Studies preceding the use of single-cell RNA sequencing (scRNAseq) focused on identifying cell types within adipose tissue utilizing fluorescent-activated cell sorting (FACS) based on the presence or absence of specific cell surface markers. Larger, clonally identical populations of cells were then propagated and characterized in downstream analyses. These studies were important for the initial identification of different cell types relating to adipogenesis in adipose tissue, including Lin-/Cd34+/Cd29+/Sca1+/Cd24+ ASPCs [
71], Pdgfrα+ mesenchymal cells [
72], Pdgfrb+ and Myh11+ mural cells [
73,
74,
75,
76,
77,
78,
79], and mature adipocytes (ASC-1 for white; PAT2 and P2RX for brown and beige) [
80]. As reviewed by Ferrero et al. [
81], the use of FACS has been important in deciphering the heterogeneity of ASPCs, particularly in recent years with the development and utilization of scRNAseq applications. In addition, the work of Astori et al. [
4] helped to further define the components of the stromal vascular fraction (SVF) including monocytes, granulocytes, hematopoietic stem cells, and endothelial cells. Lineage tracing via Cre-recombinase mouse models has been utilized to identify adipocyte precursors [
72,
76,
82], and characterize mature adipocytes [
83] in white adipose tissue of mice [
72,
76,
82]. However, one limitation of the FACS- and lineage tracing-based strategies is the lack of single markers, which makes it very challenging to identify and validate ASPC heterogeneity.
With the advantage of developing next-generation sequencing (NGS)-based single-cell technologies, we can now profile adipose tissue at single-cell resolution to provide unbiased insight into its molecular composition [
84]. scRNAseq was first developed by Tang et al. in 2009 to sequence the individual cell from a four-cell stage embryo [
85]. Since then, significant efforts have been made on advancing various scRNAseq techniques. Among those, the droplet-based microfluidic technology is the most widely used scRNA-seq platform due to its increased throughput and more automated protocols [
86,
87]. In 2016, 10× Genomics first launched a commercial droplet-microfluidics-based scRNA-seq platform, the Chromium. In Chromium, cells and beads, which contain primers, cellular barcodes, and a unique molecular identifier (UMI), in separate channels are mixed in a microfluidic device and partitioned by nanoliter-size oil droplets for further single-cell cell lysis, reverse transcription, and cDNA library construction [
87,
88,
89,
90,
91].
Studies have demonstrated scRNAseq as an extremely powerful tool in studying adipose tissue stromal cellular composition and plasticity. The high throughput of scRNAseq provides detailed information of the cell identity, gene expression profiles, and changes in cellular states that may arise under normal physiological and pathological conditions. Various scRNAseq studies have demonstrated the heterogeneities of the mesenchymal stem cells (MSCs), immune cells, mesothelial cells, and endothelial cells in white adipose tissues [
92]. Due to their high abundancy, heterogeneity, and functional importance, many scRNAseq studies focused on ASPCs defined as cells expressing the common mesenchymal markers,
Pdgfra and
Pdgfrb. Moreover, owing to the power of single-cell profiling, distinct ASPC populations expressing adipogenic markers, like
Pparg,
Lpl, and
Cd36, and those enriched for genes related to ECM remodeling and inflammation have been identified [
92].
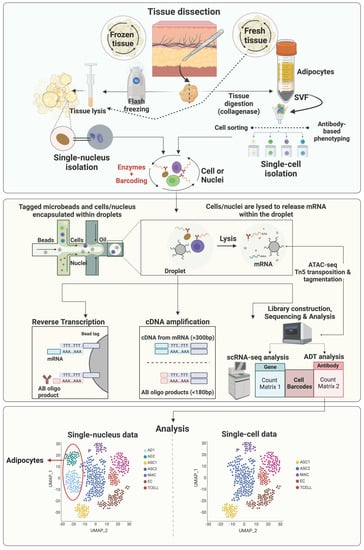
Most early scRNAseq studies of adipose tissue used freshly isolated intact cells, however, with some limitations. First, this strategy requires prolonged stressful enzymatic digestion, which might alter gene expression profiles of sensitive cell types. In addition, due to the prolonged sampling process of human and large animal tissues, it is very difficult to finish cell isolation immediately after sample collection on the same day. Moreover, mature adipocytes, the parenchymal cells of adipose tissue, are too large for most scRNAseq platforms. Alternatively, single-nuclei RNA sequencing (snRNAseq), which uses single nuclei, has overcome these obstacles [
93,
94,
95]. The general workflow of sc/snRNAseq and their differences are illustrated in
Figure 1. Single-nuclei RNA sequencing does not require tissue digestion (e.g., collagenase) to release individual viable cells and therefore can avoid the enzyme-induced cell-type biases, which may result in artificial transcriptional differences [
96,
97]. Moreover, the source of nuclei can be either fresh or frozen tissue samples, providing increased flexibility [
98]. Due to the smaller size of nuclei, nuclei of all types of cells can be identified via snRNAseq, making it possible to study adipocytes and other larger cells, such as neurons. However, snRNAseq also has its limitations. One major issue is the loss of most cytosolic content, which contains most mature mRNA, leading to low numbers of genes detected and failure to detect low abundant transcripts. Another concern is the high level of ambient RNA, which must be carefully removed via additional sample and sequencing data processing [
96,
99]. Without the protection of cytoplasm, mRNA is more susceptible to RNase-mediated degradation, which can be an issue for samples with cells expressing high levels of RNases and must be addressed by adding sufficient RNase inhibitors [
96,
99].
