 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Clarissa Strieder-Barboza | -- | 2464 | 2023-10-11 20:31:38 | | | |
| 2 | Rita Xu | -4 word(s) | 2460 | 2023-10-12 04:15:18 | | |
Video Upload Options
Adipose tissue is a major modulator of metabolic function by regulating energy storage and by acting as an endocrine organ through the secretion of adipokines. With the advantage of next-generation sequencing-based single-cell technologies, adipose tissue has been studied at single-cell resolution, thus providing unbiased insight into its molecular composition. Recent single-cell RNA sequencing studies in human and mouse models have dissected the transcriptional cellular heterogeneity of subcutaneous (SAT), visceral (VAT), and intramuscular (IMAT) white adipose tissue depots and revealed unique populations of adipose tissue progenitor cells, mature adipocytes, immune cell, vascular cells, and mesothelial cells that play direct roles on adipose tissue function and the development of metabolic disorders.
1. Introduction
2. The Single-Cell Era: Applicability of Single-Cell/Nucleus RNA Sequencing to Decode Adipose Tissue Heterogeneity
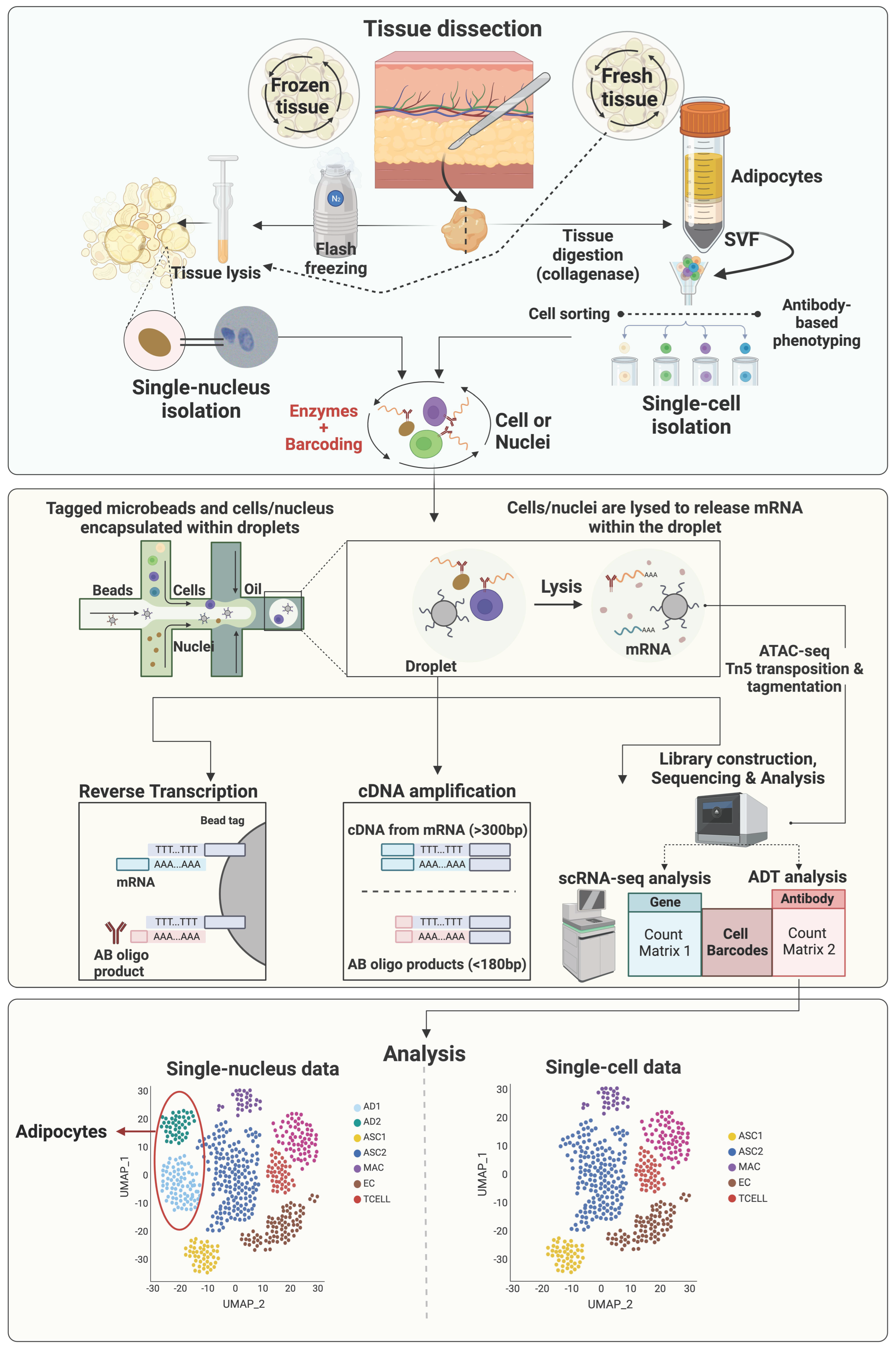
3. Transcriptional Diversity of Adipose Tissue Immune Cells
3.1. Mouse Models
3.2. Human Models
3.3. Livestock Models
References
- Harvey, I.; Boudreau, A.; Stephens, J.M. Adipose tissue in health and disease. Open Biol. 2020, 10, 200291.
- Komolka, K.; Albrecht, E.; Wimmers, K.; Michal, J.J.; Maak, S. Molecular heterogeneities of adipose depots-potential effects on adipose-muscle cross-talk in humans, mice and farm animals. J. Genom. 2014, 2, 31.
