Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Giacomo Maria Bacci | -- | 3985 | 2023-10-18 15:00:18 | | | |
| 2 | Alfred Zheng | Meta information modification | 3985 | 2023-10-19 05:07:33 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Becherucci, V.; Bacci, G.M.; Marziali, E.; Sodi, A.; Bambi, F.; Caputo, R. Pathomolecular Mechanisms of Retinitis Pigmentosa. Encyclopedia. Available online: https://encyclopedia.pub/entry/50470 (accessed on 24 July 2026).
Becherucci V, Bacci GM, Marziali E, Sodi A, Bambi F, Caputo R. Pathomolecular Mechanisms of Retinitis Pigmentosa. Encyclopedia. Available at: https://encyclopedia.pub/entry/50470. Accessed July 24, 2026.
Becherucci, Valentina, Giacomo Maria Bacci, Elisa Marziali, Andrea Sodi, Franco Bambi, Roberto Caputo. "Pathomolecular Mechanisms of Retinitis Pigmentosa" Encyclopedia, https://encyclopedia.pub/entry/50470 (accessed July 24, 2026).
Becherucci, V., Bacci, G.M., Marziali, E., Sodi, A., Bambi, F., & Caputo, R. (2023, October 18). Pathomolecular Mechanisms of Retinitis Pigmentosa. In Encyclopedia. https://encyclopedia.pub/entry/50470
Becherucci, Valentina, et al. "Pathomolecular Mechanisms of Retinitis Pigmentosa." Encyclopedia. Web. 18 October, 2023.
Copy Citation
Retinitis pigmentosa, defined more properly as cone–rod dystrophy, is a paradigm of inherited diffuse retinal dystrophies, one of the rare diseases with the highest prevalence in the worldwide population and one of the main causes of low vision in the pediatric and elderly age groups. Generally speaking, the pathomolecular mechanisms of RP involve primarily genetic mutations that disrupt the normal functioning of the retina and the retinal pigment epithelium through specific and common pathways.
inherited retinal dystrophies
retinitis pigmentosa
1. Introduction
Retinitis pigmentosa (RP) is a relatively common inherited retinal disorder, with an estimated worldwide mean prevalence of 1:4000 people. It consists of progressive retinal degeneration due to the loss of photoreceptors, leading to severe visual impairment in later stages. RP may be inherited in an autosomal-dominant, autosomal-recessive, or X-linked manner. Some digenic and mitochondrial forms have also been described. Actual RP prevalence is related to the geographic location of the reported study and, in consequence of this, it can vary between 1:750 and 1:9000 individuals [1]. Clinically, the fundus examination usually shows diffuse dystrophy of the retinal pigment epithelium (RPE), waxy optic disc pallor, vessel narrowing, and bone spicule pigmentation. However, atypical forms with an unusual clinical picture may be found in clinical practice. Diagnosis may be easy in typical cases, but it may be challenging in atypical presentations, as the disease may present overlapping features with different retinal disorders. Additionally, RP is associated with an increased risk of other ocular complications, such as cataracts and cystoid macular edema (CME), which may cause additional visual disturbances [2][3][4]. The pathophysiology of RP, with such complications that may affect vision, often leads to significant visual impairment at a young age, which can also have an impact on patients’ physical and mental well-being. Researchers have identified mutations in over 100 genes that contribute to developing non-syndromic RP. Syndromic RP is even more complex due to molecular pathways acting in multiple organs. In the past, RP was thought to be untreatable, but recent medical advancements, such as genetic therapies, offer promising possibilities for slowing down or stopping the degeneration of photoreceptors and potentially even restoring some level of visual function [5]. Due to our enhanced comprehension of the cellular mechanisms and genetic factors associated with RP, as well as the eye’s immune-privileged nature, gene therapy has emerged as a highly promising treatment option for RP [6]. Different studies on animal models have shown the potential benefits of gene therapy for RPE65-associated inherited retinal diseases. As a result, human clinical trials for gene therapy have been initiated, aiming to explore the potential benefits of this novel treatment approach [7][8][9][10]. The positive results in terms of both the safety profiles and clinical endpoints in these trials have led to the approval of Voretigene Neparvovec as the first FDA-approved gene therapy for patients with RPE65-associated retinal dystrophies, now commercially available with the name of Luxturna® [11].
2. Pathomolecular Mechanisms of Retinitis Pigmentosa
Generally speaking, the pathomolecular mechanisms of RP involve primarily genetic mutations that disrupt the normal functioning of the retina and the retinal pigment epithelium [12] through specific and common pathways.
These genes are involved in different cellular processes within the retina, including phototransduction, visual cycle, photoreceptor structure, and the maintenance of retinal integrity. The specific genes and mutations define the precise mechanisms behind RP in individual cases [13]. Nevertheless, since the genes activate different pathways and multiple mechanisms often converge to a more unspecific process, for better clarity the research first describe the major genes and their roles that are most commonly involved in the pathogenesis of RP.
2.1. Major Genes Involved in RP
2.1.1. Rhodopsin (RHO)
Mutations in the RHO gene are a common cause of autosomal dominant retinitis pigmentosa (adRP) [14]. Rhodopsin is a protein found in the rod photoreceptor cells of the retina, and it plays a critical role in the phototransduction pathway, which converts light signals into electrical signals that can be interpreted by the brain [15]. The disruption and degeneration of rod photoreceptor cells are caused by a variety of RHO gene mutations that damage the structure or function of rhodopsin. Over 150 types of mutations of the RHO gene have been described [16][17]. Most cases present point mutations that determine the substitution of an amino acid, with consequent alteration of the protein’s structure or function. In addition to these mutations, others involving abnormalities in the protein’s folding or trafficking have also been described. Mutations in the rhodopsin gene that lead to the development of autosomal dominant forms of retinitis pigmentosa are divided into three different classes [18], distinguished by the dysfunction of rhodopsin and the nature of its accumulation in cell culture. In class 1 mutations, the photopigment remains functional, and its bond with 11-cis-retinal remains intact, while the accumulation of protein in embryonic cell culture happens specifically on the cytoplasmic membrane. Class 2 mutations cause damage to the composition of the photopigment and the buildup of faulty protein in the endoplasmic reticulum. Class 3 encompasses mutations that result in the production of hyperphosphorylated rhodopsin, which exhibits a strong association with arrestin [19]. The consequent rhodopsin–arrestin complex disturbs the structure of the endosomal compartment and impairs endocyte function. All these mutations have a strong impact on rhodopsin function as they can impair the light-sensitive ability of the protein or disrupt its signaling cascade within the photoreceptor cells [20]. These abnormalities can lead to defects in phototransduction, reduced sensitivity to light, and eventually, to the death of rod photoreceptor cells.
