Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sylwia Szrok-Jurga | -- | 5605 | 2023-10-19 13:38:21 | | | |
| 2 | Sirius Huang | Meta information modification | 5605 | 2023-10-20 03:29:47 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Szrok-Jurga, S.; Czumaj, A.; Turyn, J.; Hebanowska, A.; Swierczynski, J.; Sledzinski, T.; Stelmanska, E. The Physiological and Pathological Role of Acyl-CoA Oxidation. Encyclopedia. Available online: https://encyclopedia.pub/entry/50552 (accessed on 25 July 2026).
Szrok-Jurga S, Czumaj A, Turyn J, Hebanowska A, Swierczynski J, Sledzinski T, et al. The Physiological and Pathological Role of Acyl-CoA Oxidation. Encyclopedia. Available at: https://encyclopedia.pub/entry/50552. Accessed July 25, 2026.
Szrok-Jurga, Sylwia, Aleksandra Czumaj, Jacek Turyn, Areta Hebanowska, Julian Swierczynski, Tomasz Sledzinski, Ewa Stelmanska. "The Physiological and Pathological Role of Acyl-CoA Oxidation" Encyclopedia, https://encyclopedia.pub/entry/50552 (accessed July 25, 2026).
Szrok-Jurga, S., Czumaj, A., Turyn, J., Hebanowska, A., Swierczynski, J., Sledzinski, T., & Stelmanska, E. (2023, October 19). The Physiological and Pathological Role of Acyl-CoA Oxidation. In Encyclopedia. https://encyclopedia.pub/entry/50552
Szrok-Jurga, Sylwia, et al. "The Physiological and Pathological Role of Acyl-CoA Oxidation." Encyclopedia. Web. 19 October, 2023.
Copy Citation
Fatty acid metabolism, including β-oxidation (βOX), plays an important role in human physiology and pathology. βOX is an essential process in the energy metabolism of most human cells. Moreover, βOX is also the source of acetyl-CoA, the substrate for (a) ketone bodies synthesis, (b) cholesterol synthesis, (c) phase II detoxication, (d) protein acetylation, and (d) the synthesis of many other compounds, including N-acetylglutamate—an important regulator of urea synthesis.
beta-oxidation
peroxisomal fatty acid oxidation
acyl-CoA
fatty acid metabolism
1. Introduction
Fatty acids (FAs) are critical compounds for the health control and development of the human body due to their participation in cellular metabolism, especially energy production (ATP synthesis), metabolism regulation, and cell proliferation. They are (a) building blocks for complex lipids in cellular membranes, (b) precursors for signaling molecules, such as eicosanoids, (c) allosteric regulators of metabolic pathways, (d) substrates for protein acylation, and (e) ligands for transcription factors. FAs are also responsible for lipotoxicity and contribute to the release of proinflammatory molecules, which play an important role in many diseases. Moreover, an increase in citrate, isocitrate, and malate production associated with free fatty acid (FFA) β-oxidation (βOX) leads to increased NADPH levels in some cells. Cytosolic isocitrate dehydrogenase (which catalyzes the conversion of isocitrate in the presence of NADP to α-ketoglutarate and NADPH) and a cytosolic malic enzyme (ME) (which catalyzes the conversion of malate in the presence of NADP to pyruvate and NADPH) play an important role in NADPH homeostasis.
The most important sources of FAs found in humans include dietary supply, mainly triacylglycerols, and de novo synthesis, mainly from glucose [1].
As already mentioned, FAs serve a predominant role as substrates for ATP production in many human and animal organs, including the heart, skeletal muscle, kidney, and liver. Over 20 proteins are involved in the uptake, activation, transport into the organelles (mainly mitochondria and peroxisomes), and finally, fatty acid oxidation (FAO). The most important process of FAO-βOX occurs primarily in the mitochondria of many organs and, to a lesser extent, in peroxisomes, mainly in the liver and kidney. In peroxisomes, not only βOX but also α-oxidation takes place. Alfa oxidation produces a fatty acyl CoA, one carbon shorter [2]. From a practical point of view, this process plays an important role in the oxidation of phytanic acid (a compound present in the human diet, originating mainly from ruminant animals and fish) [3]. ω-oxidation undergoes in microsomes (smooth endoplasmic reticulum) [4]. In this process, FAs are degraded starting from the end methyl group (so-called ω-carbon) of FAs, and the CYP (cytochrome P-450) family is involved. ω-oxidation is considered a rescue process for some genetic diseases in humans, in which mitochondrial and peroxisomal FA oxidation is impaired. Interestingly, phytanic acid also undergoes ω-oxidation [2].
The energy production from FAs is strictly associated with the mitochondrial βOX. The intensity of βOX is controlled by a plethora of regulatory factors, including the supply of nutrients and the action of several hormones, including insulin, glucagon, catecholamines, triiodothyronine, and cortisol. The crucial regulator of FAO is peroxisome proliferator-activated receptor α (PPARα) [5]. PPARα is a transcription factor that functions as a heterodimer in complex with the retinoid X receptor α (RXRα) and binds via the PPARα DNA-binding domain (DBD) to the PPRE (peroxisome proliferator response element) sequence in the promoter region of target genes involved mainly in hepatic and cardiac muscle FA and FAO [6]. The initiation of transcription by PPARα (similar to other PPARs) requires its activation. Briefly, in its inactive form, the PPARα-RXRα complex is associated with corepressors [7]. The complex activation occurs following ligand binding [8]. A wide range of lipophilic molecules can activate PPARα. These include natural saturated, unsaturated, and polyunsaturated fatty acids (PUFAs) and synthetic ligands, collectively called PPARα activators [7][9]. The natural ligands show different binding affinities and strengths of PPARα activation. The potent PPARα ligands are unsaturated fatty acids, including omega-3 eicosapentaenoic acid (20:5, ω3), docosahexaenoic acid (22:6, ω3), and phytanic acid [10][11]. The natural and synthetic ligands (pharmacological ligands, for instance, fibrates) directly bind to PPARα via the ligand-binding domain (LBD). The ligand binding to a nuclear receptor causes the release of corepressors and begins the recruitment of coactivator complexes to the PPARα-RXRα, which enables the activation of the expression of genes involved in FAO [7]. PPARα is expressed at the highest level in hepatocytes, cardiomyocytes, enterocytes, and kidney proximal tubule cells, which are involved in the increased FAO [12]. Other members of the PPARs family—PPARβ/δ and PPARγ—are involved in the regulation of different processes generally associated with lipid metabolism. PPARβ/δ participates in the activation of FAO [13]. It has been observed that expression of the PPARβ/δ genes increases in skeletal muscles after fasting and endurance exercises, which promotes the transition from glucose, as the primary source of energy substrate, to lipids [14][15][16][17]. In comparison, PPARγ plays an important role in adipogenesis, lipid uptake, triacylglycerols (TAG) storage, and lipid droplet formation [18].
2. Uptake and Activation of Fatty Acids
In blood, FAs are present as components of lipids in (a) cell membranes (mainly in erythrocytes and white blood cells), (b) lipoproteins (mainly in chylomicrons, VLDL, LDL, and HDL), and (c) FFAs mostly bound to albumin. The major FAs in the whole lipids in the blood are palmitic acid (C16:0), stearic acid (C18:0), oleic acid (C18:1), linoleic acid (C18:2), and arachidonic acid (C20:4) [19][20]. The concentration of FFAs in the serum increases during exercise or fasting, and they are mainly used as FAO substrates in skeletal muscles, the heart, liver, and kidney [21]. The physiological FFA concentration in blood is around 0.2–0.5 mmol/L [22]. Due to their low solubility in H2O (1–10 nmol/L, depending on FA chain length), FFAs (mainly long-chain—LCFAs and medium-chain—MCFAs) are attached to the albumin [23]. Binding the FFAs to the albumin (a) enables transport in the blood and (b) protects human organs against some pathologies, including insulin resistance, non-alcoholic fatty liver disease, atherosclerosis, and heart dysfunction [24][25]. FFAs are translocated from the albumin FFA complex into the target cell (cells where FFAs are metabolized) cytosol across the endothelial layer of the blood vessels [26]. In the liver, the sinusoidal endothelial cells are fenestrated and do not have a basement membrane, so the absorption of FFAs is much easier than in other organs [27]. The transfer of FFAs from the blood to other cells, for instance, cardiomyocytes, seems to be more complicated since the endothelial wall in the heart capillaries is not-fenestrated and the FFAs are transferred through three lipid membranes: two endothelial (in and out of the endothelial cells) and one myocyte membrane (transported into the cell). Arts et al. proposed a model of FFA translocation across heart capillaries into cardiomyocytes, where FFAs bind to compartment-specific carrier proteins [28]. According to this model, the crossing of the plasma membrane remains under the control of several proteins, including (a) cluster of differentiation-36 (also known as FA translocase—CD36), (b) FA-binding protein—FABPm, and (c) FA-transporting protein—FATP (Figure 1). These proteins enable the cell to control the inflow of FFAs precisely. They increase the uptake of FFAs at the beginning of muscle contraction, even if the concentration of FFAs in the blood is low. Moreover, they also prevent the entrance of excess FFAs into the cell and help to select FFAs according to the cell’s demand. It should be noted that FFAs may also translocate into the target cell by a flip-flop system driven by the FFA concentration gradient [28].
The delivery of FFAs to the cells and their activation before usage in several cellular processes involves many proteins, including enzymes (Figure 1). Among these proteins are FATPs, which exhibit acyl-CoA synthetase activity. These two functions of FATPs (transport and activation) enable the immediate utilization of FFAs in the cell. Other proteins, like FABPm, CD36, and a family of acyl-CoA synthetases (ACSs), form an integrated system of the transport and activation of LCFAs, MCFAs, and short-chain FAs (SCFAs). FABPc proteins are also involved in binding FFAs in the cytosol (Figure 1).
FFAs are activated by specific ACSs. After activation, some FFAs become bound by the acyl-CoA-binding proteins (ACBPs). The binding of FFAs is responsible for channeling acyl-CoA to particular cellular compartments and processes. Acyl-CoA’s minor pool is deacylated by acyl-CoA diesterases (ACOTs) [29][30]. However, the physiological significance of deacylation is unknown. Very recently, it has been reported that ACOT1 knock-out partially protects mice from high-fat diet-induced weight gain by increasing energy expenditure [31]. Thus, these results suggest that inhibition of ACOT1 could prevent obesity during caloric excess.
