Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Desirée Martín García | -- | 2936 | 2023-10-11 14:35:14 | | | |
| 2 | Rita Xu | -108 word(s) | 2828 | 2023-10-12 03:40:31 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Téllez, T.; Martin-García, D.; Redondo, M.; García-Aranda, M. Colorectal Carcinomas. Encyclopedia. Available online: https://encyclopedia.pub/entry/50134 (accessed on 03 July 2026).
Téllez T, Martin-García D, Redondo M, García-Aranda M. Colorectal Carcinomas. Encyclopedia. Available at: https://encyclopedia.pub/entry/50134. Accessed July 03, 2026.
Téllez, Teresa, Desirée Martin-García, Maximino Redondo, Marilina García-Aranda. "Colorectal Carcinomas" Encyclopedia, https://encyclopedia.pub/entry/50134 (accessed July 03, 2026).
Téllez, T., Martin-García, D., Redondo, M., & García-Aranda, M. (2023, October 11). Colorectal Carcinomas. In Encyclopedia. https://encyclopedia.pub/entry/50134
Téllez, Teresa, et al. "Colorectal Carcinomas." Encyclopedia. Web. 11 October, 2023.
Copy Citation
Colorectal cancer is the third most diagnosed cancer, behind only breast and lung cancer. In terms of overall mortality, it ranks second due to, among other factors, problems with screening programs, which means that one of the factors that directly impacts survival and treatment success is early detection of the disease. Clusterin (CLU) is a molecular chaperone that has been linked to tumorigenesis, cancer progression and resistance to anticancer treatments, which has made it a promising drug target.
clusterin
colorectal cancer
biomarker
1. Introduction
Clusterin (CLU), also known as lipid-binding protein type A2 or Apolipoprotein J (ApoJ), SPG2, TRPM-2 or CLU is a molecular chaperone that was first discovered in 1979 in rat testis, where it was observed to cause cell aggregation in vitro [1]. This protein is present in human tissues and body fluids, such as blood, saliva, cerebrospinal fluid, and synovial fluid, which have an important role in processes such as cell adhesion, immune response, and cell survival [2][3][4]. CLU has also been shown to inhibit protein aggregation under cellular stress by binding to misfolded proteins, resulting in the accumulation of inactive but stabilized proteins, which can regain their functionality with the help of other folding chaperones [5]. In stressful cellular situations, such as exposure to chemotherapeutic drugs, hypoxia, or ionizing radiation, increase clusterin levels as part of their defense mechanism [6].
The human CLU gene, located on chromosome 8p21-p12, consists of 11 exons and approximately 16 kilobases. CLU produces three main transcription products [7], Variant 1: NM_001831, Variant 2: NR_038335 and Variant 3: NR_045494, that share exons 2–11 but differ in exon 1 (1a, 1b, 1c), suggesting that each variant has a distinct transcription start site.
The first 22 amino acids of this sequence encode a signal that directs translocation of the preprotein to the endoplasmic reticulum (ER), where that sequence is removed and N-glycosylation of 6 asparagine residues occurs, as well as the formation of 4 or 5 disulfide bridges [8]. The protein then translocates to the Golgi apparatus, where it undergoes a more complex glycosylation that promotes its maturation, reaching a weight of 70 to 80 kDa. Subsequently, the highly glycosylated protein is cleaved between residues 227 and 228 and bound by a disulfide bond, leading to the formation of the mature form of the protein or secreted isoform (sCLU). This isoform can be internalized by other cells and stabilize the synthesis of the p53 protein, which is involved in the activation of cell cycle arresting genes, thus can inhibit cell proliferation [9][10] (Figure 1).