Integrating the sc/snRNAseq data with other omics data is becoming a popular strategy to gain more comprehensive and multidimensional understanding of tissue heterogeneity. The assay for transposase-accessible chromatin using sequencing (ATACseq;
Figure 1) is a powerful method for determining chromatin accessibility across the genome, an important component of the epigenome [
100], and identifying potential transcription factors regulating differential gene expression [
101]. In 2015, ATACseq was optimized for single-cell applications [
102,
103]. Single-cell and single-nuclei ATACseq facilitates the single-cell open chromatin landscaping of heterogeneous tissues like adipose tissue. It is expected that more and more studies integrating sc/snRNAseq and sc/snATACseq data to uncover the contribution of epigenetic regulation of gene expression to the heterogeneities of cells residing in adipose tissue will be published in the near future.
In the following sections, we describe recent studies utilizing sc/snRNASeq to characterize the heterogeneity of adipocyte progenitor cells, mature adipocytes, mesothelial cells, immune cells, and vascular cells in white adipose tissue. While SAT and VAT have garnered most of the research attention for their implication in metabolic diseases in both humans and livestock, recent investigations into IMAT have provided valuable insight into the mechanisms contributing to enhanced marbling in pigs and beef cattle, as well as the pathogenesis of human diseases, including muscular dystrophies and sarcopenia. Studies using sc/snRNASeq to elucidate the cellular heterogeneity of intermuscular adipose tissue are lacking but represent a novel area of study for future research endeavors in this field.
3. Transcriptional Diversity of Adipose Tissue Immune Cells
The network of immune cells in white adipose tissue represents an important link between metabolic and immune function and these cells are implicated both in the prevention and pathogenesis of metabolic disease and inflammation. Recent sc/snRNASeq approaches have provided critical insight into the specific types of immune cells present in adipose tissue as well as transcriptional changes that occur in these cells during different metabolic conditions.
3.1. Mouse Models
In their study on β3-adrenergic stimulation in mice, Burl et al. [
105] identified populations of macrophages (
C1qa,
Trem2,
Adgre1,
Spp1) and natural killer and T cells (NKT;
Nkg7), as well as a population of mixed macrophages and dendritic cells (
Cd74) in epididymal VAT. Notably, β3 stimulation resulted in an increase in the abundance of macrophages and a decrease in the abundance of NKT cells in adipose tissue. Additionally, the authors described a subpopulation of lipid-associated macrophages characterized by a high expression of
Spp1,
Cd36,
Fabp5,
Lpl, and
Lipa and suggested to be involved in the clearance of dead adipocytes and stimulation of adipogenesis by new adipocytes.
Nine distinct immune cell subpopulations were identified in VAT (epididymal adipose tissue) from obese and lean mice by Sárvári et al. [
95], six of which were classified as macrophages (
Adgre1,
Lyz2,
Ccl6), with one subpopulation of dendritic cells (
Cd244a,
Cd209a), one subpopulation of T cells (
Skap1), and one subpopulation of B cells (
Ms4a1,
Cd79a,
Cd79b). Among macrophages, the authors identified subpopulations of perivascular macrophages (
Mrc1,
Lyve1,
Cd163), lipid-associated macrophages (
Lpl,
Trem2,
Cd9), non-perivascular macrophages (
Fcrls,
Ear2), collagen-expressing macrophages (
Col5a2,
Tgfbr3,
Col3a1), proliferating lipid-associated macrophages (
Pola1,
Kif11,
Kif15) and regulatory macrophages (
Prg4,
Tgfb2,
Ltbp1). Notably, the authors found a dramatic increase in lipid-associated and proliferating lipid-associated macrophages in obese mice, and a shift in the transcriptional profile of perivascular and non-perivascular macrophages towards increased expression of lipid metabolism genes (
Pparg,
Lpl) compared to lean mice.