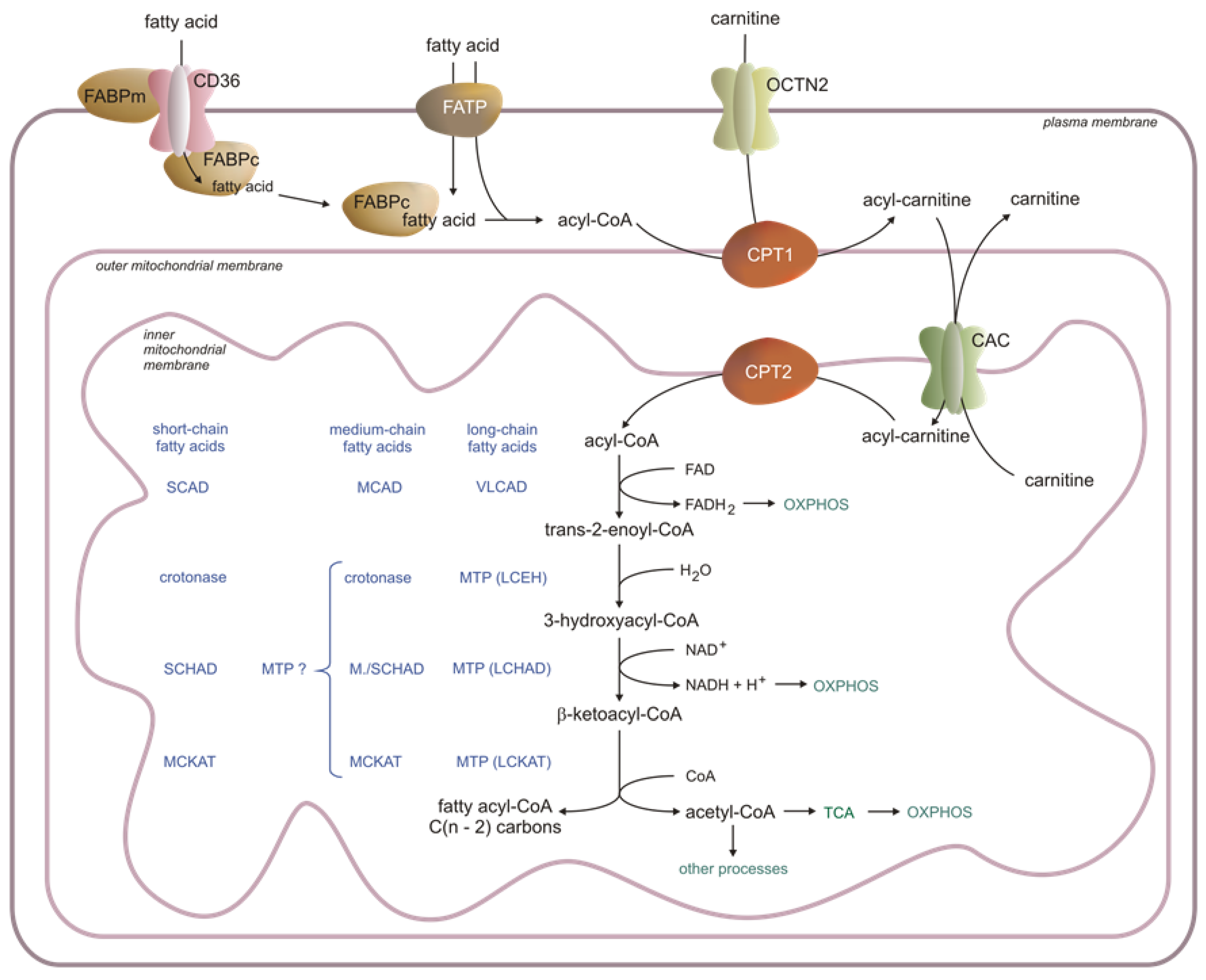
Figure 1. Fatty acid transport and metabolism in the cell. CAC—acylcarnitine translocase, CP—carnitine palmitoyltransferase, FABP—fatty acid-binding protein, LCEH—long-chain enoyl-CoA hydratase, LCHAD—long-chain fatty acid hydroxy acyl-CoA dehydrogenase, LCKAT—long-chain fatty acid β-ketothiolase, MCAD—medium-chain acyl-CoA dehydrogenase, MCKAT—medium-chain ketoacyl-CoA thiolase, OXPHOS—oxidative phosphorylation, SCAD—short-chain acyl-CoA dehydrogenase, SCHAD—short-chain hydroxy acyl-CoA dehydrogenase, TCA—Krebs cycle, VLCAD—very-long-chain acyl-CoA dehydrogenase, OCTN2—carnitine transporter, present in the heart, skeletal muscle, and kidney (hepatocytes have a different translocator with low affinity and high capacity), FABPm—membrane fatty acid-binding protein, FABPc—cytosolic fatty acid-binding protein, MTP—mitochondrial trifunctional protein, MTP ? – possible involvement of MTP protein, CD-36—fatty acid translocase, FATP—fatty acid transporting protein (the acyl-carnitines are transported across the outer mitochondrial membrane via a voltage-dependent anion channel (VDAC) [32]).
According to the chain length, influencing the hydrophobicity and water solubility of FFAs, four ACS families have been established: (a) short-chain acyl-CoA synthetases (ACSSs), (b) medium-chain acyl-CoA synthetases (ACSM), (c) long-chain acyl-CoA synthetases (ACSLs), and (d) very-long-chain acyl-CoA synthetases (ACSVLs) [33]. An overview of the characteristics of ACS isoforms is presented in Table 1.
Table 1. Characteristics of acyl-CoA synthetases. ACSL—long-chain acyl-CoA synthetase, ACSM—medium-chain acyl-CoA synthetases, ACSS—short-chain acyl-CoA synthetases, ACSVL—very-long-chain acyl-CoA synthetase, BAT—brown adipose tissue, ER—endoplasmic reticulum, WAT—white adipose tissue.
| Name/Abbreviation | Organ/Tissue Localization | Subcellular Compartment | References |
|---|---|---|---|
| ACSVL [FATP2] | Liver, intestine, kidneys, brain | Peroxisomes, ER | [34] |
| ACSVL [FATP6] | Heart | Cytosol, plasma membrane | [35] |
| ACSVL [FATP3] | Lungs, gonads, adrenals | ER, mitochondrial membrane | [34] |
| ACSVL [FATP1] | Skeletal muscles, BAT, WAT, heart | Plasma membrane | [36] |
| ACSVL [FATP4] | Skeletal muscles, BAT, WAT, intestine, skin | Peroxisomes, ER, mitochondrial membrane | [37] |
| ACSVL [FATP5] | Liver | Plasma membrane | [38] |
| ACSL1 | Liver, heart, BAT, WAT, skeletal muscles | Mitochondria (outer mitochondrial membrane on the cytosolic side), lipid droplets, microsomes, plasma membrane | [39] |
| ACSL3 | Brain, gonads, small amounts in other tissues (liver) | Lipid droplets, the cytoplasmic face of ER, the outer mitochondrial membrane | [40] |
| ACSL4 | Adrenals, ovaries, testes, liver, skeletal muscles, small amounts in the brain | Endosomes, peroxisomes, plasma membrane, secretory vesicles, ER regions in close contact with mitochondria—mitochondrial-associated membranes | [41] |
| ACSL5 | BAT, the duodenal mucosa, liver, skeletal muscles, kidneys, lungs | The outer mitochondrial membrane on the cytosolic side | [42] |
| ACSL6 | Ovaries, testes, brain, skeletal muscles, small amounts in the WAT, kidneys, the duodenal mucosa | Plasma membrane | [43] |
| ACSM | Liver, skeletal muscles, cardiomyocytes, colonocytes, kidneys | Mitochondria. All ACSMs belong to a group of enzymes called XM-ligases (xenobiotic/medium-chain fatty acid-CoA ligases) | [44][45] |
| ACSS1 | Brain, blood, testes, intestine, heart, kidneys, skeletal muscles, BAT | Mitochondria. ACSS1 activates acetate | [46] |
| ACSS2 | Liver and kidneys | Cytosol, nucleus. ACSS2 activates acetate. ACSS2 is downregulated during fasting | [46][47] |
| ACSS3 | Liver | Mitochondria. ACSS3 has a higher affinity for propionate. ACSS3 is upregulated in the fasting state | [30][46] |
3. Carnitine Shuttle
3.1. Carnitine Palmitoyltransferase 1 (CPT1)
The inner mitochondrial membrane is impermeable to the long-chain acyl-CoAs. Thus, the acyl-CoAs are converted to acylcarnitine in the reaction catalyzed by carnitine palmitoyltransferase 1 (CPT1):
acyl-CoA + carnitine → acylcarnitine + CoASH
CPT1 is a hexamer, a part of a protein complex formed and attached to the outer mitochondrial membrane. Other elements of that complex are ACSL and VDAC (voltage-dependent anion channel) [49][50]. Three isoforms of CPT1 are known: CPT1A, CPT1B, and CPT1C. CPT1A is the main CPT1 in the liver, but it is also present in minor amounts in the heart, skeletal muscles, brain, kidneys, lungs, spleen, intestine, pancreas, ovaries, and fibroblasts. It is involved in transporting LCFAs and medium-chain lauric acid (C:12) into mitochondria, though its highest activity is in lauric acid. CPT1B is the dominating form in the skeletal muscles, heart, and testes, and like CPT1A, it is an enzyme transporting LCFAs to mitochondria, with the highest activity in C12-C16 FFAs. CPT1C is a neural form attached to endoplasmic reticulum (ER) membranes. Potentially, it is involved in the neuronal control of thermogenesis in brown adipose tissue (BAT) [51][52]. CPT1C activity is significantly (20–300 times) lower than CPT1A [53][54][55]. CPT1A and B share 62% similarity in the amino acid sequence. Both isoforms differ significantly in activity and regulation [56].
A high-fat diet induces the expression of the CPT1 gene by the PPARα transcription factors in the liver and muscles [5][57][58]. Insulin, glucagon, and triiodothyronine regulate CPT1 activity in the liver, and the physiological status significantly influences that regulation [57][59][60][61]. The major regulator of CPT1 is malonyl-CoA, a negative allosteric effector of this enzyme. The intracellular level of malonyl-CoA depends on acetyl-CoA carboxylase (ACC—enzyme-synthesizing malonyl-CoA) activity and malonyl-CoA decarboxylase (MCD—enzyme-degrading malonyl-CoA) activity [62][63][64][65]. Malonyl-CoA, an intermediate in palmitate synthesis, inhibits FAO during intensive FFA synthesis. It protects the cell from the immediate oxidation of the newly synthesized FFAs [52]. At a negative energy balance, when the activity of MCD is elevated, CPT1 restores its activity, leading to efficient acylcarnitine synthesis. It should be noted that CPT1B is activated mainly by exercise and is more sensitive to changes in the malonyl-CoA level.
Both LCFAs and MCFAs stimulate CPT1 activity during the exercises [66]. A high-fat diet or fasting induces the expression of the CPT1 gene by the two independent systems involving PPARα transcription factors or the PGC1α/PPARγ complex in the liver and muscles. The binding site in the Cpt1 gene for PPARα in the rat liver is located in the second intron and PGC1α/PPARγ in the first intron [5][57][66]. Mutations in PPRE totally eliminate the induction of Cpt1 gene expression by both regulatory systems [5].
Carnitine is transported from the blood to the cells by the high-affinity OCTN2 carnitine transporter in the cell membrane of the heart, skeletal muscle, and kidney (Figure 1) [67]. It should be noted that different types of carnitine transporters with low affinity and high capacity are present in hepatocytes.
3.2. Carnitine Palmitoyltransferase 2 (CPT2) and Acylcarnitine Translocase CAC (SLC25A20)
CAC (SLC25A20) transfers acylcarnitines across the inner mitochondrial membrane [68]. CAC forms a functional complex with carnitine palmitoyltransferase 2 (CPT2) in the inner mitochondrial membrane (Figure 1), leading to the transesterification of acyl groups from acylcarnitines to mitochondrial CoAs according to the reaction:
acylcarnitine + CoA-SH → acyl-CoA + carnitine
A high acylcarnitine concentration in the intermembrane space drives its translocation into the matrix [68]. The overall role of CPT1, CAC, and CPT2 in the transport of acyl-CoA into the mitochondrial matrix is presented in Figure 1. NO, H2S, nonenzymatic acetylations, β-lactam antibiotics, omeprazole (proton pump inhibitor), and heavy metals inhibit CAC [69][70][71][72][73][74][75][76]. PPARα and other transcription factors or transcriptional coactivators (estrogen receptors, PGC1α) activate the transcription of CAC, and polyphenols (antioxidants) increase the effectiveness of βOX. Statins, drugs lowering serum cholesterol concentration, and retinoic acid also increase CAC activity [77][78][79][80].