2.1.2. Peripherin/RDS (PRPH2)
Mutations in the PRPH2 gene can cause autosomal dominant RP. The peripherin/RDS protein is involved in the structural integrity and function of photoreceptor outer segments. Specifically, PRPH2 is a transmembrane protein that is mainly expressed in rod and cone photoreceptor cells. It plays a crucial role in the structural integrity and organization of the photoreceptor outer segments, which are responsible for capturing and processing light signals [21]. Mutations occurring in the PRPH2 gene can give rise to diverse structural anomalies in the peripherin 2 protein [22]. These abnormalities can impact crucial aspects, such as protein folding, stability, and interactions with other proteins. The resultant disruption in the peripherin 2 function can hinder the formation of photoreceptor outer segments, consequently compromising the normal functionality of the photoreceptor cells [23]. One common type of mutation in PRPH2 is the missense mutation, which can affect the protein’s folding, stability, and ability to interact with other proteins in the photoreceptor cells [24]. Another type of common mutation is the frameshift mutation, mainly associated with anomalies in the protein’s structure and function [22]. Frameshift mutations often result in a truncated or non-functional peripherin/RDS protein.
Defective peripherin/RDS protein can lead to mislocalization [25], aggregation, or degradation of the protein, affecting the integrity and function of the outer segments. This disruption ultimately results in the progressive degeneration of the photoreceptor cells and the characteristic symptoms of retinitis pigmentosa, such as night blindness, peripheral vision loss, and eventually, central vision impairment.
2.1.3. Cyclic Nucleotide-Gated (CNG) Channels
Mutations in genes encoding the CNG channels, such as CNGA1 and CNGB1, are associated with autosomal recessive RP [26]. These channels are located in the outer segment of rod and cone photoreceptor cells, and they are involved in the regulation of ion influx in response to light stimulation. They are responsible for the regulation of intracellular calcium and sodium ions, which are essential for phototransduction (the process by which light signals are converted into electrical signals in the retina thanks to hyperpolarization/depolarization phenomena) [27]. Mutations in genes encoding CNG channels can impair the normal function of these channels, disrupting the phototransduction process and leading to RP [28]. Mutations in the CNGB1 and CNGA1 genes, which encode subunits of the CNG channels, have been associated with autosomal recessive RP [29]. Mutations in the GNAT2 gene, which encodes the transducin alpha-subunit involved in CNG channel regulation, have also been linked to autosomal dominant RP. Impaired CNG channel function leads to abnormalities in the phototransduction process, where the conversion of light stimuli into electrical signals is disrupted [30]. This alteration can result in reduced sensitivity to light, decreased visual acuity, and progressive vision loss, which are characteristic symptoms of RP. Additionally, dysfunctional CNG channels can lead to cellular stress and oxidative damage, and ultimately trigger photoreceptor cell death [31]. The loss of photoreceptor cells further contributes to the degeneration of the retina and the progression of RP.
2.1.4. Retinal Pigment Epithelium-Specific 65 kDa Protein (RPE65)
Mutations in the RPE65 gene are associated with autosomal recessive RP forms [32]. The RPE65 gene encodes a protein called retinoid isomerohydrolase, which is primarily expressed in the retinal pigment epithelium (RPE) cells. RPE65 is involved in the visual cycle, a process that regenerates the visual pigment rhodopsin in photoreceptor cells. It plays a crucial role in converting all-trans-retinol to 11-cis-retinal, which is essential for the proper functioning of photoreceptor cells [33]. Mutations in the RPE65 gene result in a loss or dysfunction of the RPE65 protein, disrupting the visual cycle and impairing the regeneration of 11-cis-retinal [34]. As a consequence, there is a decreased availability of 11-cis-retinal, leading to compromised phototransduction and eventual degeneration of photoreceptor cells [32][35]. Mutations in the RPE65 gene are also associated with a severe form of retinitis pigmentosa known as Leber congenital amaurosis (LCA) or severe early childhood-onset retinal dystrophy (SECORD).
2.1.5. Retinitis Pigmentosa GTPase Regulator (RPGR)
Mutations in the RPGR gene are a major cause of X-linked RP (XLRP), which primarily affects males, as the RPGR gene is located on the X chromosome and is involved in the structure and function of the photoreceptor connecting cilium [36][37]. The RPGR protein is indeed predominantly localized in the connecting cilium and outer segment of photoreceptor cells in the retina. These cellular structures play critical roles in the phototransduction cascade and the maintenance of normal vision [38]. Mutations in the RPGR gene can affect the normal function of the RPGR protein, leading to retinal degeneration in XLRP [39]. The specific pathogenic mechanisms underlying RPGR-related retinal degeneration are not fully understood. However, it is believed that the mutations result in impaired ciliary transport, altered protein–protein interactions, or disrupted signaling pathways, ultimately leading to photoreceptor cell death and vision loss [36][40].
2.1.6. Cone–Rod Homeobox Protein (CRX)
Mutations in the CRX gene are associated with the autosomal-dominant RP form [41]. The cone–rod homeobox protein is a transcription factor that plays a crucial role in the development and function of photoreceptor cells in the retina. CRX is primarily expressed in the cone and rod photoreceptor cells of the retina, where it regulates the expression of genes involved in photoreceptor development, differentiation, and maintenance [42]. CRX is essential for the proper formation and function of these specialized cells, which are responsible for capturing and processing light signals [43]. CRX regulates the expression of genes encoding various photoreceptor-specific proteins, including opsins (light-sensitive pigments), transducin, and other important components of the phototransduction pathway. It helps establish the unique characteristics and functions of cone and rod photoreceptor cells, ensuring their proper light-sensitive abilities [44]. Mutations in the CRX gene can disrupt the normal function of the CRX protein, leading to impaired development and function of photoreceptor cells [43]. This can result in the degeneration of cones and rods with different specific effects depending on the kind of CRX mutation, resulting in isolated cone dysfunction to more generalized cone-rod dystrophy [45].