3.2. Human Models
Strieder-Barboza identified macrophage (MAC;
MERTK,
CD163), dendritic cell (DC;
CD1D,
FLT3), T cell, and NK cell (
SKAP1) populations that were similarly distributed across human abdominal SAT and omental VAT [
112]. Four major MAC subtypes were identified including lipid-associated macrophages (LAM) expressing high
ITGAX,
TREM2,
CD9, and
CD52, and two
MRC1/CD206+ MAC subtypes expressing marker genes of resident macrophages such as
F13A1,
PDGFC, and
LYVE1, as previously reported [
130]. Another resident macrophage was identified by the lower
MRC1 expression and the unique
TIMD4 expression [
131]. Emont et al. [
93] also identified a range of immune cells in human SAT and VAT samples including macrophages (
MAFB,
CYBB), monocytes (
CYBB,
CSF3R), dendritic cells (
FLT3), mast cells (
CPA3), neutrophils (
CSF3R), B cells (
MS4A1), NK cells (
KLRD1), and T cells (
IL7R). While Emont et al. [
93] included mouse adipose tissue samples in their analysis, they did not identify major differences between mouse and human immune cells.
Analyzing SAT and VAT samples from human patients undergoing bariatric surgery, Vijay et al. [
20] found a similar panel of immune cells including naïve T cells (
IL7R), activated T cells (
CCL5,
IL32), NK cells (
NKG7), as well as a subpopulation of dysfunctional T cells characterized by high expression of the metallothioneins
MT1F and
MT1G. Macrophages (
CD68) were also identified in this study, including a subpopulation of lipid-associated macrophages characterized by a high expression of lipid metabolism-related genes (
LIPA,
LPL,
CD36,
FABP4), as well as subpopulations of pro-inflammatory (
CXCL3,
CXCL8,
IL1B) and anti-inflammatory (
FOLR2,
KLF4) macrophages. Dendritic cells, characterized by a high expression of
HLA genes, and B cells (
IGKC,
CD79A,
CD37) were also identified in these adipose tissue samples [
20]. Resident (
CD14,
CD8,
CD163) and pro-inflammatory (
ITGAX,
CD86,
TREM2) macrophages were also described by Whytock et al. [
111] in human SAT. Immune cells were identified in the integrated dataset of human white adipose tissue samples described by Massier et al. [
128], including B cells (
MS4A1), mast cells (
CPA3), T, NK, and NKT cells (
CD3D), and a large group of cells including macrophages, dendritic cells, and monocytes (
MRC1,
CD11c,
MRC1,
FABP4). Innate lymphoid cells (ILCs) have only recently been characterized as important components of the innate immune system and are typically associated with their roles at mucosal barriers; however, Hildreth et al. [
109] identified a subpopulation of ILCs in human SAT, characterized by high expression of
KLRB1,
KIT,
CD200R1, and
CCR6. Supporting the findings of Sárvári et al. [
95] and Vijay et al. [
20], Hildreth et al. [
109] also identified a subpopulation of lipid-associated macrophages (
TREM2,
CD9,
LPL), that were in greater abundance in SAT samples from obese patients compared to lean patients.
Analyzing fatty infiltration in human skeletal muscle, Fitzgerald et al. [
115] identified a general population of macrophages (
MRC1,
C1QA) as well as a subpopulation of inflammatory macrophages (
CD68,
PECAM1), but did not describe the presence of additional immune cell populations.
3.3. Livestock Models
Our recent snRNAseq data on dairy cows’ abdominal SAT and omental VAT revealed populations of macrophages, and natural killer and T cells, and a significant increase in the abundance of these immune cell populations in VAT vs. SAT, specifically on the proportion of lipid-associated macrophages (LAM) [
33]. Macrophages (MAC) expressing
MRC1 and
MSR1 corresponded to nearly 10% of all adipose tissue nuclei and were the most heterogenous cell type in the adipose tissue of dairy cows. We identified five distinct MAC subtypes, including two subtypes of LAMs (
FABP4,
LPL,
CD36,
FASN,
CD9), perivascular MACs, M2-like MACs (
CD163,
CD206), and a MAC subtype enriched for complement genes (
C3,
CFI,
CFB) and
S100A12 and
S100A8, suggesting these cells were likely monocytes or differentiating macrophages [
20]. Dairy cow adipose tissue MAC subpopulations were similar to the ones identified via snRNAseq across human SAT and VAT [
112]. Notably, we also identified a SAT-specific MAC subtype characterized by
ABL1,
SPTBN1,
ZBTB16, and
ASAMTSK3. In addition to MAC, our snRNAseq data also revealed a cluster of cells that were enriched for gene markers of T cells (
CCL5,
CD3E,
CD2,
CD247,
CD52) and natural killer cells (
NKG7,
CTSW) comprising nearly 3% and 1.3% of total nuclei in VAT and SAT, respectively. Our data on the immune cell abundance and heterogeneity in the SAT and VAT of dairy cows highlighted depot specificities and potential distinct functional roles for immune cell subtypes, particularly macrophages. However, the specific roles of MAC and NKT populations in the SAT and VAT of dairy cows and how they underly health and disease statuses are yet to be elucidated.
In other animal models, Li et al. [
129] did not detect any immune cells in the breast muscle of chicken at either 5 and 100 days old, whereas a distinct population of myeloid-derived cells (
MRC1) and a population of general immune cells (
CD3E) were identified in the IMAT of pigs [
117].