4. Mitochondrial β-Oxidation
A few years ago, the mitochondrial βOX was described by Hounten et al. in an excellent review [81]. Briefly, the first step of each βOX round is catalyzed by an acyl-CoA dehydrogenase (AD), producing trans-2-enoyl-CoA. In the next step, the hydration of a double-bond is catalyzed by enoyl-CoA-hydratase (ECH), and the following dehydrogenation by hydroxy-acyl-CoA dehydrogenase (HAD) leads to the production of 3-keto-acyl-CoA. The last step of the cycle is thiolysis. In each round of βOX, one FAD and NAD+ accept two electrons each and change into FADH2 and NADH, respectively. The electrons are then transferred to the mitochondrial respiratory chain, where oxidative phosphorylation (OXPHOS) occurs. The acetyl-CoA formed may enter the Krebs cycle (TCA) (mainly in the heart, kidney, and skeletal muscle) and other processes (for instance, ketogenesis in the liver) (Figure 1) [82][83]. The acyl-CoAs, which are shorter by two carbons compared to the initial substrate, enter the next round of βOX. The odd-chain FFAs (present in a small amount in human tissue) are degraded, like the even-chain acyl-CoAs, to several acetyl-CoAs (depending on FFAs). However, propionyl-CoA arises from the methyl end of the odd-chain acyl-CoA. Propionyl-CoA is converted via methylmalonyl-CoA to succinyl-CoA, metabolized in the TCA, or converted to glucose in the liver. The amount of propionyl-CoA formed from odd-chain FFAs is very small because the number of such FAs in the diet is relatively low.
Five ADs found in human cells are involved in the first step of βOX. Characteristics of ADs are presented in Table 2.
Table 2. Characteristics of acyl-CoA dehydrogenases. ACAD9—acyl-CoA dehydrogenase DH-9, BCFA—branched-chain fatty acid, LCAD—long-chain acyl-CoA dehydrogenase, LCFA—long-chain fatty acid, MCAD—medium-chain acyl-CoA dehydrogenase, MCFA—medium-chain fatty acid, SCAD—short-chain acyl-CoA dehydrogenase, SCFA—short-chain fatty acid, VLCAD—very-long-chain acyl-CoA dehydrogenase, VLCFA—very-long-chain fatty acid.
| Enzyme | Mitochondrial Compartment | Preferred Substrates (Acyl-CoAs) | Tissue/Organ/Cell | Reference |
|---|---|---|---|---|
| VLCAD | Inner mitochondrial membrane | LCFA (mainly palmitoyl-CoA) and VLCFA (C14–C22) | Muscles, heart, liver, skin fibroblasts | [84] |
| Acyl-CoA DH-9 (ACAD9) | Inner mitochondrial membrane | Unsaturated LCFA, VLCFA (C16:1, C18:1, C18:2; C22:6) | Brain, liver, heart, skeletal muscle | [85] |
| LCAD | Matrix | LCFA, unsaturated MCFA, SCFA, BCFA (in vitro) | Lungs—pulmonary surfactant | [86] |
| MCAD | Matrix | MCFA (C6:0–C12:0) | Heart, skeletal muscles, liver | [87] |
| SCAD | Matrix | SCFA (mainly butyryl-CoA); MCFA (C6:0–C12:0) |
Liver, heart, skeletal muscle | [88] |
4.1. Oxidation of Long-Chain Acyl-CoA
Oxidation of long-chain acyl-CoA is catalyzed by one of three ADs: a) very-long-chain acyl-CoA dehydrogenase (VLCAD), b) acyl-CoA dehydrogenase DH-9 (ACAD9), and c) long-chain acyl-CoA dehydrogenase (LCAD). VLCAD oxidizes most LCFAs entering mitochondria. This enzyme, bound to the inner mitochondrial membrane, oxidizes C14:0–C22:0 acyl-CoA, although the preferred substrate is palmitoyl-CoA. The presence of an unsaturated bond in FFAs decreases the efficiency of the reaction catalyzed by this enzyme. PPARα is the most important VLCAD regulator, increasing its gene expression. Sirtuins (especially sirtuin 3) may also activate VLCAD through deacetylation [84][85][86][87][88].
ACAD9 is homologous to VLCAD and uses mostly unsaturated long-chain acyl-CoAs as substrates. It is abundant in the brain and liver. Despite the homology of this enzyme with VLCAD, neither enzyme can compensate for each other in their deficiency [85][89]. LCAD is localized in the mitochondrial matrix. It is mainly present in lung alveolar cells. LCAD knockout caused pulmonary surfactant (complex substances, mainly lipids, which play important functions in the alveoli and small airways) dysfunction and increased susceptibility to lung infections [86]. An in vitro investigation showed that some unsaturated and branched-chain acyl-CoA are the principal substrates for LCAD. This enzyme is exceptional among ADs because it tends to leak electrons, producing H2O2. Its function in organs other than the lungs has not been estimated [90].
Each AD uses FAD as an electron acceptor. Formed FADH2 has to be re-oxidized, so the electrons are translocated to a flavoprotein, electron-transferring flavoprotein (ETF), and then ETF-dehydrogenase transfers them into coenzyme Q (CoQ) in the OXPHOS system (Figure 1) [91][92].
The mitochondrial trifunctional protein (MTP) complex participates in the second step of LCFA oxidation. The MTP catalyzes three different reactions in a row. The MTP enzymatic activities are long-chain enoyl-CoA hydratase (LCEH), long-chain hydroxy acyl-CoA dehydrogenase (LCHAD), and long-chain β-ketothiolase (LCKAT). The MTP complex contains “a” and “b” subunits, forming an octamer bound to the surface of the inner mitochondrial membrane due to a strong interaction with membrane phospholipids [93][94]. Subunit “a” contains the enzymatic activities of hydratase and dehydrogenase, whereas subunit “b” contains thiolase activity. This enzymatic complex binds the enoyl-CoAs containing 6–16 carbons, but in the liver, its activity is the highest for C10 and longer acyl-CoAs. The final product of MTP activity is acetyl-CoA and acyl-CoA, which is shortened by two carbons and enters the next cycle of βOX [95].
4.2. Oxidation of Monounsaturated and Polyunsaturated Long-Chain Acyl-CoA
Oxidation of monounsaturated long-chain acyl-CoA requires an additional enzyme called 3,2-trans-enoyl-CoA isomerase (ECI), which catalyzes the following reaction:
trans-3-enoyl-CoA → trans-2-enoyl-CoA
ECI exists in two isoforms: ECI1 and ECI2. ECI1 is found in mitochondria only, whereas ECI2 is present in mitochondria and peroxisomes. ECI2 has a much higher affinity for LCFAs [96][97][98]. The studies on ECI isoforms were performed using enzymes isolated from rat liver [96] and the ECI1 knock-out mice model [97], and structural studies using X-ray scattering were performed for a human ECI2 isoform [98].
The βOX of polyunsaturated FAs requires a) ECI and b) 2,4-dienoyl-CoA reductase, which catalyzes the following reaction:
trans-2,cis-4-dienoyl-CoA + NADPH + H+ → tans-3-enoyl-CoA + NADP+
Formed tans-3-enoyl-CoA by 2,4-dienoyl-CoA reductase is converted to trans-2-enoyl-CoA by ECI, as presented above.
4.3. Oxidation of Medium-Chain Fatty Acids
In the first cycle of MCFA mitochondrial FAO, medium-chain acyl-CoA dehydrogenase (MCAD) catalyzes the initial step. It is a flavoprotein cooperating with ETF and ETF-dehydrogenase. MCAD is a homotetrameric protein localized in the mitochondrial matrix. It is abundant in the human heart, skeletal muscles, and liver [99][100]. The enzymes responsible for the subsequent reactions are not well-defined in humans. It is possible that human MTP participates in the oxidation of medium-chain enoyl-CoAs. However, it is not excluded that MCFAs, which translocate from the cytosol to mitochondria, might be activated and elongated, finally becoming the substrate for MTP [101].
4.4. Oxidation of Short-Chain Fatty Acids
The first step of SCFA degradation is catalyzed by short-chain acyl-CoA dehydrogenase (SCAD), a flavoprotein cooperating with ETF/ETF-dehydrogenase. Butyryl-CoA, formed from butyrate produced by gut microbiota, is the major substrate for SCAD, and the product is crotonyl-CoA [102]. SCAD is abundant in the liver, heart, and skeletal muscles. It is a matrix-localized homotetramer. In the liver and kidneys, SCAD also displays oxidase activity, but the significance of this feature is unresolved [103][104]. The other enzymes involved in short-chain acyl-CoA oxidation are crotonase (enoyl-CoA hydratase), medium-chain hydroxy acyl-CoA dehydrogenase, short-chain hydroxy acyl-CoA dehydrogenase (SCHAD), and medium-chain ketoacyl-CoA thiolase (MCKAT), and all those activities are localized in the mitochondrial matrix. Human crotonase uses crotonyl-CoA as a substrate. It is also involved in the metabolism of some amino acids. Crotonase is present in significant amounts in the liver, less in muscles and fibroblasts, and even less in the kidneys and spleen [105][106]. Hydroxyacyl-CoA dehydrogenase is a homodimer localized in the matrix, which produces acetoacetyl-CoA and NADH. The highest activity of this enzyme is present in the heart, muscles, liver, and pancreas [107]. MCKAT catalyzes the last step of short-chain FAO. The activity of MCKATs is present in the mitochondrial matrix, peroxisomes, and cytosol. MCKATs that are present in the matrix of human mitochondria have two main substrates: methyl-acetyl-CoA (metabolized into propionyl-CoA) and acetoacetyl-CoA (metabolized into two molecules of acetyl-CoA).
5. Peroxisomal FAO
In the liver, FAO takes place both in mitochondria and peroxisomes. However, under physiological conditions, peroxisomal FAO accounts for approx. 5% of total FAO in the liver [108]. Peroxisomal βOX differs significantly from mitochondrial βOX [109][110]. In mitochondria, acyl-CoA dehydrogenases transfer the electrons to ETF, which are subsequently transferred to the mitochondrial respiratory chain and reduce oxygen to water, producing energy (ATP) [82]. In contrast, peroxisome acyl-CoA oxidase 1 (ACOX1) reduces FAD, and electrons are transported directly from FADH2 to molecular oxygen, generating hydrogen peroxide (H2O2) [110]. CoA esters of straight-chain FAs (VLCFAs, LCFAs, PUFAs, and dicarboxylic acids) are preferred substrates for ACOX1, whereas ACOX2 is responsible for the oxidation of branched-chain FAs (BCFA) and the transformation of bile acid intermediates [111]. In addition, Ferdinandusse et al. identified a novel ACOX isoform, ACOX3, which is involved, similar to ACOX2, in the degradation of BCFAs [112].
The oxidation of LCFAs in peroxisomes stops at the level of MCFA-CoAs [110]. MCFA-CoAs can be hydrolyzed to FFAs by the peroxisomal thioesterases. Then, MCFAs, via the pore-forming proteins, leave the peroxisome and are transported to the mitochondria, where βOX is completed. The second way of MCFA oxidation uses carnitine and carnitine acyltransferase with specificity for short- and medium-chain acyl-CoA. Formed acylcarnitines are transported into mitochondria via the mitochondrial CAC [113]. It should be emphasized that peroxisomal FAO needs the participation of mitochondria not only for the oxidation of acetyl-CoA (formed from MCFA-CoAs) but also for the oxidation of NADH [110][114]. For a summary of mitochondrial and peroxisomal βOX, see Table 3.