2.1.7. Usher Syndrome Genes
It has been demonstrated that some forms of RP are associated with Usher syndrome [46], which involves both hearing loss and vision impairment. Genes associated with Usher syndrome, such as USH2A, MYO7A, CLRN1, and CDH23, can cause RP, in addition to other symptoms. It is a heterogeneous condition with several genes implicated in its development. The most commonly associated genes with Usher syndrome and RP include the following:
Mutations in the MYO7A gene account for the majority of Usher syndrome type 1 (USH1) cases [47]. MYO7A encodes the protein myosin VIIA, which is involved in the structure and function of hair cells in the inner ear and the development and maintenance of photoreceptor cells in the retina. It plays an important role in the renewal of the outer photoreceptor discs, in the distribution and migration of retinal pigment epithelium melanosomes and phagosomes, and in the regulation of opsin transport in retinal photoreceptors [48]. Mutations in the USH1C gene are also associated with Usher syndrome type 1. The USH1C gene encodes harmony, a scaffolding protein involved in the organization of hair cell stereocilia and the synaptic connections in the retina [47].
Mutations in the USH2A gene are the most common cause of Usher syndrome type 2 (USH2). The USH2A gene encodes usherin, a protein involved in the maintenance of the structure and function of the photoreceptor cells and the hair cells of the inner ear [49].
Mutations in the GPR98 gene, also known as adhesion G protein-coupled receptor V1 (ADGRV1) are associated with Usher syndrome type 2. The GPR98 gene encodes the protein G protein-coupled receptor 98, which is involved in the development and function of sensory cells in the inner ear and the retina [50].
Mutations in the CLRN1 gene are associated with Usher syndrome type 3. The CLRN1 gene encodes clarin-1, a protein found in the hair cells of the inner ear and the photoreceptor cells of the retina [51], which seems to play an important role in the development and homeostasis as a regulatory element for the synapses within the retina.
2.2. Mechanisms Involved in RP
As discussed above, the pathogenesis of RP involves a complex interplay of genetic, biochemical [52], and cellular mechanisms that ultimately lead to the degeneration of photoreceptor cells in the retina [53]. Before proceeding, people think that is noteworthy to underline the extreme genetic heterogeneity of RP: the increasing number of papers describing novel mutations and novel genes implicated in the pathogenesis of RP represent evidence of the work that has yet to be carried out to define a proper genotype–phenotype relationship to predict the role of such variants in determining specific isolated or syndromic forms. For instance, a recent paper by Smirnov et al. [54] showed the wide range of presentation of the potentially life-threatening CLN3 mutations: the authors found that a genotype–phenotype relationship could be extremely useful to predict isolated retinal dystrophy from a devastating early-onset disorder with neurological involvement.
Although the exact pathogenesis can change depending on the specific genetic mutations that act as initial triggers, in the next section it describe an overview of more general and common processes that contribute to the development and progression of RP, which are also briefly illustrated in Figure 1.
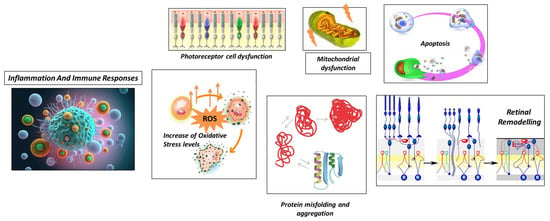
Figure 1. Schematic representation of general processes underlying retinitis pigmentosa.
2.2.1. Photoreceptor Cell Dysfunction
The first pathogenetic mechanism is photoreceptor cell dysfunction. RP primarily affects the photoreceptor cells in the retina, where mutations in the genes involved in the visual cycle, phototransduction, and photoreceptor structure can lead to impaired functioning of these cells, affecting the structure and function of photoreceptor proteins [55].
2.2.2. Protein Misfolding and Aggregation
Another mechanism that contributes to the development of retinitis pigmentosa is represented by protein misfolding and aggregation [56]. In this case, mutations in specific genes can lead to the production of misfolded or unstable proteins. Abnormal proteins can accumulate within photoreceptor cells and trigger cellular stress responses, including endoplasmic reticulum stress and unfolded protein response. The accumulation of misfolded proteins and the activation of stress pathways can contribute to photoreceptor cell dysfunction and degeneration [57].
2.2.3. Increase in Oxidative Stress Levels
Another important pathomolecular mechanism is the increase in oxidative stress levels, encouraged by damage to the normal cellular processes in the retina.
The accumulation of reactive oxygen species (ROS) can damage cellular components, including DNA, proteins, and lipids. This oxidative stress can further contribute to the degeneration of photoreceptor cells in RP [58]. In RP, oxidative stress occurs due to multiple factors related to the degenerative processes in the retina. In RP, there is evidence of decreased antioxidant capacity in the retina, which results in an imbalance between ROS production and antioxidant defenses (impaired antioxidant defense) [59]. This imbalance results in the accumulation of ROS, which can damage cellular components, including photoreceptor cells [60]. The consequences of oxidative stress in RP contribute to the ongoing degeneration of photoreceptor cells, exacerbating the visual impairment experienced by individuals with the condition [61]. Strategies aimed at reducing oxidative stress and enhancing the antioxidant defense system have been investigated as potential therapeutic approaches for RP [62]. These include the use of antioxidants, such as vitamins C and E, and other compounds that can mitigate ROS-induced damage [63][64].
2.2.4. Mitochondrial Dysfunction
In the context of retinitis pigmentosa, another important pathogenetic mechanism is mitochondrial dysfunction, as it contributes to the disease’s progression and the degeneration of photoreceptor cells in the retina [65].
Mutations in the genes associated with mitochondrial function, such as those encoding mitochondrial proteins or involved in mitochondrial DNA maintenance, can lead to mitochondrial dysfunction in RP. Impaired energy production and increased oxidative stress associated with dysfunctional mitochondria can contribute to photoreceptor cell death [66]. Mitochondria are responsible for generating energy in the form of adenosine triphosphate (ATP) through oxidative phosphorylation, and due to their role in capturing and processing light signals, photoreceptor cells have high energy demands [67]. Mitochondrial dysfunction in RP can lead to impaired ATP production, resulting in energy deficits that can compromise the survival and function of photoreceptor cells [68]. Moreover, mitochondrial dysfunction can contribute to an imbalance between the production and scavenging of reactive oxygen species, as ROS are byproducts of mitochondrial metabolism, and excessive ROS production due to mitochondrial dysfunction can lead to oxidative stress, causing damage to cellular components, including photoreceptor cells [69]. Mitochondria are also involved in regulating calcium homeostasis in cells. The disruption of calcium signaling due to mitochondrial dysfunction can impact various cellular processes, including phototransduction and cell survival [70]. Dysregulated calcium levels can trigger apoptotic pathways and contribute to the degeneration of photoreceptor cells in RP [71].