- Corvera, S. Cellular Heterogeneity in Adipose Tissues. Annu. Rev. Physiol. 2021, 83, 257–278.
- Astori, G.; Vignati, F.; Bardelli, S.; Tubio, M.; Gola, M.; Albertini, V.; Bambi, F.; Scali, G.; Castelli, D.; Rasini, V.; et al. “In vitro” and multicolor phenotypic characterization of cell subpopulations identified in fresh human adipose tissue stromal vascular fraction and in the derived mesenchymal stem cells. J. Transl. Med. 2007, 5, 55.
- Lei, Y.; Tang, R.; Xu, J.; Wang, W.; Zhang, B.; Liu, J.; Yu, X.; Shi, S. Applications of single-cell sequencing in cancer research: Progress and perspectives. J. Hematol. Oncol. 2021, 14, 91.
- Rodeheffer, M.S.; Birsoy, K.; Friedman, J.M. Identification of white adipocyte progenitor cells in vivo. Cell 2008, 135, 240–249.
- Lee, Y.-H.; Emilio, A.; Granneman, J.G. In Vivo Identification of Bipotential Adipocyte Progenitors Recruited by β3-Adrenoceptor Activation and High-Fat Feeding. Cell Metab. 2012, 15, 480–491.
- Long, J.Z.; Svensson, K.J.; Tsai, L.; Zeng, X.; Roh, H.C.; Kong, X.; Rao, R.R.; Lou, J.; Lokurkar, I.; Baur, W.; et al. A Smooth Muscle-Like Origin for Beige Adipocytes. Cell Metab. 2014, 19, 810–820.
- Shao, M.; Vishvanath, L.; Busbuso, N.C.; Hepler, C.; Shan, B.; Sharma, A.X.; Chen, S.; Yu, X.; An, Y.A.; Zhu, Y.; et al. De novo adipocyte differentiation from Pdgfrbeta(+) preadipocytes protects against pathologic visceral adipose expansion in obesity. Nat. Commun. 2018, 9, 890.