Table 3. Comparison between peroxisomal and mitochondrial β-oxidation. ABCD1–4—ATP-binding cassette sub-family D 1–4, ACADs—acyl-CoA dehydrogenases, ACOXs—acyl-CoA oxidases, BCFA—branched-chain fatty acid, CPT1—carnitine palmitoyltransferase 1, CPT2—carnitine palmitoyltransferase 2, CAC—acylcarnitine translocase, FAs—fatty acids, VLCADs—very-long-chain fatty acids, LCFAs—long-chain fatty acids, MCFAs—medium-chain fatty acids, PUFAs—polyunsaturated fatty acids, SCFAs—short-chain fatty acids, H2O—hydrogen peroxide, ETF—electron-transferring flavoprotein, OXPHOS—oxidative phosphorylation.
| Peroxisomal β-Oxidation | Mitochondrial β-Oxidation | References | |
|---|---|---|---|
| Proteins involved in the transport of FAs to peroxisomes/mitochondria | ABCD1, ABCD2, and ABCD3 | Carnitine transport system (CPT1, CPT2, CAC) | [115][116] |
| Substrates | VLCFAs (>C22), BCFAs (e.g., pristanic acid), PUFA, 2-hydroxy FAs, long-chain dicarboxylic acids, bile acid intermediates, and a number of prostanoids | VLCFAs (≤22), LCFAs, MCFAs, and SCFAs | [117][118] |
| Enzyme catalyzing the first reaction | ACOXs The transfer of electrons from FADH2 to oxygen results in the production of H2O2, which is subsequently cleaved by peroxisomal catalase |
ACADs The electrons that originate from FADH2 are transported to ETF, the ETF dehydrogenase, and transferred to OXPHOS. Finally, they reduce oxygen to water, which results in the production of energy in the form of ATP |
[82][110] |
| β-oxidation end products | Acetyl-CoA, NADH, MCFAs, and FADH2 | Acetyl-CoA, NADH, and FADH2 | [94][110] |
It has been shown that during peroxisomal βOX (both dicarboxylic and monocarboxylic acids), free acetate is formed, which is preferentially exported from the hepatocyte and used as an energy substrate in other organs [113]. It has been postulated that acetate is formed from acetyl-CoA in a reaction catalyzed by acetyl-CoA hydrolase [92].
5.1. Peroxisomal α-Oxidation—Role in Phytol and Phytanic Acid Metabolism
The average Western diet contains approx. (a) 50–100 mg per day of phytanic acid, (b) 10–30 mg per day of pristanic acid, and (c) 10 mg per day of phytol [119]. Phytol mostly comes from nuts [120]. Phytanic acid and pristanic acid are derived primarily from lipids found in beef, dairy products, and fish. [119]. The phytanic acid present in the diet is derived mainly from phytol [121]. Phytol is widely distributed as a constituent of chlorophyll present in the green leaves of plants and trees [3]. Bacteria present in the rumen of ruminant animals cleave the phytol from the porphyrin ring of chlorophyll (the human alimentary tract cannot do this). The released phytol can be oxidized to phytanic acid in the ruminants [3]. Thus, it is clear that phytanic acid is present in meat and dairy products from grass-fed cattle or other ruminants. Phytanic acid can also be derived from vegetables (as phytol bound to chlorophyll) [122]. Moreover, phytyl FA esters are also present in the leaves of some plants, fruits, and vegetables. These compounds are hydrolyzed in the human gastrointestinal tract, providing phytol [123].
Subjects consuming products rich in phytol and phytanic acid oxidize these compounds via α-oxidation because BCFAs containing a methyl group in the 3-position (like phytanic acid) are not metabolized by βOX. First, phytol is oxidized to phytenal in the reaction catalyzed by alcohol dehydrogenase. Formed phytenal is oxidized by aldehyde dehydrogenase to phytenic acid, which in turn is converted to phytenoyl-CoA by acyl-CoA synthetase. In the reaction catalyzed by enoyl-CoA reductase, phytenoyl-CoA is converted to phytanoyl-CoA. Phytanoyl-CoA can also be formed from phytanic acid in the reaction catalyzed by acyl-CoA synthetase. Formed phytanoyl-CoA undergo α-oxidation to 2-hydroxyphytanoylo-CoA, catalyzed by phytanoyl-CoA 2-hydroxylase. This process requires 2-oxoglutarate and Fe2+, and O2. 2-hydroxyphytanoilo-CoA is converted with the participation of hydroxy acyl-CoA and aldehyde dehydrogenase to pristanic acid, which is activated to pristanoyl-CoA by acyl-CoA synthetase. Next, pristanoyl-CoA undergoes peroxisomal βOX to 4,8-dimethyl nonaoyl-CoA, which in turn is metabolized in mitochondria (Figure 2) [123].
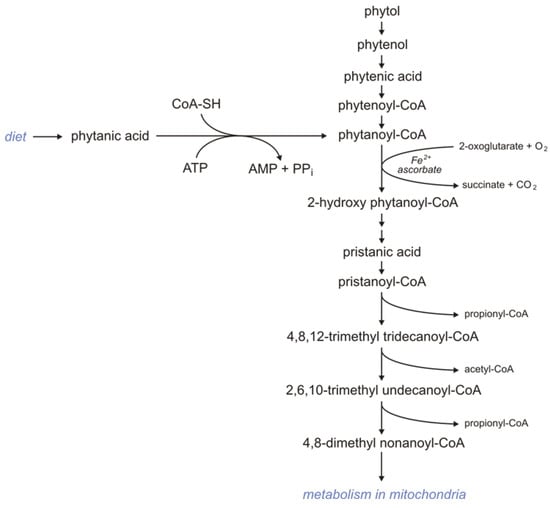
Figure 2. Metabolism of phytanic acid.
Deficiency of the phytanoyl-CoA 2-hydroxylase impairs the conversion of phytanic acid to pristanic acid (2-methyl BCFAs) and leads to Refsum disease (type IV motor and sensory neuropathy) [124][125]. The only therapy available for that disorder is a diet low in phytanic acid.
5.2. Peroxisomes Are Essential for the Degradation of Dicarboxylic Acid Formed during ω-Oxidation in Microsomes
VLCFAs are also oxidized in microsomes via ω-oxidation. In humans, the first step of ω-oxidation is catalyzed by CYP (CYP4F2 or CYP4F3B). Omega-hydroxy-VLCFAs, formed by CYP4F2 or CYP4F3B, can be oxidized to ω-HOOC-VLCFA (dicarboxylic-VLCFA) by alcohol dehydrogenase and subsequently by aldehyde dehydrogenase. Formed HOOC-VLCFA is then oxidized by βOX in peroxisomes. Importantly, the βOX of HOOC-VLCFA is not affected in X-ALD (X-linked adrenoleukodystrophy) patients [2]. Thus, it has been suggested that the peroxisomal βOX of dicarboxylic-VLCFA (formed during ω-oxidation) can provide an alternative route of VLCFA oxidation in X-ALD patients (Figure 3) [2].
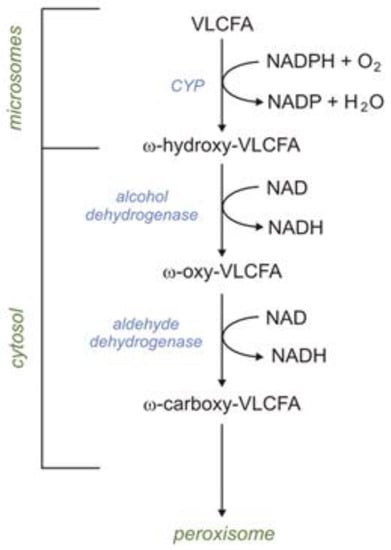
Figure 3. Omega oxidation of very-long-chain fatty acids (VLCFAs).
5.3. Peroxisomal FAO—Potential Role in the Utilization of Toxic FFAs
Peroxisomal βOX is necessary for the oxidation of VLCFAs (≥22 carbons), both saturated and mono- and polyunsaturated [110][113]. These FFAs need to be degraded not because of their role in providing energy but due to the toxic effect of their excessive accumulation (for instance, monounsaturated erucic acid C22:1, present in commonly used canola oil) [113][126]. The βOX of VLCFAs, notably C26:0 and longer-chain FFAs, occurs exclusively in peroxisomes [113].
Abnormalities in the biogenesis of peroxisomes are the cause of Zellweger syndrome. This rare familial disease is characterized by muscle weakness, hepatomegaly, and brain and kidney dysfunction. Goldfischer et al. reported that peroxisomes are absent in the liver and kidney of patients with this syndrome [127]. Consequently, significant amounts of VLCFAs and bile acid synthesis intermediates are accumulated in plasma [125][127][128][129].
Subfamily D of ABC transporters (ATP-binding cassette transporters) in mammals comprises four distinct proteins, namely ABCD1 (adrenoleukodystrophy protein), ABCD2 (adrenoleukodystrophy-related protein), ABCD3 (70 kDa peroxisomal membrane protein), and ABCD4 (peroxisomal membrane protein 69). Three of these, ABCD1-3, are localized solely in peroxisomes and mediate the uptake of substrates into the peroxisome for βOX [115].
ABCD1 and ABCD2 facilitate the transport of VLCFAs or their CoA derivatives into peroxisomes. Interestingly, ABCD1 has a higher specificity for saturated VLCFA-CoA. In contrast, ABCD2 prefers to transport PUFAs, such as C22:6-CoA and C24:6-CoA [130]. However, it is worth adding that the main substrate for ABCD2 in humans is still not completely defined [131]. The ABCD3 transporter is important in transporting branched chain acyl-CoA and bile acid intermediates, e.g., di- and tri-hydroxycholestanoyl-CoA (DHCA and THCA) [132]. Abcd genes are under complex regulation at the transcriptional level. The transcription of Abcd1, Abcd2, and Abcd3 genes is regulated by PPARα [133][134]. Leclercq et al. demonstrated that the hepatic expression of Abcd2 and Abcd3, but not Abcd1 and Abcd4, exhibits a high degree of sensitivity toward dietary PUFA intake [135].
5.4. Peroxisomal FAO Related to the Synthesis of Cholesterol and Phospholipids
Acetyl-CoA formed during FAO in peroxisomes can be used for synthesizing cholesterol and phospholipids (mainly plasmalogen) [136]. For instance, the first two steps of plasmalogen biosynthesis occur in peroxisomes from the acetyl-CoA derived from peroxisomal FAO [137]. Recent studies indicate that peroxisomal βOX stimulates cholesterol biosynthesis in the liver of diabetic mice [138]. Moreover, it has been reported that the inhibition of peroxisomal βOX suppresses cholesterol biosynthesis and consequently lowers plasma cholesterol concentration. Based on these data, the authors suggest that the upregulation of peroxisomal cholesterol biosynthesis related to βOX may contribute to diabetes hypercholesterolemia [138].
5.5. Peroxisomal FAO—Inhibition of Lipophagy
Lipophagy involves the encapsulation of lipid droplets into the autophagosome, which fuses with the lysosome, resulting in the hydrolysis of triacylglycerols catalyzed by lysosomal acid lipase A [110][139][140][141]. Peroxisomal FAO in the liver promotes hepatic steatosis by inhibiting lipophagy [141]. Supplied by FAO, acetyl-CoA is involved in the acetylation of Raptor, a component of mTORC1, a metabolic regulatory complex that inhibits autophagy [141].
