | 2183 | 2023-10-20 03:09:19 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Dadgar, N.; Edlukudige Keshava, V.; Raj, M.S.; Wagner, P.L. Microbiome on Immunotherapy for Gastroesophageal Cancer. Encyclopedia. Available online: https://encyclopedia.pub/entry/50564 (accessed on 24 July 2026).
Dadgar N, Edlukudige Keshava V, Raj MS, Wagner PL. Microbiome on Immunotherapy for Gastroesophageal Cancer. Encyclopedia. Available at: https://encyclopedia.pub/entry/50564. Accessed July 24, 2026.
Dadgar, Neda, Vinay Edlukudige Keshava, Moses S. Raj, Patrick L. Wagner. "Microbiome on Immunotherapy for Gastroesophageal Cancer" Encyclopedia, https://encyclopedia.pub/entry/50564 (accessed July 24, 2026).
Dadgar, N., Edlukudige Keshava, V., Raj, M.S., & Wagner, P.L. (2023, October 19). Microbiome on Immunotherapy for Gastroesophageal Cancer. In Encyclopedia. https://encyclopedia.pub/entry/50564
Dadgar, Neda, et al. "Microbiome on Immunotherapy for Gastroesophageal Cancer." Encyclopedia. Web. 19 October, 2023.
Copy Citation
Esophageal cancer (EC) is a highly aggressive malignancy, comprising two main subtypes, esophageal squamous-cell carcinoma/ESCC and esophageal adenocarcinoma/EAC. Gastric cancer (GC) has been identified as a disease caused by a combination of genetic, molecular, and environmental factors, with H. pylori infection being the most common factor among them.
gastroesophageal cancers
PD1 blockade
immunotherapy
resistance
1. Introduction
Although progress has been made in reducing the incidence of esophageal cancer (EC) and gastric cancer (GC), they still contribute significantly to global cancer mortality [1][2]. Reducing exposure to known risk factors, regular screening of high-risk individuals, and early detection can help decrease the incidence of GC and EC, but most patients continue to present with advanced disease, for which conventional treatments offer little chance of a cure [3][4]. Thus, there has been strong interest in adapting novel treatment strategies to EC and GC, with significant progress in the integration of immunotherapy into modern treatment algorithms [5]. As a result, current NCCN guidelines now endorse immunotherapy as an option for several key clinical scenarios in EC and GC, including first-line treatment for metastatic esophageal or gastroesophageal junction (GEJ) adenocarcinoma (in combination with conventional cytotoxic therapy), second-line treatment of unresectable or metastatic esophageal squamous-cell carcinoma, and post-operative treatment for selected high-risk patients with esophageal or GEJ adenocarcinoma following neoadjuvant chemo-radiation and surgery [6].
Since only a minority of patients are eligible for or responsive to immunotherapy, there is an intense need to develop new strategies for expanding the pool of patients in whom immunotherapy is expected to provide a survival benefit. One emerging strategy involves harnessing the tumor and host microbiome to promote anti-tumor immunity and immunotherapy, based on several key lines of evidence linking the microbiome with cancer biology and immune response. First, dysbiosis and reduced microbial diversity have been associated with poor response rates and treatment resistance in gastroesophageal cancer patients, while specific microbial profiles have shown correlation with improved outcomes [7][8]. Second, microbiome-based biomarkers have shown promise in predicting patient response to immunotherapy [9]. Third, interventions targeting the microbiome, such as microbiota transfer techniques, prebiotics, and dietary modifications, are gaining increasing traction due to their proven ability to enhance immunotherapy efficacy [10]. Understanding the complex interactions between the microbiome, tumor biology, and immunotherapy is thus essential in order to improve upon the benefits that immunotherapy has brought to patients with EC/GC.
2. Tumor and Host Microbiome on the Pathogenesis of EC and GC
2.1. Microbiome in Esophageal Cancer
Esophageal cancer (EC) is a highly aggressive malignancy, comprising two main subtypes, esophageal squamous-cell carcinoma/ESCC and esophageal adenocarcinoma/EAC [11]. EAC is linked to risk factors such as a Western diet, obesity, gastroesophageal reflux disease (GERD), and Barrett’s esophagus (BE), which can lead to EAC through progressive dysplasia [12][13]. By contrast, ESCC is more strongly associated with smoking and alcohol use [14][15]. A growing body of research has documented the correlation of ESCC and EAC incidence with unique microbial signatures from the oral mucosa, esophageal lumen, tumor tissue, or gut (Figure 1). These data provide compelling epidemiologic evidence of an association between the microbiome and EC, and may open new pathways for research into microbiome-based prevention or treatment strategies.