Mitochondria play a critical role in apoptosis, and their dysfunction can trigger the release of pro-apoptotic factors, such as cytochrome c, leading to the activation of apoptotic pathways and the subsequent death of photoreceptor cells [65]. Finally, mitochondrial dysfunction can result in the accumulation of toxic reactive metabolites, such as reactive aldehydes and lipid peroxidation products. These metabolites can damage cellular components, including lipids, proteins, and DNA, further contributing to the degeneration of photoreceptor cells in RP [65]. The exact mechanisms underlying mitochondrial dysfunction in RP can vary depending on the specific genetic mutations involved. Several genes associated with RP are known to affect mitochondrial function directly or indirectly [55]. Understanding the specific mitochondrial defects and their impact on retinal cells is essential for developing targeted therapeutic strategies to mitigate mitochondrial dysfunction and potentially slow down the progression of RP [72].
2.2.5. Apoptosis
Apoptosis represents another pathogenetic mechanism underlying RP [73]. Mutations in genes, such as FASLG, FOS, and NRL, have been implicated in regulating apoptosis in photoreceptor cells. The dysregulation of these genes can lead to increased cell death and accelerate the progression of RP [74]. The principal mechanism underlying the activation of the apoptosis process in retinitis pigmentosa is photoreceptor cell stress. As described before, photoreceptor cell stress can arise from factors such as oxidative stress, mitochondrial dysfunction, calcium dysregulation, and protein misfolding underlying molecular defects, which can activate apoptotic pathways [75]. Classic apoptotic pathways are activated by cellular stress in RP, involving the activation of pro-apoptotic proteins and the release of cytochrome c from the mitochondria, finally leading to caspase activation [73]. The progressive loss of photoreceptor cells in the retina occurs as the apoptotic process in RP progresses over time. At first, the rod photoreceptor cells are more severely affected, resulting in night blindness and peripheral vision loss [76]. Later, cone photoreceptor cells may also undergo apoptosis, resulting in further visual impairment, including central vision loss and color vision defects.
2.2.6. Retinal Remodeling
Another remarkable mechanism, characteristic of retinitis pigmentosa, is called retinal remodeling. Retinal remodeling is a phenomenon that refers to the structural and functional changes that take place in the retina in response to the progressive degeneration of photoreceptor cells [77]. These alterations include the synaptic connections and gene expression profiles of various retinal cell types, contributing to the overall dysfunction of the retina. The pathogenetic mechanism underlying retinal remodeling can be classified into four different kinds of alterations, involving different kinds of retinal cells [78]:
-
Neuronal Rearrangement: As photoreceptor cells degenerate, the remaining retinal neurons, including bipolar cells, horizontal cells, and amacrine cells, undergo reorganization. These neurons undergo structural changes and establish new connections with each other to compensate for the loss of photoreceptor input [79].
-
Bipolar Cell Dystrophy: The second-order neurons in the visual pathway, bipolar cells, also undergo structural and functional changes in RP. Abnormal dendritic sprouting or retraction may occur, leading to the formation of ectopic synapses [80]. These changes can result in altered signal processing and contribute to visual abnormalities in RP [81].
-
Müller Cell Gliosis: Müller cells are the major glial cells in the retina and play a crucial role in maintaining retinal homeostasis. In response to photoreceptor cell degeneration, Müller cells undergo gliotic changes, becoming activated and hypertrophic [82]. This gliosis involves changes in gene expression, increased production of glial fibrillary acidic protein (GFAP), and alterations to their structural morphology. Müller cell gliosis can have both protective and detrimental effects on retinal function and can influence the survival and function of the remaining retinal neurons [83].
-
Synaptic Remodeling: Synaptic connections in the retina are reorganized in RP. As photoreceptor cells degenerate, the synaptic connections between photoreceptor cells and downstream neurons, such as bipolar cells and horizontal cells, alter. As a consequence, bipolar cells and surviving cones or bipolar cells and other retinal neurons may form new synaptic connections [84]. This synaptic remodeling can lead to altered signal processing and contribute to the rewiring of the retinal circuitry [85].
Retinal remodeling in RP has severe functional consequences. Rewiring the retinal circuitry can enable surviving neurons to receive input from a wider area of the retina, potentially improving their sensitivity to light. The remodeling processes can also interfere with normal signal processing and cause visual dysfunction, resulting in changes in visual acuity, contrast sensitivity, and color vision [86][87]. Understanding retinal remodeling in RP is important for the development of potential therapeutic interventions. Strategies aimed at modulating or harnessing the adaptive aspects of retinal remodeling while minimizing the maladaptive changes are being investigated as potential approaches to slow down the progression of the disease and restore visual function in RP patients [88].
2.2.7. Inflammation and Immune Responses
Inflammation and immune responses [71] are the final important pathogenetic mechanisms that significantly contribute to the onset and progression of retinitis pigmentosa. While genetic mutations are the primary cause of RP, inflammation and immune responses can exacerbate degenerative processes in the retina. They can be classified into five different mechanisms:
-
Microglial Activation: In response to photoreceptor cell death and degeneration, microglia, the resident immune cells of the retina, become activated. Activated microglia release pro-inflammatory cytokines, chemokines, and reactive oxygen species. While microglial activation initially aims to clear debris and promote tissue repair, chronic or excessive activation can lead to neuroinflammation and further damage to the retina [89][90].
-
Infiltration of Immune Cells: Immune cells from the bloodstream can infiltrate the retina in some cases of RP, which further contributes to the inflammatory response. The release of inflammatory mediators by immune cells, including macrophages and T cells, can worsen retinal damage [91].
-
Cytokine Imbalance: In RP, there is evidence of an imbalance in cytokine signaling in the retina. Pro-inflammatory cytokines, such as tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), and interleukin-6 (IL-6), are upregulated, while anti-inflammatory cytokines, such as interleukin-10 (IL-10) and transforming growth factor-beta (TGF-β), are downregulated. This imbalance can perpetuate the inflammatory response and contribute to the degeneration of photoreceptor cells [92][93].
-
Complement System Activation: Activation of the complement system can lead to the deposition of complement proteins on photoreceptor cells and subsequent immune-mediated damage [91].
-
Oxidative Stress and Inflammation: The imbalance between reactive oxygen species production and antioxidant defense mechanisms can lead to oxidative stress, which can further cause inflammation in RP. The inflammatory cascade and retinal damage can be exacerbated by ROS activating various intracellular signaling pathways involved in inflammatory responses [94].
Modulating immune responses through anti-inflammatory strategies, immunomodulatory therapies, or targeted interventions to regulate specific immune cell functions are areas of active research for potential therapeutic approaches in RP [95].
References
- Hamel, C. Retinitis pigmentosa. Orphanet J. Rare Dis. 2006, 1, 40.