5.6. Peroxisomal FAO—Regulation of Mitochondrial β-Oxidation
Peroxisomal βOX increases the cellular NADH/NAD+ ratio, which inhibits the SIRT1/AMPK pathway. The inhibition of that pathway leads to increased ACC activity. It causes elevation of malonyl-CoA levels in the cytosol, inhibiting CPT1 and the transport of LCFAs into mitochondria, decreasing mitochondrial βOX [110][142].
5.7. Peroxisomal FAO As a Process Associated with the Production of H2O2—An Important Signaling Molecule and Toxic Substance
As mentioned above, peroxisomal FADH2 formed during βOX is involved in H2O2 production. H2O2 is an important signaling molecule that regulates many cellular processes by modulating the activity of several proteins via cysteine oxidation [143]. Under physiological conditions, catalase converts most of the H2O2 formed during peroxisomal βOX to H2O and O2 [144]. However, when catalase activity is decreasing, for instance, during aging, part of H2O2 formed via peroxisomal βOX diffuses out the peroxisome (it is a relatively stable ROS) and may modulate the activity of redox-sensitive protein, which in turn triggers a complex network of signaling processes leading to regulation of (a) NF-ϰB activation, (b) E cadherin expression, (c) the secretion of matrix metalloproteinases, (d) mTORC activity, and (e) autophagy [144][145]. However, it is generally believed that reactive oxygen species (ROS) play a dual role. At physiological conditions, they are required for many signaling processes, affecting proliferation, differentiation, and aging, but there are also toxic byproducts of aerobic metabolism, including products of FFA oxidation [146]. H2O2 can be converted to highly reactive hydroxyl radicals, causing damage to proteins, lipids, and DNA, leading to many diseases, including atherosclerosis, cancer, diabetes, and rheumatoid arthritis [147]. Thus, it is tempting to speculate that microsomal βOX, via H2O2 production, may affect aging processes and aging-related diseases.
5.8. Microsomal Fatty Acid ω-Oxidation
Under physiological conditions, FA ω-oxidation accounts for no more than 10% of total fatty oxidation in the liver [2]. In this process, the terminal methyl group (ω carbon) of FFAs is oxidized to the carboxyl group. The first step of ω-oxidation is catalyzed by the CYP family present in the microsome (including CYP4F2 and CYP4F3B), which requires NADPH and O2. Formed ω-hydroxy-FFAs are oxidized to ω-oxo-FFAs by cytosolic alcohol dehydrogenase. Finally, ω-oxo-FFAs are oxidized by cytosolic aldehyde dehydrogenase to carboxy-FFAs. Formed carboxy-FFAs (dicarboxylic-FAs) can be excreted into the urine or transported into mitochondria or peroxisomes, where they are metabolized via βOX. It should be noted that phytanic acid (described above) can also be oxidized via ω-oxidation [2]. Moreover, it has also been postulated that microsomal ω-hydroxylase is involved in (a) the synthesis of ω-hydroxylated arachidonic acid in the human liver and kidney, which regulates cardiovascular function (as vasoconstrictor), (b) ω-oxidation, and consequently the inactivation of leukotriene B4 (LTB4) in human leukocytes, and (c) the ω-oxidation of MCFAs and some xenobiotics [2].
References
- He, Q.; Chen, Y.; Wang, Z.; He, H.; Yu, P. Cellular Uptake, Metabolism and Sensing of Long-Chain Fatty Acids. Front. Biosci. Landmark 2023, 28, 10.
- Wanders, R.J.A.; Komen, J.; Kemp, S. Fatty acid omega-oxidation as a rescue pathway for fatty acid oxidation disorders in humans. FEBS J. 2011, 278, 182–194.
- Wanders, R.J.A.; Komen, J.; Ferdinandusse, S. Phytanic acid metabolism in health and disease. Biochim. Biophys. Acta 2011, 1811, 498–507.
- Sanders, R.-J.; Ofman, R.; Valianpour, F.; Kemp, S.; Wanders, R.J.A. Evidence for two enzymatic pathways for omega-oxidation of docosanoic acid in rat liver microsomes. J. Lipid Res. 2005, 46, 1001–1008.
- Brandt, J.M.; Djouadi, F.; Kelly, D.P. Fatty acids activate transcription of the muscle carnitine palmitoyltransferase I gene in cardiac myocytes via the peroxisome proliferator-activated receptor alpha. J. Biol. Chem. 1998, 273, 23786–23792.
- Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptors: Nuclear control of metabolism. Endocr. Rev. 1999, 20, 649–688.
- Tahri-Joutey, M.; Andreoletti, P.; Surapureddi, S.; Nasser, B.; Cherkaoui-Malki, M.; Latruffe, N. Mechanisms Mediating the Regulation of Peroxisomal Fatty Acid Beta-Oxidation by PPARα. Int. J. Mol. Sci. 2021, 22, 8969.
- Maciejewska-Skrendo, A.; Buryta, M.; Czarny, W.; Król, P.; Stastny, P.; Petr, M.; Safranow, K.; Sawczuk, M. The Polymorphisms of the Peroxisome-Proliferator Activated Receptors’ Alfa Gene Modify the Aerobic Training Induced Changes of Cholesterol and Glucose. J. Clin. Med. 2019, 8, 1043.
- Forman, B.M.; Chen, J.; Evans, R.M. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors alpha and delta. Proc. Natl. Acad. Sci. USA 1997, 94, 4312–4317.
- Ellinghaus, P.; Wolfrum, C.; Assmann, G.; Spener, F.; Seedorf, U. Phytanic acid activates the peroxisome proliferator-activated receptor α (PPARα) in sterol carrier protein 2-/sterol carrier protein x-deficient mice. J. Biol. Chem. 1999, 274, 2766–2772.
- Duszka, K.; Gregor, A.; Guillou, H.; König, J.; Wahli, W. Peroxisome Proliferator-Activated Receptors and Caloric Restriction-Common Pathways Affecting Metabolism, Health, and Longevity. Cells 2020, 9, 1708.
- Mirza, A.Z.; Althagafi, I.I.; Shamshad, H. Role of PPAR receptor in different diseases and their ligands: Physiological importance and clinical implications. Eur. J. Med. Chem. 2019, 166, 502–513.
- Muoio, D.M.; MacLean, P.S.; Lang, D.B.; Li, S.; Houmard, J.A.; Way, J.M.; Winegar, D.A.; Corton, J.C.; Dohm, G.L.; Kraus, W.E. Fatty acid homeostasis and induction of lipid regulatory genes in skeletal muscles of peroxisome proliferator-activated receptor (PPAR) alpha knock-out mice. Evidence for compensatory regulation by PPAR delta. J. Biol. Chem. 2002, 277, 26089–26097.
- de Lange, P.; Farina, P.; Moreno, M.; Ragni, M.; Lombardi, A.; Silvestri, E.; Burrone, L.; Lanni, A.; Goglia, F. Sequential changes in the signal transduction responses of skeletal muscle following food deprivation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2006, 20, 2579–2581.
- Luquet, S.; Lopez-Soriano, J.; Holst, D.; Fredenrich, A.; Melki, J.; Rassoulzadegan, M.; Grimaldi, P.A. Peroxisome proliferator-activated receptor delta controls muscle development and oxidative capability. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2003, 17, 2299–2301.
- Manickam, R.; Wahli, W. Roles of Peroxisome Proliferator-Activated Receptor β/δ in skeletal muscle physiology. Biochimie 2017, 136, 42–48.
- Neels, J.G.; Grimaldi, P.A. Physiological functions of peroxisome proliferator-activated receptor β. Physiol. Rev. 2014, 94, 795–858.
- Wang, Y.; Nakajima, T.; Gonzalez, F.J.; Tanaka, N. PPARs as Metabolic Regulators in the Liver: Lessons from Liver-Specific PPAR-Null Mice. Int. J. Mol. Sci. 2020, 21, 2061.
- Abdelmagid, S.A.; Clarke, S.E.; Nielsen, D.E.; Badawi, A.; El-Sohemy, A.; Mutch, D.M.; Ma, D.W.L. Comprehensive profiling of plasma fatty acid concentrations in young healthy Canadian adults. PLoS ONE 2015, 10, e0116195.
- Janczy, A.; Szymanski, M.; Stankiewicz, M.; Kaska, L.; Waleron, K.; Stelmanska, E.; Sledzinski, T.; Mika, A. Increased Amount of Polyunsaturated Fatty Acids in the Intestinal Contents of Patients with Morbid Obesity. Obes. Surg. 2023, 33, 1228–1236.
- Huber, A.H.; Kleinfeld, A.M. Unbound free fatty acid profiles in human plasma and the unexpected absence of unbound palmitoleate. J. Lipid Res. 2017, 58, 578–585.
- Rojek, L.; Hebanowska, A.; Stojek, M.; Jagielski, M.; Goyke, E.; Szrok-Jurga, S.; Smoczynski, M.; Swierczynski, J.; Sledzinski, T.; Adrych, K. High levels of reactive oxygen species in pancreatic necrotic fluid of patients with walled-off pancreatic necrosis. Gastroenterol. Rev. Gastroenterol. 2020, 16, 56–61.
- Jupin, M.; Michiels, P.J.; Girard, F.C.; Spraul, M.; Wijmenga, S.S. NMR identification of endogenous metabolites interacting with fatted and non-fatted human serum albumin in blood plasma: Fatty acids influence the HSA-metabolite interaction. J. Magn. Reson. 2013, 228, 81–94.
- Roden, M.; Stingl, H.; Chandramouli, V.; Schumann, W.C.; Hofer, A.; Landau, B.R.; Nowotny, P.; Waldhäusl, W.; Shulman, G.I. Effects of free fatty acid elevation on postabsorptive endogenous glucose production and gluconeogenesis in humans. Diabetes 2000, 49, 701–707.
- Pavićević, I.D.; Jovanović, V.B.; Takić, M.M.; Aćimović, J.M.; Penezić, A.Z.; Mandić, L.M. Quantification of total content of non-esterified fatty acids bound to human serum albumin. J. Pharm. Biomed. Anal. 2016, 129, 43–49.
- Schwenk, R.W.; Holloway, G.P.; Luiken, J.J.F.P.; Bonen, A.; Glatz, J.F.C. Fatty acid transport across the cell membrane: Regulation by fatty acid transporters. Prostaglandins Leukot. Essent. Fat. Acids 2010, 82, 149–154.
- De Leeuw, A.M.; Brouwer, A.; Knook, D.L. Sinusoidal endothelial cells of the liver: Fine structure and function in relation to age. J. Electron Microsc. Tech. 1990, 14, 218–236.
- Arts, T.; Reneman, R.S.; Bassingthwaighte, J.B.; van der Vusse, G.J. Modeling Fatty Acid Transfer from Artery to Cardiomyocyte. PLoS Comput. Biol. 2015, 11, e1004666.
- Glatz, J.F.C.; Nabben, M.; Luiken, J.J.F.P. CD36 (SR-B2) as master regulator of cellular fatty acid homeostasis. Curr. Opin. Lipidol. 2022, 33, 103.
- Ma, Y.; Nenkov, M.; Chen, Y.; Press, A.T.; Kaemmerer, E.; Gassler, N. Fatty acid metabolism and acyl-CoA synthetases in the liver-gut axis. World J. Hepatol. 2021, 13, 1512.
- Heden, T.D.; Franklin, M.P.; Dailey, C.; Mashek, M.T.; Chen, C.; Mashek, D.G. ACOT1 deficiency attenuates high-fat diet induced fat mass gain by increasing energy expenditure. J. Clin. Investig. 2023, 8, e160987.