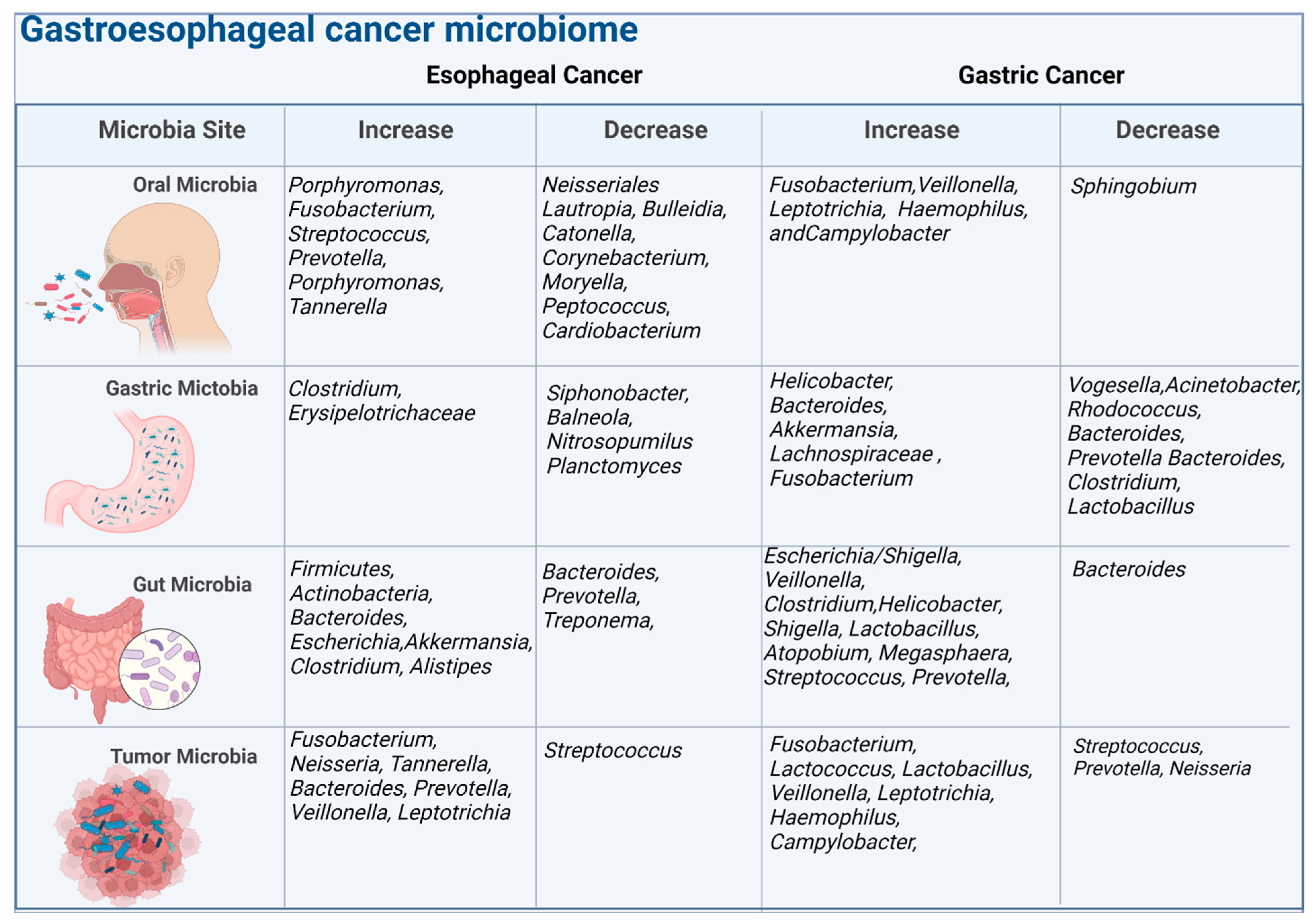
Figure 1. The figure illustrates the microbiome composition at different body sites, including the oral cavity, gastric region, gut, and tumor. It highlights the associations between the microbiome and esophageal cancer and gastric cancer, indicating increases or decreases in specific bacteria. The analysis is conducted at the genus level, representing groups of related bacteria.
The oral cavity’s normal bacterial flora are primarily composed of six major phyla, namely, Firmicutes, Bacteroidetes, Proteobacteria, Actinobacteria, Spirochaetes, and Fusobacteria [16], with the most frequent species being Streptococcus spp. (S. mitis, S. sanguis, S. gordonii), Gemella spp. (G. sanguinis, G. haemolysans), Granulicatella spp., and Neisseria spp. [17]. Recent studies suggest an association between ESCC and increased oral abundance of Firmicutes, Prevotella, Streptococcus, and Porphyromonas, as well as a decrease in Proteobacteria, Neisseriales, Lautropia, Bulleidia, Catonella, Corynebacterium, Moryella, Peptococcus, and Cardiobacterium [18][19][20][21][22]. Similar but distinct changes have been observed in EAC, where Tannerella forsythia was associated with higher risk, while depletion of Neisseria and Streptococcus pneumoniae was associated with lower risk [22][23]. These differences in the oral microbiome could serve as a biomarker for predicting esophageal cancer risk or directly influence cancer risk by mechanisms that remain to be discovered [24].