- Verbakel, S.K.; van Huet, R.A.C.; Boon, C.J.F.; den Hollander, A.I.; Collin, R.W.J.; Klaver, C.C.W.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-syndromic retinitis pigmentosa. Prog. Retin. Eye Res. 2018, 66, 157–186.
- Fishman, G.A.; Anderson, R.J.; Lourenco, P. Prevalence of posterior subcapsular lens opacities in patients with retinitis pigmentosa. Br. J. Ophthalmol. 1985, 69, 263–266.
- Hagiwara, A.; Yamamoto, S.; Ogata, K.; Sugawara, T.; Hiramatsu, A.; Shibata, M.; Mitamura, Y. Macular abnormalities in patients with retinitis pigmentosa: Prevalence on OCT examination and outcomes of vitreoretinal surgery. Acta Ophthalmol. 2011, 89, e122–e125.
- Kumaran, N.; Moore, A.T.; Weleber, R.G.; Michaelides, M. Leber congenital amaurosis/early-onset severe retinal dystrophy: Clinical features, molecular genetics and therapeutic interventions. Br. J. Ophthalmol. 2017, 101, 1147–1154.
- Wu, K.Y.; Kulbay, M.; Toameh, D.; Xu, A.Q.; Kalevar, A.; Tran, S.D. Retinitis Pigmentosa: Novel Therapeutic Targets and Drug Development. Pharmaceutics 2023, 15, 685.
- Maguire, A.M.; High, K.A.; Auricchio, A.; Wright, J.F.; Pierce, E.A.; Testa, F.; Mingozzi, F.; Bennicelli, J.L.; Ying, G.-S.; Rossi, S.; et al. Age-dependent effects of RPE65 gene therapy for Leber’s congenital amaurosis: A phase 1 dose-escalation trial. Lancet 2009, 374, 1597–1605.
- Bainbridge, J.W.; Smith, A.J.; Barker, S.S.; Robbie, S.; Henderson, R.; Balaggan, K.; Viswanathan, A.; Holder, G.E.; Stockman, A.; Tyler, N.; et al. Effect of Gene Therapy on Visual Function in Leber’s Congenital Amaurosis. N. Engl. J. Med. 2008, 358, 2231–2239.
- Sodi, A.; Banfi, S.; Testa, F.; Della Corte, M.; Passerini, I.; Pelo, E.; Rossi, S.; Simonelli, F.; Italian IRD Working Group. RPE65-associated inherited retinal diseases: Consensus recommendations for eligibility to gene therapy. Orphanet J. Rare Dis. 2021, 16, 257.
- Acland, G.M.; Aguirre, G.D.; Ray, J.; Zhang, Q.; Aleman, T.S.; Cideciyan, A.V.; Pearce-Kelling, S.E.; Anand, V.; Zeng, Y.; Maguire, A.M.; et al. Gene therapy restores vision in a canine model of childhood blindness. Nat. Genet. 2001, 28, 92–95.
- Pierce, E.A.; Bennett, J. The Status of RPE65 Gene Therapy Trials: Safety and Efficacy. Cold Spring Harb. Perspect. Med. 2015, 5, a017285.
- Shu, X.; Pang, J.-J.; Zhang, H.; Mansfield, D. Retinitis Pigmentosa: Disease Mechanisms, Diagnosis, and Therapies. J. Ophthalmol. 2015, 2015, 819452.
- Daiger, S.P.; Sullivan, L.S.; Bowne, S.J. Genes and mutations causing retinitis pigmentosa. Clin. Genet. 2013, 84, 132–141.
- Athanasiou, D.; Aguila, M.; Bellingham, J.; Li, W.; McCulley, C.; Reeves, P.J.; Cheetham, M.E. The molecular and cellular basis of rhodopsin retinitis pigmentosa reveals potential strategies for therapy. Prog. Retin. Eye Res. 2018, 62, 1–23.
- Sakmar, T.P.; Huber, T. Rhodopsin. In Encyclopedia of Neuroscience; Squire, L.R., Ed.; Academic Press: Cambridge, MA, USA, 2009; pp. 365–372. ISBN 9780080450469.
- Xiao, T.; Xu, K.; Zhang, X.; Xie, Y.; Li, Y. Sector Retinitis Pigmentosa caused by mutations of the RHO gene. Eye 2019, 33, 592–599.
- Lewin, A.S.; Rossmiller, B.; Mao, H. Gene Augmentation for adRP Mutations in RHO. Cold Spring Harb. Perspect. Med. 2014, 4, a017400.
- Chuang, J.-Z.; Vega, C.; Jun, W.; Sung, C.-H. Structural and functional impairment of endocytic pathways by retinitis pigmentosa mutant rhodopsin-arrestin complexes. J. Clin. Investig. 2004, 114, 131–140.
- Trofimova, S. Molecular Mechanisms of Retina Pathology and Ways of Its Correction; Springer: Berlin/Heidelberg, Germany, 2021.
- Liu, X.; Feng, B.; Vats, A.; Tang, H.; Seibel, W.; Swaroop, M.; Tawa, G.; Zheng, W.; Byrne, L.; Schurdak, M.; et al. Pharmacological clearance of misfolded rhodopsin for the treatment of RHO-associated retinitis pigmentosa. FASEB J. 2020, 34, 10146–10167.
- Tebbe, L.; Sakthivel, H.; Makia, M.S.; Kakakhel, M.; Conley, S.M.; Al-Ubaidi, M.R.; Naash, M.I. Prph2 disease mutations lead to structural and functional defects in the RPE. FASEB J. 2022, 36, e22284.
- Peeters, M.H.; Khan, M.; Rooijakkers, A.A.; Mulders, T.; Haer-Wigman, L.; Boon, C.J.; Klaver, C.C.; van den Born, L.I.; Hoyng, C.B.; Cremers, F.P.; et al. PRPH2 mutation update: In silico assessment of 245 reported and 7 novel variants in patients with retinal disease. Hum. Mutat. 2021, 42, 1521–1547.
- Boon, C.J.; den Hollander, A.I.; Hoyng, C.B.; Cremers, F.P.; Klevering, B.J.; Keunen, J.E. The spectrum of retinal dystrophies caused by mutations in the peripherin/RDS gene. Prog. Retin. Eye Res. 2008, 27, 213–235.
- Coco-Martin, R.M.; Sanchez-Tocino, H.T.; Desco, C.; Usategui-Martín, R.; Tellería, J.J. PRPH2-Related Retinal Diseases: Broadening the Clinical Spectrum and Describing a New Mutation. Genes 2020, 11, 773.
- Stuck, M.W.; Conley, S.M.; Naash, M.I. PRPH2/RDS and ROM-1: Historical context, current views and future considerations. Prog. Retin. Eye Res. 2016, 52, 47–63.