- Angelini, A.; Saha, P.K.; Jain, A.; Jung, S.Y.; Mynatt, R.L.; Pi, X.; Xie, L. PHDs/CPT1B/VDAC1 axis regulates long-chain fatty acid oxidation in cardiomyocytes. Cell Rep. 2021, 37, 109767.
- Mashek, D.G.; Bornfeldt, K.E.; Coleman, R.A.; Berger, J.; Bernlohr, D.A.; Black, P.; DiRusso, C.C.; Farber, S.A.; Guo, W.; Hashimoto, N.; et al. Revised nomenclature for the mammalian long-chain acyl-CoA synthetase gene family. J. Lipid Res. 2004, 45, 1958–1961.
- Pei, Z.; Fraisl, P.; Berger, J.; Jia, Z.; Forss-Petter, S.; Watkins, P.A. Mouse very long-chain Acyl-CoA synthetase 3/fatty acid transport protein 3 catalyzes fatty acid activation but not fatty acid transport in MA-10 cells. J. Biol. Chem. 2004, 279, 54454–54462.
- Gimeno, R.E.; Ortegon, A.M.; Patel, S.; Punreddy, S.; Ge, P.; Sun, Y.; Lodish, H.F.; Stahl, A. Characterization of a heart-specific fatty acid transport protein. J. Biol. Chem. 2003, 278, 16039–16044.
- Richards, M.R.; Harp, J.D.; Ory, D.S.; Schaffer, J.E. Fatty acid transport protein 1 and long-chain acyl coenzyme A synthetase 1 interact in adipocytes. J. Lipid Res. 2006, 47, 665–672.
- Gimeno, R.E. Fatty acid transport proteins. Curr. Opin. Lipidol. 2007, 18, 271–276.
- Doege, H.; Baillie, R.A.; Ortegon, A.M.; Tsang, B.; Wu, Q.; Punreddy, S.; Hirsch, D.; Watson, N.; Gimeno, R.E.; Stahl, A. Targeted Deletion of FATP5 Reveals Multiple Functions in Liver Metabolism: Alterations in Hepatic Lipid Homeostasis. Gastroenterology 2006, 130, 1245–1258.
- Lewin, T.M.; Kim, J.H.; Granger, D.A.; Vance, J.E.; Coleman, R.A. Acyl-CoA synthetase isoforms 1, 4, and 5 are present in different subcellular membranes in rat liver and can be inhibited independently. J. Biol. Chem. 2001, 276, 24674–24679.
- Poppelreuther, M.; Rudolph, B.; Du, C.; Großmann, R.; Becker, M.; Thiele, C.; Ehehalt, R.; Füllekrug, J. The N-terminal region of acyl-CoA synthetase 3 is essential for both the localization on lipid droplets and the function in fatty acid uptake. J. Lipid Res. 2012, 53, 888–900.
- Kuwata, H.; Hara, S. Role of acyl-CoA synthetase ACSL4 in arachidonic acid metabolism. Prostaglandins Other Lipid Mediat. 2019, 144, 106363.
- Bu, S.Y.; Mashek, D.G. Hepatic long-chain acyl-CoA synthetase 5 mediates fatty acid channeling between anabolic and catabolic pathways. J. Lipid Res. 2010, 51, 3270–3280.
- Marszalek, J.R.; Kitidis, C.; DiRusso, C.C.; Lodish, H.F. Long-chain acyl-CoA synthetase 6 preferentially promotes DHA metabolism. J. Biol. Chem. 2005, 280, 10817–10826.
- Vessey, D.A.; Kelley, M.; Warren, R.S. Characterization of the CoA ligases of human liver mitochondria catalyzing the activation of short- and medium-chain fatty acids and xenobiotic carboxylic acids. Biochim. Biophys. Acta Gen. Subj. 1999, 1428, 455–462.
- Miyagawa, Y.; Mori, T.; Goto, K.; Kawahara, I.; Fujiwara-Tani, R.; Kishi, S.; Sasaki, T.; Fujii, K.; Ohmori, H.; Kuniyasu, H. Intake of medium-chain fatty acids induces myocardial oxidative stress and atrophy. Lipids Health Dis. 2018, 17, 1–7.
- Moffett, J.R.; Puthillathu, N.; Vengilote, R.; Jaworski, D.M.; Namboodiri, A.M. Acetate Revisited: A Key Biomolecule at the Nexus of Metabolism, Epigenetics and Oncogenesis—Part 1: Acetyl-CoA, Acetogenesis and Acyl-CoA Short-Chain Synthetases. Front. Physiol. 2020, 11, 580167.
- Yoshimura, Y.; Araki, A.; Maruta, H.; Takahashi, Y.; Yamashita, H. Molecular cloning of rat acss3 and characterization of mammalian propionyl-CoA synthetase in the liver mitochondrial matrix. J. Biochem. 2017, 161, 279–289.
- Montgomery, M.K.; Osborne, B.; Brown, S.H.J.; Small, L.; Mitchell, T.W.; Cooney, G.J.; Turner, N. Contrasting metabolic effects of medium-versus long-chain fatty acids in skeletal muscle. J. Lipid Res. 2013, 54, 3322–3333.
- Faye, A.; Esnous, C.; Price, N.T.; Onfray, M.A.; Girard, J.; Prip-Buus, C. Rat liver carnitine palmitoyltransferase 1 forms an oligomeric complex within the outer mitochondrial membrane. J. Biol. Chem. 2007, 282, 26908–26916.
- Lee, K.; Kerner, J.; Hoppel, C.L. Mitochondrial carnitine palmitoyltransferase 1a (CPT1a) is part of an outer membrane fatty acid transfer complex. J. Biol. Chem. 2011, 286, 25655–25662.
- Rufer, A.C.; Thoma, R.; Hennig, M. Structural insight into function and regulation of carnitine palmitoyltransferase. Cell. Mol. Life Sci. 2009, 66, 2489–2501.
- Schlaepfer, I.R.; Joshi, M. CPT1A-mediated Fat Oxidation, Mechanisms, and Therapeutic Potential. Endocrinology 2020, 161, bqz046.
- Wolfgang, M.J.; Kurama, T.; Dai, Y.; Suwa, A.; Asaumi, M.; Matsumoto, S.I.; Cha, S.H.; Shimokawa, T.; Lane, M.D. The brain-specific carnitine palmitoyltransferase-1c regulates energy homeostasis. Proc. Natl. Acad. Sci. USA 2006, 103, 7282–7287.
- Carrasco, P.; Sahún, I.; McDonald, J.; Ramírez, S.; Jacas, J.; Gratacós, E.; Sierra, A.Y.; Serra, D.; Herrero, L.; Acker-Palmer, A.; et al. Ceramide levels regulated by carnitine palmitoyltransferase 1C control dendritic spine maturation and cognition. J. Biol. Chem. 2012, 287, 21224–21232.
- Taïb, B.; Bouyakdan, K.; Hryhorczuk, C.; Rodaros, D.; Fulton, S.; Alquier, T. Glucose regulates hypothalamic long-chain fatty acid metabolism via AMP-activated kinase (AMPK) in neurons and astrocytes. J. Biol. Chem. 2013, 288, 37216–37229.
- Van Weeghel, M.; Abdurrachim, D.; Nederlof, R.; Argmann, C.A.; Houtkooper, R.H.; Hagen, J.; Nabben, M.; Denis, S.; Ciapaite, J.; Kolwicz, S.C.; et al. Increased cardiac fatty acid oxidation in a mouse model with decreased malonyl-CoA sensitivity of CPT1B. Cardiovasc. Res. 2018, 114, 1324–1334.
- Louet, J.-F.F.; Le May, C.; Pégorier, J.-P.P.; Decaux, J.-F.F.; Girard, J. Regulation of liver carnitine palmitoyltransferase I gene expression by hormones and fatty acids. Biochem. Soc. Trans. 2001, 29, 310–316.
- Bruce, C.R.; Hoy, A.J.; Turner, N.; Watt, M.J.; Allen, T.L.; Carpenter, K.; Cooney, G.J.; Febbraio, M.A.; Kraegen, E.W. Overexpression of carnitine palmitoyltransferase-1 in skeletal muscle is sufficient to enhance fatty acid oxidation and improve high-fat diet-induced insulin resistance. Diabetes 2009, 58, 550–558.
- Park, E.A.; Mynatt, R.L.; Cook, G.A.; Kashfi, K. Insulin regulates enzyme activity, malonyl-CoA sensitivity and mRNA abundance of hepatic carnitine palmitoyltransferase-I. Biochem. J. 1995, 310, 853–858.
- Faye, A.; Borthwick, K.; Esnous, C.; Price, N.T.; Gobin, S.; Jackson, V.N.; Zammit, V.A.; Girard, J.; Prip-Buus, C. Demonstration of N- and C-terminal domain intramolecular interactions in rat liver carnitine palmitoyltransferase 1 that determine its degree of malonyl-CoA sensitivity. Biochem. J. 2005, 387, 67.
- Akkaoui, M.; Cohen, I.; Esnous, C.; Lenoir, V.; Sournac, M.; Girard, J.; Prip-Buus, C. Modulation of the hepatic malonyl-CoA-carnitine palmitoyltransferase 1A partnership creates a metabolic switch allowing oxidation of de novo fatty acids. Biochem. J. 2009, 420, 429–438.
- Zhu, H.; Shi, J.; De Vries, Y.; Arvidson, D.N.; Cregg, J.M.; Woldegiorgis, G. Functional Studies of Yeast-Expressed Human Heart Muscle Carnitine Palmitoyltransferase I. Arch. Biochem. Biophys. 1997, 347, 53–61.
- Roepstorff, C.; Halberg, N.; Hillig, T.; Saha, A.K.; Ruderman, N.B.; Wojtaszewski, J.F.P.; Richter, E.A.; Kiens, B. Malonyl-CoA and carnitine in regulation of fat oxidation in human skeletal muscle during exercise. Am. J. Physiol. Endocrinol. Metab. 2005, 288, 133–142.
- Lefort, N.; Glancy, B.; Bowen, B.; Willis, W.T.; Bailowitz, Z.; De Filippis, E.A.; Brophy, C.; Meyer, C.; Højlund, K.; Yi, Z.; et al. Increased Reactive Oxygen Species Production and Lower Abundance of Complex I Subunits and Carnitine Palmitoyltransferase 1B Protein Despite Normal Mitochondrial Respiration in Insulin-Resistant Human Skeletal Muscle. Diabetes 2010, 59, 2444–2452.
- Maples, J.M.; Brault, J.J.; Witczak, C.A.; Park, S.; Hubal, M.J.; Weber, T.M.; Houmard, J.A.; Shewchuk, B.M. Differential epigenetic and transcriptional response of the skeletal muscle carnitine palmitoyltransferase 1B (CPT1B) gene to lipid exposure with obesity. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E345.
- Song, S.; Attia, R.R.; Connaughton, S.; Niesen, M.I.; Ness, G.C.; Elam, M.B.; Hori, R.T.; Cook, G.A.; Park, E.A. Peroxisome proliferator activated receptor alpha (PPARalpha) and PPAR gamma coactivator (PGC-1alpha) induce carnitine palmitoyltransferase IA (CPT-1A) via independent gene elements. Mol. Cell. Endocrinol. 2010, 325, 54–63.
- Longo, N.; Amat Di San Filippo, C.; Pasquali, M. Disorders of carnitine transport and the carnitine cycle. Am. J. Med. Genet. C Semin. Med. Genet. 2006, 142C, 77–85.