The gastric microbiome may also impact or predict esophageal cancer, and special mention is warranted for Helciobacter pylori in any discussion of microbial influences on upper gastrointestinal cancer. A meta-analysis aimed at clarifying the link between H. pylori infection and esophageal carcinoma found no significant association with ESCC in the overall population. However, significant associations were found in Eastern subjects, suggesting a decreased risk of ESCC in individuals infected with cagA-positive H. pylori strains [25]. Another study aimed to compare the pattern of gastric corpus microbiota in early ESCC and esophageal squamous dysplasia with normal esophagus. Clostridiales and Erysipelotrichales orders were more abundant among cases. No such difference was observed between esophagitis and healthy controls, suggesting that the composition of gastric corpus mucosal microbiota may differ along the spectrum of dysplasia and cancer in ESCC [26]. In another study, the gastric juice samples of EC patients showed a significant reduction in the abundance of genera Siphonobacter, Balneola, Nitrosopumilus, and Planctomyces [27].
The gut microbiome, consisting predominantly of the phyla Firmicutes, Bacteriodetes, Proteobacteria, and Actinobacteria [28], may also influence EC pathogenesis. Microbiota can stimulate carcinogenesis through immune pathways, as Münch et al. showed in a mouse model of Barrett’s esophagus. High-fat diet-induced changes in gut microbiota led to proinflammatory cytokines, immune cells, and a pro-tumor immune phenotype, highlighting the critical role of gut microbiota in transmitting dietary influences via inflammatory mechanisms [29]. In ESCC patients, Yang et al. found that the levels of Bacteroidetes, Fusobacteria, and Spirochaetes in the gut microbiome are decreased relative to controls [30]. Another study showed that the abundance of short-chain fatty acid-producing bacteria is reduced, while the amount of lipopolysaccharide-producing bacteria increases in EC patients [31]. In a similar study, the fecal quantities of Bacteroides fragilis, Escherichia coli, Akkermansia muciniphila, Clostridium hathaway, and Alistipes finegoldii distinguished cancer patients versus control subjects [32]. In a study of 783 patients who underwent oncologic esophagectomy, researchers investigated the gut microbiota detected by fecal culture tests and its association with patient outcomes, finding that Bacillus species had a better response to preoperative treatment, lower modified Glasgow prognostic score, and improved survival, whereas patients with P. mirabilis had higher systemic inflammation scores, increased postoperative pneumonia incidence, and unfavorable survival [33]. The mechanisms accounting for these intriguing differences remain unknown.
The intratumoral microbiome present within malignant ESCC, EAC, and BE tissue has uniformly been found to show less diversity relative to normal esophageal tissue [27][34]. EAC was associated with increased abundance of intratumoral Streptococcus and Neisseria, and the microbiome composition of EAC differed from that of Barrett’s esophagus and GERD, suggesting that local dysbiosis may be associated with neoplastic progression along the Barrett’s sequence [35]. Interestingly, some studies have found that certain bacterial species associated with periodontal disease, such as Fusobacterium nucleatum and Tannerella forsythia, have been detected in esophageal cancer tissues, suggesting that oral microbiota may contribute to esophageal cancer pathogenesis [36][37][38]. The analysis of microbial composition has been examined as a means of stratifying cancer risk, with studies indicating certain bacteria as strong predictors for the development of ESCC and EAC. For ESCC, bacteria such as P gingivalis, Streptococcus, Neisseria, Actinomyces, and Atopobium are associated with a higher risk, whereas Prevotella oral taxon 306 and Aggregatibacter paraphrophilus suggest a lower risk [39]. EAC exhibited a remarkable decrease in Streptococcus, accompanied by an increase in Prevotella, Veillonella, and Leptotrichia [40]. In the case of EAC, the periodontal pathogen Tannerella forsythia and oral species such as Actinomyces cardiffensis, Selenomonas oral taxon 134, and Veillonella oral taxon 917 are potential factors for a higher risk, while certain bacteria such as Lachnoanaerobaculum umeaense, Oribacterium parvum, Solobacterium moorei (Firmicutes), Neisseria sicca, Neisseria flavescens, and Haemophilus oral taxon 908 (Proteobacteria), Corynebacterium durum, Prevotella nanceiensis, and S. pneumoniae are associated with a lower risk. However, it remains unclear whether these microbial changes play a causative role in cancer development or are a consequence of the presence of cancer [23][41].