- Gerhardt, M.J.; Priglinger, S.G.; Biel, M.; Michalakis, S. Biology, Pathobiology and Gene Therapy of CNG Channel-Related Retinopathies. Biomedicines 2023, 11, 269.
- Napolitano, L.M.R.; Torre, V.; Marchesi, A. CNG channel structure, function, and gating: A tale of conformational flexibility. Pflügers Arch. -Eur. J. Physiol. 2021, 473, 1423–1435.
- Michalakis, S.; Becirovic, E.; Biel, M. Retinal Cyclic Nucleotide-Gated Channels: From Pathophysiology to Therapy. Int. J. Mol. Sci. 2018, 19, 749.
- Gerhardt, M.J.; Petersen-Jones, S.M.; Michalakis, S. CNG channel-related retinitis pigmentosa. Vis. Res. 2023, 208, 108232.
- Dryja, T.P.; Finn, J.T.; Peng, Y.W.; McGee, T.L.; Berson, E.L.; Yau, K.W. Mutations in the gene encoding the alpha subunit of the rod cGMP-gated channel in autosomal recessive retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 1995, 92, 10177–10181.
- Duricka, D.L.; Brown, R.L.; Varnum, M.D. Defective trafficking of cone photoreceptor CNG channels induces the unfolded protein response and ER-stress-associated cell death. Biochem. J. 2011, 441, 685–696.
- Morimura, H.; Fishman, G.A.; Grover, S.A.; Fulton, A.B.; Berson, E.L.; Dryja, T.P. Mutations in the RPE65 gene in patients with autosomal recessive retinitis pigmentosa or leber congenital amaurosis. Proc. Natl. Acad. Sci. USA 1998, 95, 3088–3093.
- Wolf, G. Function of the Protein RPE65 in the Visual Cycle. Nutr. Rev. 2005, 63, 97–100.
- Sallum, J.M.F.; Kaur, V.P.; Shaikh, J.; Banhazi, J.; Spera, C.; Aouadj, C.; Viriato, D.; Fischer, M.D. Epidemiology of Mutations in the 65-kDa Retinal Pigment Epithelium (RPE65) Gene-Mediated Inherited Retinal Dystrophies: A Systematic Literature Review. Adv. Ther. 2022, 39, 1179–1198.
- Thompson, D.A.; Gyürüs, P.; Fleischer, L.L.; Bingham, E.L.; McHenry, C.L.; Apfelstedt-Sylla, E.; Zrenner, E.; Lorenz, B.; Richards, J.E.; Jacobson, S.G.; et al. Genetics and phenotypes of RPE65 mutations in inherited retinal degeneration. Investig. Ophthalmol. Vis. Sci. 2000, 41, 4293–4299.
- He, S.; Parapuram, S.K.; Hurd, T.W.; Behnam, B.; Margolis, B.; Swaroop, A.; Khanna, H. Retinitis Pigmentosa GTPase Regulator (RPGR) protein isoforms in mammalian retina: Insights into X-linked Retinitis Pigmentosa and associated ciliopathies. Vis. Res. 2008, 48, 366–376.
- Vinikoor-Imler, L.C.; Simpson, C.; Narayanan, D.; Abbasi, S.; Lally, C. Prevalence of RPGR-mutated X-linked retinitis pigmentosa among males. Ophthalmic Genet. 2022, 43, 581–588.
- Khanna, H. Photoreceptor Sensory Cilium: Traversing the Ciliary Gate. Cells 2015, 4, 674–686.
- Murga-Zamalloa, C.A.; Atkins, S.J.; Peranen, J.; Swaroop, A.; Khanna, H. Interaction of retinitis pigmentosa GTPase regulator (RPGR) with RAB8A GTPase: Implications for cilia dysfunction and photoreceptor degeneration. Hum. Mol. Genet. 2010, 19, 3591–3598.
- Murga-Zamalloa, C.; Swaroop, A.; Khanna, H. Multiprotein Complexes of Retinitis Pigmentosa GTPase Regulator (RPGR), a Ciliary Protein Mutated in X-Linked Retinitis Pigmentosa (XLRP). Adv. Exp. Med. Biol. 2010, 664, 105–114.
- Sun, C.; Chen, S. Gene Augmentation for Autosomal Dominant CRX-Associated Retinopathies. Adv. Exp. Med. Biol. 2023, 1415, 135–141.
- Clanor, P.B.; Buchholz, C.N.; Hayes, J.E.; Friedman, M.A.; White, A.M.; Enke, R.A.; Berndsen, C.E. Structural and functional analysis of the human cone-rod homeobox transcription factor. Proteins Struct. Funct. Bioinform. 2022, 90, 1584–1593.
- Swain, P.K.; Chen, S.; Wang, Q.-L.; Affatigato, L.M.; Coats, C.L.; Brady, K.D.; Fishman, G.A.; Jacobson, S.G.; Swaroop, A.; Stone, E.; et al. Mutations in the Cone-Rod Homeobox Gene Are Associated with the Cone-Rod Dystrophy Photoreceptor Degeneration. Neuron 1997, 19, 1329–1336.
- Freund, C.L.; Gregory-Evans, C.Y.; Furukawa, T.; Papaioannou, M.; Looser, J.; Ploder, L.; Bellingham, J.; Ng, D.; Herbrick, J.-A.S.; Duncan, A.; et al. Loutradis-Anagnostou A, Jacobson SG, Cepko CL, Bhattacharya SS, McInnes RR. Cone-rod dystrophy due to mutations in a novel photoreceptor-specific homeobox gene (CRX) essential for maintenance of the photoreceptor. Cell 1997, 91, 543–553.
- Chen, S.; Wang, Q.-L.; Xu, S.; Liu, I.; Li, L.Y.; Wang, Y.; Zack, D.J. Functional analysis of cone-rod homeobox (CRX) mutations associated with retinal dystrophy. Hum. Mol. Genet. 2002, 11, 873–884.
- Fuster-García, C.; García-Bohórquez, B.; Rodríguez-Muñoz, A.; Aller, E.; Jaijo, T.; Millán, J.M.; García-García, G. Usher Syndrome: Genetics of a Human Ciliopathy. Int. J. Mol. Sci. 2021, 22, 6723.
- Lenassi, E.; Saihan, Z.; Cipriani, V.; Stabej, P.L.Q.; Moore, A.T.; Luxon, L.M.; Bitner-Glindzicz, M.; Webster, A.R. Natural History and Retinal Structure in Patients with Usher Syndrome Type 1 Owing to MYO7A Mutation. Ophthalmology 2013, 121, 580–587.