- Palmieri, F.; Scarcia, P.; Monné, M. Diseases caused by mutations in mitochondrial carrier genes SLC25: A review. Biomolecules 2020, 10, 655.
- Pochini, L.; Galluccio, M.; Scumaci, D.; Giangregorio, N.; Tonazzi, A.; Palmieri, F.; Indiveri, C. Interaction of β-lactam antibiotics with the mitochondrial carnitine/acylcarnitine transporter. Chem. Biol. Interact. 2008, 173, 187–194.
- Doulias, P.T.; Tenopoulou, M.; Greene, J.L.; Raju, K.; Ischiropoulos, H. Nitric oxide regulates mitochondrial fatty acid metabolism through reversible protein S-nitrosylation. Sci. Signal. 2013, 6, rs1.
- Tonazzi, A.; Eberini, I.; Indiveri, C. Molecular mechanism of inhibition of the mitochondrial carnitine/acylcarnitine transporter by omeprazole revealed by proteoliposome assay, mutagenesis and bioinformatics. PLoS ONE 2013, 8, e82286.
- Branco, V.; Godinho-Santos, A.; Gonçalves, J.; Lu, J.; Holmgren, A.; Carvalho, C. Mitochondrial thioredoxin reductase inhibition, selenium status, and Nrf-2 activation are determinant factors modulating the toxicity of mercury compounds. Free Radic. Biol. Med. 2014, 73, 95–105.
- Soni, M.S.; Rabaglia, M.E.; Bhatnagar, S.; Shang, J.; Ilkayeva, O.; Mynatt, R.; Zhou, Y.P.; Schadt, E.E.; Thornberry, N.A.; Muoio, D.M.; et al. Downregulation of carnitine acyl-carnitine translocase by miRNAs 132 and 212 amplifies glucose-stimulated insulin secretion. Diabetes 2014, 63, 3805–3814.
- Tonazzi, A.; Giangregorio, N.; Console, L.; Scalise, M.; La Russa, D.; Notaristefano, C.; Brunelli, E.; Barca, D.; Indiveri, C. Mitochondrial Carnitine/Acylcarnitine Transporter, a Novel Target of Mercury Toxicity. Chem. Res. Toxicol. 2015, 28, 1015–1022.
- Giangregorio, N.; Tonazzi, A.; Console, L.; Lorusso, I.; De Palma, A.; Indiveri, C. The mitochondrial carnitine/acylcarnitine carrier is regulated by hydrogen sulfide via interaction with C136 and C155. Biochim. Biophys. Acta 2016, 1860, 20–27.
- Giangregorio, N.; Tonazzi, A.; Console, L.; Indiveri, C. Post-translational modification by acetylation regulates the mitochondrial carnitine/acylcarnitine transport protein. Mol. Cell. Biochem. 2017, 426, 65–73.
- Huizing, M.; Ruitenbeek, W.; Van den Heuvel, L.P.; Dolce, V.; Iacobazzi, V.; Smeitink, J.A.M.; Palmieri, F.; Frans Trijbels, J.M. Human mitochondrial transmembrane metabolite carriers: Tissue distribution and its implication for mitochondrial disorders. J. Bioenerg. Biomembr. 1998, 30, 277–284.
- Iacobazzi, V.; Convertini, P.; Infantino, V.; Scarcia, P.; Todisco, S.; Palmieri, F. Statins, fibrates and retinoic acid upregulate mitochondrial acylcarnitine carrier gene expression. Biochem. Biophys. Res. Commun. 2009, 388, 643–647.
- Iacobazzi, V.; Infantino, V.; Palmieri, F. Transcriptional regulation of the mitochondrial citrate and carnitine/acylcarnitine transporters: Two genes involved in fatty acid biosynthesis and β-oxidation. Biology 2013, 2, 284–303.
- Lara, C.; Nicola, G.; Saverio, C.; Isabella, B.; Marino, P.; Cesare, I.; Giovanna, I.; Sabrina, C.; Annamaria, T. Human mitochondrial carnitine acylcarnitine carrier: Molecular target of dietary bioactive polyphenols from sweet cherry (Prunus avium L.). Chem. Biol. Interact. 2019, 307, 179–185.
- Houten, S.M.; Violante, S.; Ventura, F.V.; Wanders, R.J.A. The Biochemistry and Physiology of Mitochondrial Fatty Acid β-Oxidation and Its Genetic Disorders. Annu. Rev. Physiol. 2016, 78, 23–44.
- Adeva-Andany, M.M.; Carneiro-Freire, N.; Seco-Filgueira, M.; Fernández-Fernández, C.; Mouriño-Bayolo, D. Mitochondrial β-oxidation of saturated fatty acids in humans. Mitochondrion 2019, 46, 73–90.
- Czumaj, A.; Szrok-Jurga, S.; Hebanowska, A.; Turyn, J.; Swierczynski, J.; Sledzinski, T.; Stelmanska, E. The pathophysiological role of CoA. Int. J. Mol. Sci. 2020, 21, 9057.
- Aoyama, T.; Souri, M.; Ushikubo, S.; Kamijo, T.; Yamaguchi, S.; Kelley, R.I.; Rhead, W.J.; Uetake, K.; Tanaka, K.; Hashimoto, T. Purification of human very-long-chain acyl-coenzyme A dehydrogenase and characterization of its deficiency in seven patients. J. Clin. Investig. 1995, 95, 2465–2473.
- Sinsheimer, A.; Mohsen, A.W.; Bloom, K.; Karunanidhi, A.; Bharathi, S.; Wu, Y.L.; Schiff, M.; Wang, Y.; Goetzman, E.S.; Ghaloul-Gonzalez, L.; et al. Development and Characterization of a Mouse Model for Acad9 deficiency. Mol. Genet. Metab. 2021, 134, 156.
- Goetzman, E.S.; Alcorn, J.F.; Bharathi, S.S.; Uppala, R.; McHugh, K.J.; Kosmider, B.; Chen, R.; Zuo, Y.Y.; Beck, M.E.; McKinney, R.W.; et al. Long-chain Acyl-CoA dehydrogenase deficiency as a cause of pulmonary surfactant dysfunction. J. Biol. Chem. 2014, 289, 10668–10679.
- Horowitz, J.F.; Klein, S. Lipid metabolism during endurance exercise. Am. J. Clin. Nutr. 2000, 72, 558S–563S.
- Nochi, Z.; Olsen, R.K.J.; Gregersen, N. Short-chain acyl-CoA dehydrogenase deficiency: From gene to cell pathology and possible disease mechanisms. J. Inherit. Metab. Dis. 2017, 40, 641–655.
- Xia, C.; Lou, B.; Fu, Z.; Mohsen, A.W.; Shen, A.L.; Vockley, J.; Kim, J.J.P. Molecular mechanism of interactions between ACAD9 and binding partners in mitochondrial respiratory complex I assembly. iScience 2021, 24, 103153.
- Beck, M.E.; Zhang, Y.; Bharathi, S.S.; Kosmider, B.; Bahmed, K.; Dahmer, M.K.; Nogee, L.M.; Goetzman, E.S. The common K333Q polymorphism in long-chain acyl-CoA dehydrogenase (LCAD) reduces enzyme stability and function. Mol. Genet. Metab. 2020, 131, 83–89.
- Henriques, B.J.; Katrine Jentoft Olsen, R.; Gomes, C.M.; Bross, P. Electron transfer flavoprotein and its role in mitochondrial energy metabolism in health and disease. Gene 2021, 776, 145407.
- Salerno, K.M.; Domenico, J.; Le, N.Q.; Stiles, C.D.; Solov’Yov, I.A.; Martino, C.F. Long-Time Oxygen Localization in Electron Transfer Flavoprotein. J. Chem. Inf. Model. 2022, 62, 4191–4199.
- Fould, B.; Garlatti, V.; Neumann, E.; Fenel, D.; Gaboriaud, C.; Arlaud, G.J. Structural and functional characterization of the recombinant human mitochondrial trifunctional protein. Biochemistry 2010, 49, 8608–8617.
- Xia, C.; Fu, Z.; Battaile, K.P.; Kim, J.J.P. Crystal structure of human mitochondrial trifunctional protein, a fatty acid β-oxidation metabolon. Proc. Natl. Acad. Sci. USA 2019, 116, 6069–6074.
- Dagher, R.; Massie, R.; Gentil, B.J. MTP deficiency caused by HADHB mutations: Pathophysiology and clinical manifestations. Mol. Genet. Metab. 2021, 133, 1–7.
- Zhang, D.; Yu, W.; Geisbrecht, B.V.; Gould, S.J.; Sprecher, H.; Schulz, H. Functional characterization of Delta3,Delta2-enoyl-CoA isomerases from rat liver. J. Biol. Chem. 2002, 277, 9127–9132.
- Van Weeghel, M.; Te Brinke, H.; Van Lenthe, H.; Kulik, W.; Minkler, P.E.; Stoll, M.S.K.; Sass, J.O.; Janssen, U.; Stoffel, W.; Schwab, K.O.; et al. Functional redundancy of mitochondrial enoyl-CoA isomerases in the oxidation of unsaturated fatty acids. FASEB J. 2012, 26, 4316–4326.
- Onwukwe, G.U.; Kursula, P.; Koski, M.K.; Schmitz, W.; Wierenga, R.K. Human Δ3,Δ2-enoyl-CoA isomerase, type 2: A structural enzymology study on the catalytic role of its ACBP domain and helix-10. FEBS J. 2015, 282, 746–768.
- Horowitz, J.F.; Leone, T.C.; Feng, W.; Kelly, D.P.; Klein, S. Effect of endurance training on lipid metabolism in women: A potential role for PPARalpha in the metabolic response to training. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E348–E355.
- Toogood, H.S.; Van Thiel, A.; Basran, J.; Sutcliffe, M.J.; Scrutton, N.S.; Leys, D. Extensive domain motion and electron transfer in the human electron transferring flavoprotein.medium chain Acyl-CoA dehydrogenase complex. J. Biol. Chem. 2004, 279, 32904–32912.
- Jones, P.M.; Butt, Y.; Messmer, B.; Boriak, R.; Bennett, M.J. Medium-chain fatty acids undergo elongation before beta-oxidation in fibroblasts. Biochem. Biophys. Res. Commun. 2006, 346, 193–197.
- Houten, S.M.; Wanders, R.J.A.A. A general introduction to the biochemistry of mitochondrial fatty acid β-oxidation. J. Inherit. Metab. Dis. 2010, 33, 469–477.
- Vanhove, G.; Veldhoven, P.P.V.; Eyssen, H.J.; Mannaerts, G.P. Mitochondrial short-chain acyl-CoA dehydrogenase of human liver and kidney can function as an oxidase. Biochem. J. 1993, 292 Pt 1, 23–30.
- Corydon, T.J.; Bross, P.; Jensen, T.G.; Corydon, M.J.; Lund, T.B.; Jensen, U.B.; Kim, J.J.P.; Gregersen, N.; Bolund, L. Rapid degradation of short-chain acyl-CoA dehydrogenase variants with temperature-sensitive folding defects occurs after import into mitochondria. J. Biol. Chem. 1998, 273, 13065–13071.
- Kanazawa, M.; Ohtake, A.; Abe, H.; Yamamoto, S.; Satoh, Y.; Takayanagi, M.; Niimi, H.; Mori, M.; Hashimoto, T. Molecular cloning and sequence analysis of the cDNA for human mitochondrial short-chain enoyl-CoA hydratase. Enzyme Protein 1993, 47, 9–13.