A few studies focusing on surgical resection specimens have examined the esophageal microbiome and metabolic changes before and after esophagectomy. One such study aimed to characterize the esophageal microbiome of patients with esophageal squamous-cell carcinoma (ESCC) and GEJ cancer, as well as post-esophagectomy patients, versus healthy controls (HC). Microbial diversity was significantly lower in the ESCC, GEJ, and post-ESCC groups than in the HC group. The abundance of Fusobacteria was higher, and the abundance of Actinobacteria was lower in the ESCC group than in the HC group. Significant differences were found in the abundance of Bacteroidetes and Fusobacteria between the ESCC and post-ESCC group. Microbial sequencing results from 19 pairs of tissues revealed that Proteobacteria, Firmicutes, Bacteroidetes, Deinococcus-Thermus, and Actinobacteria were the predominant bacteria in both tumor and adjacent non-tumor tissues. Additionally, the group found that Streptococcus had the highest relative proportion in tumor tissues, while Labrys was more abundant in adjacent non-tumor tissues at the genus level. It was also observed that the microbial interactions in tumor tissues were less complex than those in adjacent non-tumor tissues, providing valuable insights into the potential relationship between intratumoral microorganisms and esophageal carcinogenesis [42][43].
2.2. Microbiome in Gastric Cancer
Gastric cancer (GC) has been identified as a disease caused by a combination of genetic, molecular, and environmental factors, with H. pylori infection being the most common factor among them [44][45][46]. Recent discoveries challenge previous assumptions about the uniformity of microhabitats within the stomach, revealing significant variations in pH, mucin distribution, nutrients, ions, and chemical levels between tumor and adjacent tumor-free tissue [47]. In GC patients, five oral bacteria were found to accurately distinguish GC from non-atrophic gastritis with good performance. The increase in oral abundance of H. pylori, Shigella spp., Lactobacillus spp., Atopobium spp., Megasphaera spp., Streptococcus spp., Veillonella spp., Prevotella spp., and Clostridium spp. flora in GC, and significant enrichment of Escherichia coli, Prevotella spp., Clostridium spp., and Bacteroides fragilis have also been associated with gastric tumorigenesis. The abundance of Sphingobium yanoikuyae was significantly reduced in GC, and higher relative abundances of several bacterial genera commonly found in the oral cavity, including Fusobacterium, Veillonella, Leptotrichia, Haemophilus Campylobacter, have been observed in gastric cancer patients [48].
These variations impact the diversity and composition of the gastric microbiome, which is primarily made up of five phyla, namely, Proteobacteria, Firmicutes, Bacteroidetes, Actinobacteria, and Fusobacteria. Notably, H. pylori has been identified as the most prevalent species among GC tumor samples. However, following surgery, there is a considerable shift in the gastric microbiome’s community composition at the phylum level, with a reduction in Proteobacteria and Actinobacteria and an increase in Firmicutes and Bacteroidetes [49]. A study conducted by Park et al. analyzed gastric secretions from patients with gastritis, gastric adenoma, or gastric cancer, finding that microbial diversity decreased continuously along the spectrum of gastric carcinogenesis, and the microbial composition differed significantly by disease status. The composition of the gastric microbiome in patients with gastric cancer was characterized by reduced levels of Verrucomicrobia and Deferribacteres, whereas Akkermansia and Lachnospiraceae NK4A136 group were significantly more abundant in patients with gastritis. The study also predicted functional pathways related to carcinogenesis using Tax4Fun, suggesting that gastric cancer is associated with microbial dysbiosis and functional changes that could promote carcinogenesis [50].
A study conducted by Liang et al. compared the fecal microbiota of GC patients before and after radical distal gastrectomy with that of healthy individuals. The study found that during the perioperative period, the relative abundances of certain genera, such as Akkermansia, Esherichia/Shigella, Lactobacillus, and Dialister, showed significant changes. Moreover, GC patients exhibited higher abundances of Escherichia/Shigella, Veillonella, and Clostridium XVIII and lower abundances of Bacteroides when compared to healthy controls. Another study showed increased abundance of H. pylori, Shigella spp., Lactobacillus spp., Atopobium spp., Megasphaera spp., Streptococcus spp., Veillonella spp., Prevotella spp., and Clostridium spp. flora in gut microbia in the patients with gastric cancer, and significant enrichment of Escherichia coli, Prevotella spp., Clostridium spp., and Bacteroides fragilis [51]. Together, these studies provide valuable insights into the complex interplay between the microbiome and the development of GC, suggesting that microbial dysbiosis and functional changes may play a role in promoting gastric carcinogenesis [52].
The intratumoral microbiome of gastric cancer tissue has also been shown to be enriched with oral bacteria, such as Streptococcus and Fusobacterium, whereas adjacent non-cancerous tissue has higher levels of bacteria that produce lactic acid, such as Lactococcus lactis and Lactobacillus brevis [53], along with oral species such as Fusobacterium nucleatum, Veillonella, Leptotrichia, Haemophilus, and Campylobacter [54][55][56]. A microbial dysbiosis index was calculated based on 6 enriched and 18 depleted bacterial genera in GC, and successfully discriminated between GC and NAG patient samples, an AUC of 87% with sensitivity rate of 97%, and provided a false-positive rate of 7.7%. Additionally, bacterial biomarkers such as P. gingivalis and Streptococcus anginosus have been linked to increased risk of GC and worse clinical outcomes, while Lactobacillus has been associated with better prognosis and longer survival [57]. This emerging body of work highlights the potential contribution of microbiome-based biomarker assays that could aid in the diagnosis and treatment planning of upper digestive tract malignancies [58][59].