- Vishvanath, L.; MacPherson, K.A.; Hepler, C.; Wang, Q.A.; Shao, M.; Spurgin, S.B.; Wang, M.Y.; Kusminski, C.M.; Morley, T.S.; Gupta, R.K. Pdgfrbeta+ Mural Preadipocytes Contribute to Adipocyte Hyperplasia Induced by High-Fat-Diet Feeding and Prolonged Cold Exposure in Adult Mice. Cell Metab. 2016, 23, 350–359.
- Berry, R.; Rodeheffer, M.S. Characterization of the adipocyte cellular lineage in vivo. Nat. Cell Biol. 2013, 15, 302–308.
- Tran, K.-V.; Gealekman, O.; Frontini, A.; Zingaretti, M.C.; Morroni, M.; Giordano, A.; Smorlesi, A.; Perugini, J.; De Matteis, R.; Sbarbati, A.; et al. The Vascular Endothelium of the Adipose Tissue Gives Rise to Both White and Brown Fat Cells. Cell Metab. 2012, 15, 222–229.
- Tang, W.; Zeve, D.; Suh, J.M.; Bosnakovski, D.; Kyba, M.; Hammer, R.E.; Tallquist, M.D.; Graff, J.M. White fat progenitor cells reside in the adipose vasculature. Science 2008, 322, 583–586.
- Gupta, R.K.; Mepani, R.J.; Kleiner, S.; Lo, J.C.; Khandekar, M.J.; Cohen, P.; Frontini, A.; Bhowmick, D.C.; Ye, L.; Cinti, S.; et al. Zfp423 expression identifies committed preadipocytes and localizes to adipose endothelial and perivascular cells. Cell Metab. 2012, 15, 230–239.
- Ussar, S.; Lee, K.Y.; Dankel, S.N.; Boucher, J.; Haering, M.F.; Kleinridders, A.; Thomou, T.; Xue, R.; Macotela, Y.; Cypess, A.M.; et al. ASC-1, PAT2, and P2RX5 are cell surface markers for white, beige, and brown adipocytes. Sci. Transl. Med. 2014, 6, 247ra103.
- Ferrero, R.; Rainer, P.; Deplancke, B. Toward a Consensus View of Mammalian Adipocyte Stem and Progenitor Cell Heterogeneity. Trends Cell Biol. 2020, 30, 937–950.
- Krueger, K.C.; Costa, M.J.; Du, H.; Feldman, B.J. Characterization of Cre recombinase activity for in vivo targeting of adipocyte precursor cells. Stem Cell Rep. 2014, 3, 1147–1158.
- Eguchi, J.; Wang, X.; Yu, S.; Kershaw, E.E.; Chiu, P.C.; Dushay, J.; Estall, J.L.; Klein, U.; Maratos-Flier, E.; Rosen, E.D. Transcriptional control of adipose lipid handling by IRF4. Cell Metab. 2011, 13, 249–259.
- Hwang, B.; Lee, J.H.; Bang, D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 2018, 50, 96.
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382.
- Chen, G.; Ning, B.; Shi, T. Single-Cell RNA-Seq Technologies and Related Computational Data Analysis. Front. Genet. 2019, 10, 317.
- Zhang, X.; Li, T.; Liu, F.; Chen, Y.; Yao, J.; Li, Z.; Huang, Y.; Wang, J. Comparative Analysis of Droplet-Based Ultra-High-Throughput Single-Cell RNA-Seq Systems. Mol. Cell 2019, 73, 130–142.e135.
- Klein, A.M.; Mazutis, L.; Akartuna, I.; Tallapragada, N.; Veres, A.; Li, V.; Peshkin, L.; Weitz, D.A.; Kirschner, M.W. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 2015, 161, 1187–1201.
- Zilionis, R.; Nainys, J.; Veres, A.; Savova, V.; Zemmour, D.; Klein, A.M.; Mazutis, L. Single-cell barcoding and sequencing using droplet microfluidics. Nat. Protoc. 2017, 12, 44–73.
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214.
- Zheng, G.X.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8, 14049.