- Well, D.; Blanchard, S.; Kaplan, J.; Guilford, P.; Gibson, F.; Walsh, J.; Mburu, P.; Varela, A.; Levilliers, J.; Weston, M.D.; et al. Defective myosin VIIA gene responsible for Usher syndrome type IB. Nature 1995, 374, 60–61.
- Nagel-Wolfrum, K.; Fadl, B.R.; Becker, M.M.; Wunderlich, K.A.; Schäfer, J.; Sturm, D.; Fritze, J.; Gür, B.; Kaplan, L.; Andreani, T.; et al. Expression and subcellular localization of USH1C/harmonin in human retina provides insights into pathomechanisms and therapy. Hum. Mol. Genet. 2022, 32, 431–449.
- Sun, J.-P.; Li, R.; Ren, H.-Z.; Xu, A.-T.; Yu, X.; Xu, Z.-G. The Very Large G Protein Coupled Receptor (Vlgr1) in Hair Cells. J. Mol. Neurosci. 2012, 50, 204–214.
- Ratnam, K.; Västinsalo, H.; Roorda, A.; Sankila, E.-M.K.; Duncan, J.L. Cone Structure in Patients with Usher Syndrome Type III and Mutations in the Clarin 1 Gene. JAMA Ophthalmol. 2013, 131, 67–74.
- Liu, W.; Liu, S.; Li, P.; Yao, K. Retinitis Pigmentosa: Progress in Molecular Pathology and Biotherapeutical Strategies. Int. J. Mol. Sci. 2022, 23, 4883.
- Manley, A.; Meshkat, B.I.; Jablonski, M.M.; Hollingsworth, T. Cellular and Molecular Mechanisms of Pathogenesis Underlying Inherited Retinal Dystrophies. Biomolecules 2023, 13, 271.
- Smirnov, V.M.; Nassisi, M.; Hernandez, C.S.; Méjécase, C.; El Shamieh, S.; Condroyer, C.; Antonio, A.; Meunier, I.; Andrieu, C.; Defoort-Dhellemmes, S.; et al. Retinal Phenotype of Patients with Isolated Retinal Degeneration Due to CLN3 Pathogenic Variants in a French Retinitis Pigmentosa Cohort. JAMA Ophthalmol. 2021, 139, 278–291.
- Ferrari, S.; Di Iorio, E.; Barbaro, V.; Ponzin, D.; Sorrentino, F.S.; Parmeggiani, F. Retinitis pigmentosa: Genes and disease mechanisms. Curr. Genom. 2011, 12, 238–249.
- Lin, J.H.; LaVail, M.M. Misfolded Proteins and Retinal Dystrophies. Adv. Exp. Med. Biol. 2010, 664, 115–121.
- Tzekov, R.; Stein, L.; Kaushal, S. Protein Misfolding and Retinal Degeneration. Cold Spring Harb. Perspect. Biol. 2011, 3, a007492.
- Vingolo, E.M.; Casillo, L.; Contento, L.; Toja, F.; Florido, A. Retinitis Pigmentosa (RP): The Role of Oxidative Stress in the Degenerative Process Progression. Biomedicines 2022, 10, 582.
- Wang, J.; Li, M.; Geng, Z.; Khattak, S.; Ji, X.; Wu, D.; Dang, Y. Role of Oxidative Stress in Retinal Disease and the Early Intervention Strategies: A Review. Oxidative Med. Cell. Longev. 2022, 2022, 7836828.
- Gallenga, C.E.; Lonardi, M.; Pacetti, S.; Violanti, S.S.; Tassinari, P.; Di Virgilio, F.; Tognon, M.; Perri, P. Molecular Mechanisms Related to Oxidative Stress in Retinitis Pigmentosa. Antioxidants 2021, 10, 848.
- Domènech, E.B.; Marfany, G. The Relevance of Oxidative Stress in the Pathogenesis and Therapy of Retinal Dystrophies. Antioxidants 2020, 9, 347.
- Plafker, S.M.; O’Mealey, G.B.; Szweda, L.I. Mechanisms for countering oxidative stress and damage in retinal pigment epithelium. Int. Rev. Cell Mol. Biol. 2012, 298, 135–177.
- Komeima, K.; Rogers, B.S.; Lu, L.; Campochiaro, P.A. Antioxidants reduce cone cell death in a model of retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 2006, 103, 11300–11305.
- Ren, X.; Léveillard, T. Modulating antioxidant systems as a therapeutic approach to retinal degeneration. Redox Biol. 2022, 57, 102510.
- Lefevere, E.; Toft-Kehler, A.K.; Vohra, R.; Kolko, M.; Moons, L.; Van Hove, I. Mitochondrial dysfunction underlying outer retinal diseases. Mitochondrion 2017, 36, 66–76.
- Barot, M.; Gokulgandhi, M.R.; Mitra, A.K. Mitochondrial Dysfunction in Retinal Diseases. Curr. Eye Res. 2011, 36, 1069–1077.
- Jaiswal, M.; Haelterman, N.A.; Sandoval, H.; Xiong, B.; Donti, T.; Kalsotra, A.; Yamamoto, S.; Cooper, T.A.; Graham, B.H.; Bellen, H.J. Correction: Impaired Mitochondrial Energy Production Causes Light-Induced Photoreceptor Degeneration Independent of Oxidative Stress. PLoS Biol. 2018, 16, e1002622.
- Yang, J.; Chen, X.; Gao, A.L.H.; Zhao, M.; Ge, L.; Li, M.; Yang, C.; Gong, Y.; Gu, Z.; Xu, H. Alleviation of Photoreceptor Degeneration Based on Fullerenols in rd1 Mice by Reversing Mitochondrial Dysfunction via Modulation of Mitochondrial DNA Transcription and Leakage. Small 2023, 5, e2205998.
- Vlachantoni, D.; Bramall, A.N.; Murphy, M.P.; Taylor, R.W.; Shu, X.; Tulloch, B.; Van Veen, T.; Turnbull, D.M.; McInnes, R.R.; Wright, A.F. Evidence of severe mitochondrial oxidative stress and a protective effect of low oxygen in mouse models of inherited photoreceptor degeneration. Hum. Mol. Genet. 2011, 20, 322–335.
- Marigo, V.; Kutluer, M.; Huang, L.; Comitato, A.; Schiroli, D.; Schwede, F.; Rentsch, A.; Ekstrom, P.A.R.; Paquet-Durand, F. Decrease of intracellular calcium to restrain rod cell death in retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2019, 60, 4866.