References
- Hyland, P.L.; Hu, N.; Rotunno, M.; Su, H.; Wang, C.; Wang, L.; Pfeiffer, R.M.; Gherman, B.; Giffen, C.; Dykes, C.; et al. Global changes in gene expression of Barrett’s esophagus compared to normal squamous esophagus and gastric cardia tissues. PLoS ONE 2014, 9, e93219.
- Jardim, S.R.; de Souza, L.M.P.; de Souza, H.S.P. The Rise of Gastrointestinal Cancers as a Global Phenomenon: Unhealthy Behavior or Progress? Int. J. Environ. Res. Public Health 2023, 20, 3640.
- Sheikh, M.; Roshandel, G.; McCormack, V.; Malekzadeh, R. Current Status and Future Prospects for Esophageal Cancer. Cancers 2023, 15, 765.
- Rawla, P.; Barsouk, A. Epidemiology of gastric cancer: Global trends, risk factors and prevention. Gastroenterol. Rev./Przegląd Gastroenterol. 2019, 14, 26–38.
- Wagner, A.D.; Lordick, F.; Grabsch, H.I.; Terashima, M.; Terada, M.; Yoshikawa, T.; Boku, N.; Kataoka, K.; Smyth, E.C.; Mauer, M.; et al. Multidisciplinary management of stage II-III gastric and gastro-oesophageal junction cancer. Eur. J. Cancer 2019, 124, 67–76.
- Ajani, J.A.; D’Amico, T.A.; Bentrem, D.J.; Chao, J.; Cooke, D.; Corvera, C.; Das, P.; Enzinger, P.C.; Enzler, T.; Fanta, P.; et al. Gastric cancer, version 2.2022, NCCN clinical practice guidelines in oncology. J. Natl. Compr. Cancer Netw. 2022, 20, 167–192.
- Lau, H.C.H.; Sung, J.J.-Y.; Yu, J. Gut microbiota: Impacts on gastrointestinal cancer immunotherapy. Gut Microbes 2021, 13, 1869504.
- Naqash, A.R.; Kihn-Alarcón, A.J.; Stavraka, C.; Kerrigan, K.; Vareki, S.M.; Pinato, D.J.; Puri, S. The role of gut microbiome in modulating response to immune checkpoint inhibitor therapy in cancer. Ann. Transl. Med. 2021, 9, 1034.
- Schupack, D.A.; Mars, R.A.T.; Voelker, D.H.; Abeykoon, J.P.; Kashyap, P.C. The promise of the gut microbiome as part of individualized treatment strategies. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 7–25.
- Rahman, M.; Islam, R.; Shohag, S.; Ahasan, T.; Sarkar, N.; Khan, H.; Hasan, A.M.; Cavalu, S.; Rauf, A. Microbiome in cancer: Role in carcinogenesis and impact in therapeutic strategies. BioMedicine 2022, 149, 112898.
- Kim, N. Sex Difference of Esophageal Cancer: Esophageal Squamous Cell Carcinoma vs. Esophageal Adenocarcinoma. In Sex/Gender-Specific Medicine in the Gastrointestinal Diseases; Springer: Berlin/Heidelberg, Germany, 2022; pp. 69–92.
- Arnold, M.; Soerjomataram, I.; Ferlay, J.; Forman, D. Global incidence of oesophageal cancer by histological subtype in 2012. Gut 2015, 64, 381–387.
- Gregson, E.M.; Bornschein, J.; Fitzgerald, R. Genetic progression of Barrett’s oesophagus to oesophageal adenocarcinoma. Br. J. Cancer 2018, 115, 403–410.
- Yang, X.; Chen, X.; Zhuang, M.; Yuan, Z.; Nie, S.; Lu, M.; Jin, L.; Ye, W. Smoking and alcohol drinking in relation to the risk of esophageal squamous cell carcinoma: A population-based case-control study in China. Sci. Rep. 2017, 7, 17249.
- Simba, H.; Menya, D.; Mmbaga, B.T.; Dzamalala, C.; Finch, P.; Mlombe, Y.; Mremi, A.; Narh, C.T.; Schüz, J.; McCormack, V. The contribution of smoking and smokeless tobacco to oesophageal squamous cell carcinoma risk in the African oesophageal cancer corridor: Results from the ESCCAPE multicentre case-control studies. Int. J. Cancer 2023, 152, 2269–2282.