- Maniyadath, B.; Zhang, Q.; Gupta, R.K.; Mandrup, S. Adipose tissue at single-cell resolution. Cell Metab. 2023, 35, 386–413.
- Emont, M.P.; Jacobs, C.; Essene, A.L.; Pant, D.; Tenen, D.; Colleluori, G.; Di Vincenzo, A.; Jorgensen, A.M.; Dashti, H.; Stefek, A.; et al. A single-cell atlas of human and mouse white adipose tissue. Nature 2022, 603, 926–933.
- Sun, W.; Dong, H.; Balaz, M.; Slyper, M.; Drokhlyansky, E.; Colleluori, G.; Giordano, A.; Kovanicova, Z.; Stefanicka, P.; Balazova, L.; et al. snRNA-seq reveals a subpopulation of adipocytes that regulates thermogenesis. Nature 2020, 587, 98–102.
- Sárvári, A.K.; Van Hauwaert, E.L.; Markussen, L.K.; Gammelmark, E.; Marcher, A.B.; Ebbesen, M.F.; Nielsen, R.; Brewer, J.R.; Madsen, J.G.S.; Mandrup, S. Plasticity of Epididymal Adipose Tissue in Response to Diet-Induced Obesity at Single-Nucleus Resolution. Cell Metab. 2021, 33, 437–453.e435.
- Denisenko, E.; Guo, B.B.; Jones, M.; Hou, R.; de Kock, L.; Lassmann, T.; Poppe, D.; Clement, O.; Simmons, R.K.; Lister, R.; et al. Systematic assessment of tissue dissociation and storage biases in single-cell and single-nucleus RNA-seq workflows. Genome Biol. 2020, 21, 130.
- van den Brink, S.C.; Sage, F.; Vertesy, A.; Spanjaard, B.; Peterson-Maduro, J.; Baron, C.S.; Robin, C.; van Oudenaarden, A. Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nat. Methods 2017, 14, 935–936.
- Van Hauwaert, E.L.; Gammelmark, E.; Sarvari, A.K.; Larsen, L.; Nielsen, R.; Madsen, J.G.S.; Mandrup, S. Isolation of nuclei from mouse white adipose tissues for single-nucleus genomics. STAR Protoc. 2021, 2, 100612.
- Alvarez, M.; Rahmani, E.; Jew, B.; Garske, K.M.; Miao, Z.; Benhammou, J.N.; Ye, C.J.; Pisegna, J.R.; Pietilainen, K.H.; Halperin, E.; et al. Enhancing droplet-based single-nucleus RNA-seq resolution using the semi-supervised machine learning classifier DIEM. Sci. Rep. 2020, 10, 11019.
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218.
- Liu, Q.; Li, C.; Deng, B.; Gao, P.; Wang, L.; Li, Y.; Shiri, M.; Alkaifi, F.; Zhao, J.; Stephens, J.M.; et al. Tcf21 marks visceral adipose mesenchymal progenitors and functions as a rate-limiting factor during visceral adipose tissue development. Cell Rep. 2023, 42, 112166.
- Buenrostro, J.D.; Wu, B.; Litzenburger, U.M.; Ruff, D.; Gonzales, M.L.; Snyder, M.P.; Chang, H.Y.; Greenleaf, W.J. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 2015, 523, 486–490.
- Cusanovich, D.A.; Daza, R.; Adey, A.; Pliner, H.A.; Christiansen, L.; Gunderson, K.L.; Steemers, F.J.; Trapnell, C.; Shendure, J. Multiplex single-cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 2015, 348, 910–914.
- Burl, R.B.; Ramseyer, V.D.; Rondini, E.A.; Pique-Regi, R.; Lee, Y.-H.; Granneman, J.G. Deconstructing adipogenesis induced by β3-adrenergic receptor activation with single-cell expression profiling. Cell Metab. 2018, 28, 300–309.e304.