- Das, S.; Chen, Y.; Yan, J.; Christensen, G.; Belhadj, S.; Tolone, A.; Paquet-Durand, F. The role of cGMP-signalling and calcium-signalling in photoreceptor cell death: Perspectives for therapy development. Pflugers Arch. Eur. J. Physiol. 2021, 473, 1411–1421.
- Beeson, C.; Peterson, Y.K.; Perron, N.; Bandyopadhyay, M.; Nasarre, C.; Beeson, G.; Comer, R.F.; Lindsey, C.C.; Schnellmann, R.G.; Rohrer, B. Newly Identified Chemicals Preserve Mitochondrial Capacity and Decelerate Loss of Photoreceptor Cells in Murine Retinal Degeneration Models. J. Ocul. Pharmacol. Ther. 2021, 37, 367–378.
- Li, Z.-Y.; Milam, A.H. Apoptosis in Retinitis Pigmentosa. In Degenerative Diseases of the Retina; Anderson, R.E., LaVail, M.M., Hollyfield, J.G., Eds.; Springer: Boston, MA, USA, 1995.
- Wong, P. Apoptosis, retinitis pigmentosa, and degeneration. Biochem. Cell Biol. 1994, 72, 489–498.
- Newton, F.; Megaw, R. Mechanisms of Photoreceptor Death in Retinitis Pigmentosa. Genes 2020, 11, 1120.
- Remé, C.E.; Grimm, C.; Hafezi, F.; Wenzel, A.; Williams, T.P. Apoptosis in the Retina: The Silent Death of Vision. News Physiol. Sci. 2000, 15, 120–124.
- Jones, B.W.; Kondo, M.; Terasaki, H.; Lin, Y.; McCall, M.; Marc, R.E. Retinal remodeling. Jpn. J. Ophthalmol. 2012, 56, 289–306.
- Jones, B.W.; Pfeiffer, R.L.; Ferrell, W.D.; Watt, C.B.; Marmor, M.; Marc, R.E. Retinal remodeling in human retinitis pigmentosa. Exp. Eye Res. 2016, 150, 149–165.
- Marc, R.E.; Jones, B.W.; Watt, C.B.; Strettoi, E. Neural remodeling in retinal degeneration. Prog. Retin. Eye Res. 2003, 22, 607–655.
- Gilhooley, M.J.; Hickey, D.G.; Lindner, M.; Palumaa, T.; Hughes, S.; Peirson, S.N.; MacLaren, R.E.; Hankins, M.W. ON-bipolar cell gene expression during retinal degeneration: Implications for optogenetic visual restoration. Exp. Eye Res. 2021, 207, 108553.
- Martínez-Gil, N.; Maneu, V.; Kutsyr, O.; Fernández-Sánchez, L.; Sánchez-Sáez, X.; Sánchez-Castillo, C.; Campello, L.; Lax, P.; Pinilla, I.; Cuenca, N. Cellular and molecular alterations in neurons and glial cells in inherited retinal degeneration. Front. Neuroanat. 2022, 16, 984052.
- Gao, H.; Huang, X.; He, J.; Zou, T.; Chen, X.; Xu, H. The roles of microglia in neural remodeling during retinal degeneration. Histol. Histopathol. 2021, 37, 1–10.
- Gordon, W.C.; Knott, E.J.; Sheets, K.G.; Regan, C.E., Jr.; Bazan, N.G. Müller Cell Reactive Gliosis Contributes to Retinal Degeneration in Ccl2-/-/Cx3cr1-/- Mice. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1375.
- Phillips, M.J.; Otteson, D.C.; Sherry, D.M. Progression of neuronal and synaptic remodeling in the rd10 mouse model of retinitis pigmentosa. J. Comp. Neurol. 2010, 518, 2071–2089.
- Soto, F.; Kerschensteiner, D. Synaptic remodeling of neuronal circuits in early retinal degeneration. Front. Cell. Neurosci. 2015, 9, 395.
- Pfeiffer, R.L.; Marc, R.E.; Jones, B.W. Persistent remodeling and neurodegeneration in late-stage retinal degeneration. Prog. Retin Eye Res. 2020, 74, 100771.
- Telias, M.; Nawy, S.; Kramer, R.H. Degeneration-Dependent Retinal Remodeling: Looking for the Molecular Trigger. Front. Neurosci. 2020, 14, 618019.
- Pfeiffer, R.L.; Jones, B.W. Current perspective on retinal remodeling: Implications for therapeutics. Front. Neuroanat. 2022, 16, 1099348.
- Gupta, N.; Brown, K.E.; Milam, A.H. Activated microglia in human retinitis pigmentosa, late-onset retinal degeneration, and age-related macular degeneration. Exp. Eye Res. 2003, 76, 463–471.
- Peng, B.; Xiao, J.; Wang, K.; So, K.-F.; Tipoe, G.L.; Lin, B. Suppression of microglial activation is neuroprotective in a mouse model of human retinitis pigmentosa. J. Neurosci. 2014, 34, 8139–8150.
- Mohan, K.V.; Mishra, A.; Muniyasamy, A.; Sinha, P.; Sahu, P.; Kesarwani, A.; Jain, K.; Nagarajan, P.; Scaria, V.; Agarwal, M.; et al. Immunological consequences of compromised ocular immune privilege accelerate retinal degeneration in retinitis pigmentosa. Orphanet J. Rare Dis. 2022, 17, 378.
- Olivares-González, L.; Velasco, S.; Campillo, I.; Rodrigo, R. Retinal Inflammation, Cell Death and Inherited Retinal Dystrophies. Int. J. Mol. Sci. 2021, 22, 2096.
- Okita, A.; Murakami, Y.; Shimokawa, S.; Funatsu, J.; Fujiwara, K.; Nakatake, S.; Koyanagi, Y.; Akiyama, M.; Takeda, A.; Hisatomi, T.; et al. Changes of Serum Inflammatory Molecules and Their Relationships with Visual Function in Retinitis Pigmentosa. Investig. Opthalmol. Vis. Sci. 2020, 61, 30.
- Murakami, Y.; Nakabeppu, Y.; Sonoda, K.-H. Oxidative Stress and Microglial Response in Retinitis Pigmentosa. Int. J. Mol. Sci. 2020, 21, 7170.
- Zhao, L.; Hou, C.; Yan, N. Neuroinflammation in retinitis pigmentosa: Therapies targeting the innate immune system. Front. Immunol. 2022, 13, 1059947.
More
Information
Subjects:
Ophthalmology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
547
Revisions:
2 times
(View History)
Update Date:
24 Oct 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No