- Dewhirst, F.E.; Chen, T.; Izard, J.; Paster, B.J.; Tanner, A.C.; Yu, W.H.; Lakshmanan, A.; Wade, W.G. The human oral microbiome. J. Bacteriol. 2010, 192, 5002–5017.
- La Rosa, G.R.; Gattuso, G.; PEduLLà, E.; Rapisarda, E.; Nicolosi, D.; Salmeri, M. Association of oral dysbiosis with oral cancer development. Oncol. Lett. 2020, 19, 3045–3058.
- Muszyński, D.; Kudra, A.; Sobocki, B.K.; Folwarski, M.; Vitale, E.; Filetti, V.; Dudzic, W.; Kaźmierczak-Siedlecka, K.; Połom, K. Esophageal cancer and bacterial part of gut microbiota—A multidisciplinary point of view. Front. Cell. Infect. Microbiol. 2022, 12, 1057668.
- Yano, Y.; Etemadi, A.; Abnet, C.C. Microbiome and Cancers of the Esophagus: A Review. Microorganisms 2021, 9, 1764.
- Snider, E.J.; Compres, G.; Freedberg, D.E.; Giddins, M.J.; Khiabanian, H.; Lightdale, C.J.; Nobel, Y.R.; Toussaint, N.C.; Uhlemann, A.-C.; Abrams, J.A. Barrett’s esophagus is associated with a distinct oral microbiome. Clin. Transl. Gastroenterol. 2018, 9, e135.
- Li, H.; Luo, Z.; Zhang, H.; Huang, N.; Li, D.; Luo, C.; Wang, T. Characteristics of Oral Microbiota in Patients with Esophageal Cancer in China. BioMed Res. Int. 2021, 2021, 2259093.
- Chen, X.; Winckler, B.; Lu, M.; Cheng, H.; Yuan, Z.; Yang, Y.; Jin, L.; Ye, W. Oral Microbiota and Risk for Esophageal Squamous Cell Carcinoma in a High-Risk Area of China. PLoS ONE 2015, 10, e0143603.
- Peters, B.A.; Wu, J.; Pei, Z.; Yang, L.; Purdue, M.P.; Freedman, N.D.; Jacobs, E.J.; Gapstur, S.M.; Hayes, R.B.; Ahn, J. Oral Microbiome Composition Reflects Prospective Risk for Esophageal Cancers. Cancer Res. 2017, 77, 6777–6787.
- Zhao, Q.; Yang, T.; Yan, Y.; Zhang, Y.; Li, Z.; Wang, Y.; Yang, J.; Xia, Y.; Xiao, H.; Han, H.; et al. Alterations of Oral Microbiota in Chinese Patients with Esophageal Cancer. Front. Cell. Infect. Microbiol. 2020, 10, 541144.
- Xie, F.J.; Zhang, Y.P.; Zheng, Q.Q.; Jin, H.C.; Wang, F.L.; Chen, M.; Shao, L.; Zou, D.H.; Yu, X.M.; Mao, W.M. Helicobacter pylori infection and esophageal cancer risk: An updated meta-analysis. World J. Gastroenterol. 2013, 19, 6098.
- Nasrollahzadeh, D.; Malekzadeh, R.; Ploner, A.; Shakeri, R.; Sotoudeh, M.; Fahimi, S.; Nasseri-Moghaddam, S.; Kamangar, F.; Abnet, C.C.; Winckler, B.; et al. Variations of gastric corpus microbiota are associated with early esophageal squamous cell carcinoma and squamous dysplasia. Sci. Rep. 2015, 5, srep08820.
- Liu, A.Q.; Vogtmann, E.; Shao, D.T.; Abnet, C.C.; Dou, H.Y.; Qin, Y.; Su, Z.; Wei, W.Q.; Chen, W. A Comparison of Biopsy and Mucosal Swab Specimens for Examining the Microbiota of Upper Gastrointestinal CarcinomaSpecimen Comparison for Upper Gastrointestinal Microbiota. Cancer Epidemiol. Biomark. Prev. 2019, 28, 2030–2037.
- Lv, J.; Guo, L.; Liu, J.J.; Zhao, H.P.; Zhang, J.; Wang, J.H. Alteration of the esophageal microbiota in Barrett’s esophagus and esophageal adenocarcinoma. World J. Gastroenterol. 2019, 25, 2149.
- Münch, N.S.; Fang, H.-Y.; Ingermann, J.; Maurer, H.C.; Anand, A.; Kellner, V.; Sahm, V.; Wiethaler, M.; Baumeister, T.; Wein, F.; et al. High-Fat Diet Accelerates Carcinogenesis in a Mouse Model of Barrett’s Esophagus via Interleukin 8 and Alterations to the Gut Microbiome. Gastroenterology 2019, 157, 492–506.e2.