- Barboza, C.S.; Flesher, C.; Geletka, L.; Delproposto, J.; Eichler, T.; Akinleye, O.; Ky, A.; Ehlers, A.; O’Rourke, R.; Lumeng, C.N. Single-nuclei transcriptome of human adipose tissue reveals metabolically distinct depot-specific adipose progenitor subpopulations. bioRxiv 2022, arXiv:2022.2006.2029.496888.
- Muir, L.A.; Cho, K.W.; Geletka, L.M.; Baker, N.A.; Flesher, C.G.; Ehlers, A.P.; Kaciroti, N.; Lindsly, S.; Ronquist, S.; Rajapakse, I.; et al. Human CD206+ macrophages associate with diabetes and adipose tissue lymphoid clusters. JCI Insight 2022, 7, e146563.
- Dick, S.A.; Wong, A.; Hamidzada, H.; Nejat, S.; Nechanitzky, R.; Vohra, S.; Mueller, B.; Zaman, R.; Kantores, C.; Aronoff, L. Three tissue resident macrophage subsets coexist across organs with conserved origins and life cycles. Sci. Immunol. 2022, 7, eabf7777.
- Vijay, J.; Gauthier, M.-F.; Biswell, R.L.; Louiselle, D.A.; Johnston, J.J.; Cheung, W.A.; Belden, B.; Pramatarova, A.; Biertho, L.; Gibson, M.J.N.M. Single-cell analysis of human adipose tissue identifies depot- and disease-specific cell types. Nat. Metab. 2019, 2, 97–109.
- Whytock, K.L.; Sun, Y.; Divoux, A.; Yu, G.; Smith, S.R.; Walsh, M.J.; Sparks, L.M. Single cell full-length transcriptome of human subcutaneous adipose tissue reveals unique and heterogeneous cell populations. Iscience 2022, 25, 104772.
- Massier, L.; Jalkanen, J.; Elmastas, M.; Zhong, J.; Wang, T.; Nono Nankam, P.A.; Frendo-Cumbo, S.; Bäckdahl, J.; Subramanian, N.; Sekine, T. An integrated single cell and spatial transcriptomic map of human white adipose tissue. Nat. Commun. 2023, 14, 1438.
- Hildreth, A.D.; Ma, F.; Wong, Y.Y.; Sun, R.; Pellegrini, M.; O’Sullivan, T.E. Single-cell sequencing of human white adipose tissue identifies new cell states in health and obesity. Nat. Immunol. 2021, 22, 639–653.
- Fitzgerald, G.; Turiel, G.; Gorski, T.; Soro-Arnaiz, I.; Zhang, J.; Casartelli, N.C.; Masschelein, E.; Maffiuletti, N.A.; Sutter, R.; Leunig, M.; et al. MME(+) fibro-adipogenic progenitors are the dominant adipogenic population during fatty infiltration in human skeletal muscle. Commun. Biol. 2023, 6, 111.
- Michelotti, T.C.; Kisby, B.R.; Flores, L.S.; Tegeler, A.P.; Fokar, M.; Crasto, C.; Menarim, B.C.; Loux, S.C.; Strieder-Barboza, C. Single-nuclei analysis reveals depot-specific transcriptional heterogeneity and depot-specific cell types in adipose tissue of dairy cows. Front. Cell Dev. Biol. 2022, 10, 1025240.
- Li, J.; Xing, S.; Zhao, G.; Zheng, M.; Yang, X.; Sun, J.; Wen, J.; Liu, R. Identification of diverse cell populations in skeletal muscles and biomarkers for intramuscular fat of chicken by single-cell RNA sequencing. BMC Genom. 2020, 21, 752.
- Wang, L.; Zhao, X.; Liu, S.; You, W.; Huang, Y.; Zhou, Y.; Chen, W.; Zhang, S.; Wang, J.; Zheng, Q.; et al. Single-nucleus and bulk RNA sequencing reveal cellular and transcriptional mechanisms underlying lipid dynamics in high marbled pork. NPJ Sci. Food 2023, 7, 23.