- Nakagawa, J.; Waldner, H.; Meyer-Monard, S.; Hofsteenge, J.; Jenö, P.; Moroni, C. AUH, a gene encoding an AU-specific RNA binding protein with intrinsic enoyl-CoA hydratase activity. Proc. Natl. Acad. Sci. USA 1995, 92, 2051–2055.
- Vredendaal, P.J.C.M.; Van Den Berg, I.E.T.; Malingré, H.E.M.; Stroobants, A.K.; OldeWeghuis, D.E.M.; Berger, R. Human short-chain L-3-hydroxyacyl-CoA dehydrogenase: Cloning and characterization of the coding sequence. Biochem. Biophys. Res. Commun. 1996, 223, 718–723.
- Mannaerts, G.P.; Debeer, L.J.; Thomas, J.; De Schepper, P.J. Mitochondrial and peroxisomal fatty acid oxidation in liver homogenates and isolated hepatocytes from control and clofibrate-treated rats. J. Biol. Chem. 1979, 254, 4585–4595.
- Lazarow, P.B.; De Duve, C. A fatty acyl-CoA oxidizing system in rat liver peroxisomes; enhancement by clofibrate, a hypolipidemic drug. Proc. Natl. Acad. Sci. USA 1976, 73, 2043–2046.
- Kleiboeker, B.; Lodhi, I.J. Peroxisomal regulation of energy homeostasis: Effect on obesity and related metabolic disorders. Mol. Metab. 2022, 65, 101577.
- Vilarinho, S.; Sari, S.; Mazzacuva, F.; Bilgüvar, K.; Esendagli-Yilmaz, G.; Jain, D.; Akyol, G.; Dalgiç, B.; Günel, M.; Clayton, P.T.; et al. ACOX2 deficiency: A disorder of bile acid synthesis with transaminase elevation, liver fibrosis, ataxia, and cognitive impairment. Proc. Natl. Acad. Sci. USA 2016, 113, 11289–11293.
- Ferdinandusse, S.; Denis, S.; van Roermund, C.W.T.; Preece, M.A.; Koster, J.; Ebberink, M.S.; Waterham, H.R.; Wanders, R.J.A. A novel case of ACOX2 deficiency leads to recognition of a third human peroxisomal acyl-CoA oxidase. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 952–958.
- Wanders, R.J.A.; Baes, M.; Ribeiro, D.; Ferdinandusse, S.; Waterham, H.R. The physiological functions of human peroxisomes. Physiol. Rev. 2023, 103, 957–1024.
- Westin, M.A.K.; Hunt, M.C.; Alexson, S.E.H. Short- and medium-chain carnitine acyltransferases and acyl-CoA thioesterases in mouse provide complementary systems for transport of beta-oxidation products out of peroxisomes. Cell. Mol. Life Sci. 2008, 65, 982–990.
- Tawbeh, A.; Gondcaille, C.; Trompier, D.; Savary, S. Peroxisomal ABC Transporters: An Update. Int. J. Mol. Sci. 2021, 22, 6093.
- Wang, M.; Wang, K.; Liao, X.; Hu, H.; Chen, L.; Meng, L.; Gao, W.; Li, Q. Carnitine Palmitoyltransferase System: A New Target for Anti-Inflammatory and Anticancer Therapy? Front. Pharmacol. 2021, 12, 76058.
- Kawaguchi, K.; Morita, M. ABC Transporter Subfamily D: Distinct Differences in Behavior between ABCD1-3 and ABCD4 in Subcellular Localization, Function, and Human Disease. Biomed Res. Int. 2016, 2016, 1–11.
- Wang, Y.; Palmfeldt, J.; Gregersen, N.; Makhov, A.M.; Conway, J.F.; Wang, M.; McCalley, S.P.; Basu, S.; Alharbi, H.S.; Croix, C. Mitochondrial fatty acid oxidation and the electron transport chain comprise a multifunctional mitochondrial protein complex. J. Biol. Chem. 2019, 294, 12380–12391.
- Roca-Saavedra, P.; Mariño-Lorenzo, P.; Miranda, J.M.; Porto-Arias, J.J.; Lamas, A.; Vazquez, B.I.; Franco, C.M.; Cepeda, A. Phytanic acid consumption and human health, risks, benefits and future trends: A review. Food Chem. 2017, 221, 237–247.
- Steinberg, D.; Vroom, F.Q.; Engel, W.K.; Cammermeyer, J.; Mize, C.E.; Avigan, J. Refsum’s disease--a recently characterized lipidosis involving the nervous system. Combined clinical staff conference at the National Institutes of Health. Ann. Intern. Med. 1967, 66, 365–395.
- Durrett, T.P.; Welti, R. The tail of chlorophyll: Fates for phytol. J. Biol. Chem. 2021, 296, 100802.
- Wills, A.J.; Manning, N.J.; Reilly, M.M. Refsum’s disease. QJM 2001, 94, 403–406.
- Krauß, S.; Vetter, W. Phytol and Phytyl Fatty Acid Esters: Occurrence, Concentrations, and Relevance. Eur. J. Lipid Sci. Technol. 2018, 120, 1700387.
- Wanders, R.J.A.; Vreken, P.; Ferdinandusse, S.; Jansen, G.A.; Waterham, H.R.; van Roermund, C.W.T.; Van Grunsven, E.G. Peroxisomal fatty acid alpha- and beta-oxidation in humans: Enzymology, peroxisomal metabolite transporters and peroxisomal diseases. Biochem. Soc. Trans. 2001, 29, 250.
- Wanders, R.J.A.; Komen, J.C. Peroxisomes, Refsum’s disease and the alpha- and omega-oxidation of phytanic acid. Biochem. Soc. Trans. 2007, 35, 865–869.
- Chen, M.H.; Raffield, L.M.; Mousas, A.; Sakaue, S.; Huffman, J.E.; Moscati, A.; Trivedi, B.; Jiang, T.; Akbari, P.; Vuckovic, D.; et al. Trans-ethnic and Ancestry-Specific Blood-Cell Genetics in 746,667 Individuals from 5 Global Populations. Cell 2020, 182, 1198–1213.
- Goldfischer, S.; Johnson, A.B.; Essner, E.; Moore, C.; Ritch, R.H. Peroxisomal abnormalities in metabolic diseases. J. Histochem. Cytochem. 1973, 21, 972–977.
- Monnens, L.; Bakkeren, J.; Parmentier, G.; Janssen, G.; van Haelst, U.; Trijbels, F.; Eyssen, H. Disturbances in bile acid metabolism of infants with the Zellweger (cerebro-hepato-renal) syndrome. Eur. J. Pediatr. 1980, 133, 31–35.
- Cheillan, D. Zellweger Syndrome Disorders: From Severe Neonatal Disease to Atypical Adult Presentation. Adv. Exp. Med. Biol. 2020, 1299, 71–80.
- Alam, A.; Locher, K.P. Structure and Mechanism of Human ABC Transporters. Annu. Rev. Biophys. 2023, 52, 275–300.
- Chen, Z.-P.; Xu, D.; Wang, L.; Mao, Y.-X.; Li, Y.; Cheng, M.-T.; Zhou, C.-Z.; Hou, W.-T.; Chen, Y. Structural basis of substrate recognition and translocation by human very long-chain fatty acid transporter ABCD1. Nat. Commun. 2022, 13, 3299.
- van Roermund, C.W.T.; IJlst, L.; Wagemans, T.; Wanders, R.J.A.; Waterham, H.R. A role for the human peroxisomal half-transporter ABCD3 in the oxidation of dicarboxylic acids. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2014, 1841, 563–568.
- Kersten, S. Integrated physiology and systems biology of PPARα. Mol. Metab. 2014, 3, 354–371.
- Fourcade, S.; Savary, S.; Albet, S.; Gauthé, D.; Gondcaille, C.; Pineau, T.; Bellenger, J.; Bentejac, M.; Holzinger, A.; Berger, J.; et al. Fibrate induction of the adrenoleukodystrophy-related gene (ABCD2): Promoter analysis and role of the peroxisome proliferator-activated receptor PPARα. Eur. J. Biochem. 2001, 268, 3490–3500.
- Leclercq, S.; Skrzypski, J.; Courvoisier, A.; Gondcaille, C.; Bonnetain, F.; André, A.; Chardigny, J.-M.; Bellenger, S.; Bellenger, J.; Narce, M.; et al. Effect of dietary polyunsaturated fatty acids on the expression of peroxisomal ABC transporters. Biochimie 2008, 90, 1602–1607.
- Hayashi, H.; Takahata, S. Role of peroxisomal fatty acyl-CoA beta-oxidation in phospholipid biosynthesis. Arch. Biochem. Biophys. 1991, 284, 326–331.
- Hayashi, H.; Oohashi, M. Incorporation of acetyl-CoA generated from peroxisomal beta-oxidation into ethanolamine plasmalogen of rat liver. Biochim. Biophys. Acta 1995, 1254, 319–325.
- Zhang, X.; Wang, Y.; Yao, H.; Deng, S.; Gao, T.; Shang, L.; Chen, X.; Cui, X.; Zeng, J. Peroxisomal β-oxidation stimulates cholesterol biosynthesis in the liver in diabetic mice. J. Biol. Chem. 2022, 298, 101572.
- Mariño, G.; Pietrocola, F.; Eisenberg, T.; Kong, Y.; Malik, S.A.; Andryushkova, A.; Schroeder, S.; Pendl, T.; Harger, A.; Niso-Santano, M.; et al. Regulation of Autophagy by Cytosolic Acetyl-Coenzyme A. Mol. Cell 2014, 53, 710–725.
- Schulze, R.J.; Sathyanarayan, A.; Mashek, D.G. Breaking fat: The regulation and mechanisms of lipophagy. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1178–1187.
- He, A.; Chen, X.; Tan, M.; Chen, Y.; Lu, D.; Zhang, X.; Dean, J.M.; Razani, B.; Lodhi, I.J. Acetyl-CoA Derived from Hepatic Peroxisomal β-Oxidation Inhibits Autophagy and Promotes Steatosis via mTORC1 Activation. Mol. Cell 2020, 79, 30.
- Chen, X.; Shang, L.; Deng, S.; Li, P.; Chen, K.; Gao, T.; Zhang, X.; Chen, Z.; Zeng, J. Peroxisomal oxidation of erucic acid suppresses mitochondrial fatty acid oxidation by stimulating malonyl-CoA formation in the rat liver. J. Biol. Chem. 2020, 295, 10168–10179.
- Fransen, M.; Lismont, C. Redox Signaling from and to Peroxisomes: Progress, Challenges, and Prospects. Antioxid. Redox Signal. 2019, 30, 95–112.
- Terlecky, S.R.; Koepke, J.I.; Walton, P.A. Peroxisomes and aging. Biochim. Biophys. Acta 2006, 1763, 1749–1754.
- Lismont, C.; Nordgren, M.; Van Veldhoven, P.P.; Fransen, M. Redox interplay between mitochondria and peroxisomes. Front. Cell Dev. Biol. 2015, 3, 35.
- Vallejo, M.J.; Salazar, L.; Grijalva, M. Oxidative stress modulation and ROS-mediated toxicity in cancer: A review on in vitro models for plant-derived compounds. Oxid. Med. Cell. Longev. 2017, 2017, 1–9.
- Mahaseth, T.; Kuzminov, A. Potentiation of hydrogen peroxide toxicity: From catalase inhibition to stable DNA-iron complexes. Mutat. Res. Rev. Mutat. Res. 2017, 773, 274–281.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.6K
Revisions:
2 times
(View History)
Update Date:
20 Oct 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No