- Yang, W.; Chen, C.-H.; Jia, M.; Xing, X.; Gao, L.; Tsai, H.-T.; Zhang, Z.; Liu, Z.; Zeng, B.; Yeung, S.-C.J.; et al. Tumor-Associated Microbiota in Esophageal Squamous Cell Carcinoma. Front. Cell Dev. Biol. 2021, 9, 641270.
- Deng, Y.; Tang, D.; Hou, P.; Shen, W.; Li, H.; Wang, T.; Liu, R. Dysbiosis of gut microbiota in patients with esophageal cancer. Microb. Pathog. 2021, 150, 104709.
- Li, N.; Bai, C.; Zhao, L.; Sun, Z.; Ge, Y.; Li, X. The Relationship Between Gut Microbiome Features and Chemotherapy Response in Gastrointestinal Cancer. Front. Oncol. 2021, 11, 781697.
- Maruyama, S.; Okamura, A.; Kanie, Y.; Sakamoto, K.; Fujiwara, D.; Kanamori, J.; Imamura, Y.; Takeda, K.; Watanabe, M. Fecal microbes associated with the outcomes after esophagectomy in patients with esophageal cancer. Ann. Surg. Oncol. 2022, 29, 7448–7457.
- Li, M.; Shao, D.; Zhou, J.; Gu, J.; Qin, J.; Chen, W.; Wei, W. Signatures within esophageal microbiota with progression of esophageal squamous cell carcinoma. Chin. J. Cancer Res. 2020, 32, 755–767.
- Neto, A.G.; Whitaker, A.; Pei, Z. Microbiome and potential targets for chemoprevention of esophageal adenocarcinoma. Semin. Oncol. 2016, 43, 86–96.
- Liu, N.; Ando, T.; Ishiguro, K.; Maeda, O.; Watanabe, O.; Funasaka, K.; Nakamura, M.; Miyahara, R.; Ohmiya, N.; Goto, H. Characterization of bacterial biota in the distal esophagus of Japanese patients with reflux esophagitis and Barrett’s esophagus. BMC Infect. Dis. 2013, 13, 130.
- Yang, L.; Lu, X.; Nossa, C.W.; Francois, F.; Peek, R.M.; Pei, Z. Inflammation and Intestinal Metaplasia of the Distal Esophagus Are Associated with Alterations in the Microbiome. Gastroenterology 2009, 137, 588–597.
- Sharma, T.; Gupta, A.; Chauhan, R.; Bhat, A.A.; Nisar, S.; Hashem, S.; Akhtar, S.; Ahmad, A.; Haris, M.; Singh, M.; et al. Cross-talk between the microbiome and chronic inflammation in esophageal cancer: Potential driver of oncogenesis. Cancer Metastasis Rev. 2022, 41, 281–299.
- Liu, Y.; Lin, Z.; Lin, Y.; Chen, Y.; Peng, X.-E.; He, F.; Liu, S.; Yan, S.; Huang, L.; Lu, W.; et al. Streptococcus and Prevotella are associated with the prognosis of oesophageal squamous cell carcinoma. J. Med. Microbiol. 2018, 67, 1058–1068.
- Lopetuso, L.R.; Severgnini, M.; Pecere, S.; Ponziani, F.R.; Boskoski, I.; Larghi, A.; Quaranta, G.; Masucci, L.; Ianiro, G.; Camboni, T.; et al. Esophageal microbiome signature in patients with Barrett’s esophagus and esophageal adenocarcinoma. PLoS ONE 2020, 15, e0231789.
- Wang, Q.; Rao, Y.; Guo, X.; Liu, N.; Liu, S.; Wen, P.; Li, S.; Li, Y. Oral Microbiome in Patients with Oesophageal Squamous Cell Carcinoma. Sci. Rep. 2019, 9, 19055.
- Li, D.; He, R.; Hou, G.; Ming, W.; Fan, T.; Chen, L.; Zhang, L.; Jiang, W.; Wang, W.; Lu, Z.; et al. Characterization of the Esophageal Microbiota and Prediction of the Metabolic Pathways Involved in Esophageal Cancer. Front. Cell. Infect. Microbiol. 2020, 10, 268.
- Shen, W.; Tang, D.; Wan, P.; Peng, Z.; Sun, M.; Guo, X.; Liu, R. Identification of tissue-specific microbial profile of esophageal squamous cell carcinoma by full-length 16S rDNA sequencing. Appl. Microbiol. Biotechnol. 2022, 106, 3215–3229.
- Liao, O.; Ye, G.; Du, Q.; Ye, J. Gastric microbiota in gastric cancer and precancerous stages: Mechanisms of carcinogenesis and clinical value. Helicobacter 2023, 28, e12964.
- Kupcinskas, J.; Wex, T.; Link, A.; Bartuseviciute, R.; Dedelaite, M.; Kevalaite, G.; Leja, M.; Skieceviciene, J.; Kiudelis, G.; Jonaitis, L. PSCA and MUC1 gene polymorphisms are linked with gastric cancer and pre-malignant gastric conditions. Anticancer Res. 2014, 34, 7167–7175.
- Petkevicius, V.; Salteniene, V.; Juzenas, S.; Wex, T.; Link, A.; Leja, M.; Steponaitiene, R.; Skieceviciene, J.; Kupcinskas, L.; Jonaitis, L. Polymorphisms of microRNA target genes IL12B, INSR, CCND1 and IL10 in gastric cancer. World J. Gastroenterol. 2017, 23, 3480.
- Liu, X.; Shao, L.; Liu, X.; Ji, F.; Mei, Y.; Cheng, Y.; Liu, F.; Yan, C.; Li, L.; Ling, Z. Alterations of gastric mucosal microbiota across different stomach microhabitats in a cohort of 276 patients with gastric cancer. EBioMedicine 2019, 40, 336–348.
- Castaño-Rodríguez, N.; Goh, K.-L.; Fock, K.M.; Mitchell, H.M.; Kaakoush, N.O. Dysbiosis of the microbiome in gastric carcinogenesis. Sci. Rep. 2017, 7, 15957.
- Tseng, C.-H.; Lin, J.-T.; Ho, H.J.; Lai, Z.-L.; Wang, C.-B.; Tang, S.-L.; Wu, C.-Y. Gastric microbiota and predicted gene functions are altered after subtotal gastrectomy in patients with gastric cancer. Sci. Rep. 2016, 6, 20701.
- Park, J.Y.; Seo, H.; Kang, C.S.; Shin, T.S.; Kim, J.W.; Park, J.M.; Kim, J.G.; Kim, Y.K. Dysbiotic change in gastric microbiome and its functional implication in gastric carcinogenesis. Sci. Rep. 2022, 12, 4285.
- Chen, C.; Chen, L.; Lin, L.; Jin, D.; Du, Y.; Lyu, J. Research progress on gut microbiota in patients with gastric cancer, esophageal cancer, and small intestine cancer. Appl. Microbiol. Biotechnol. 2021, 105, 4415–4425.
- Liang, W.; Yang, Y.; Wang, H.; Wang, H.; Yu, X.; Lu, Y.; Shen, S.; Teng, L. Gut microbiota shifts in patients with gastric cancer in perioperative period. Medicine 2019, 98, e16626.
- Chen, X.-H.; Wang, A.; Chu, A.-N.; Gong, Y.-H.; Yuan, Y. Mucosa-Associated Microbiota in Gastric Cancer Tissues Compared With Non-cancer Tissues. Front. Microbiol. 2019, 10, 1261.
- Gao, J.-J.; Zhang, Y.; Gerhard, M.; Mejias-Luque, R.; Zhang, L.; Vieth, M.; Ma, J.-L.; Bajbouj, M.; Suchanek, S.; Liu, W.-D.; et al. Association Between Gut Microbiota and Helicobacter pylori-Related Gastric Lesions in a High-Risk Population of Gastric Cancer. Front. Cell. Infect. Microbiol. 2018, 8, 202.
- Coker, O.O.; Dai, Z.; Nie, Y.; Zhao, G.; Cao, L.; Nakatsu, G.; Wu, W.K.; Wong, S.H.; Chen, Z.; Sung, J.J.Y.; et al. Mucosal microbiome dysbiosis in gastric carcinogenesis. Gut 2018, 67, 1024–1032.
- Hsieh, Y.-Y.; Tung, S.-Y.; Pan, H.-Y.; Yen, C.-W.; Xu, H.-W.; Lin, Y.-J.; Deng, Y.-F.; Hsu, W.-T.; Wu, C.-S.; Li, C. Increased Abundance of Clostridium and Fusobacterium in Gastric Microbiota of Patients with Gastric Cancer in Taiwan. Sci. Rep. 2018, 8, 158.
- Sun, J.; Li, X.; Yin, J.; Li, Y.; Hou, B.; Zhang, Z. A screening method for gastric cancer by oral microbiome detection. Oncol. Rep. 2018, 39, 2217–2224.
- Wu, Z.-F.; Zou, K.; Wu, G.-N.; Jin, Z.-J.; Xiang, C.-J.; Xu, S.; Wang, Y.-H.; Wu, X.-Y.; Chen, C.; Xu, Z.; et al. A Comparison of Tumor-Associated and Non-Tumor-Associated Gastric Microbiota in Gastric Cancer Patients. Dig. Dis. Sci. 2021, 66, 1673–1682.
- Liu, C.; Ng, S.-K.; Ding, Y.; Lin, Y.; Liu, W.; Wong, S.H.; Sung, J.J.-Y.; Yu, J. Meta-analysis of mucosal microbiota reveals universal microbial signatures and dysbiosis in gastric carcinogenesis. Oncogene 2022, 41, 3599–3610.
More
Information
Subjects:
Physiology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
566
Revisions:
2 times
(View History)
Update Date:
20 Oct 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No