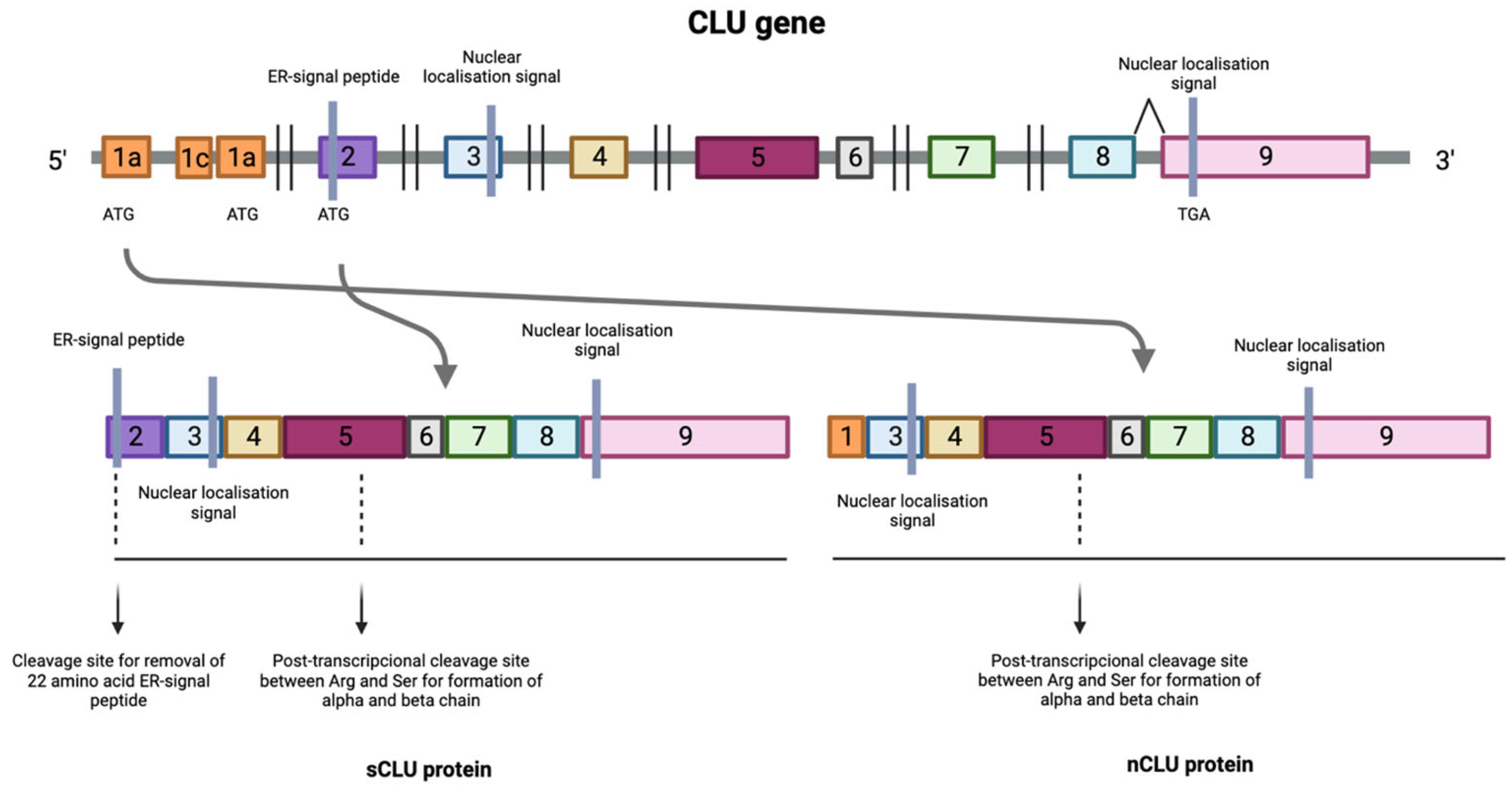
Figure 1. Transcription process of the CLU gene where the numbers indicate the different exons that make up the CLU gene. Transcription of the protein can be initiated at the mRNA synthesis initiation codon in exon 2, resulting in the formation of the mature form of the protein or secreted isoform (sCLU). In situations where this maturation pathway does not occur, a process known as alternative splicing occurs between exons 1 and 3, resulting in the deletion of exon 2 generating a nuclear and functional isoform (nCLU).
In situations where this maturation pathway does not occur, a process known as alternative splicing occurs between exons 1 and 3, resulting in the deletion of exon 2. This produces a protein that lacks the ER translocation signal, generating a non-functional 49 kDa prenuclear isoform (pnCLU). However, under stress, this isoform transforms into a nuclear and functional 55 kDa isoform (nCLU) [8]. The nuclear isoform translocates to the nucleus and, through its binding to the Ku70 protein, promotes cell apoptosis [11][12][13][14] (Figure 1).
On the other hand, there may also be a nearly mature, presecreted 53 KDa isoform (psCLU), which binds to the GRP78 (Bip) chaperone in the ER under stress and translocates to the mitochondrion. There, it binds to the activated form of the Bax protein, which modulates its ability to form homodimers and, in turn, inhibits the formation of Bax-Bak complexes [15]. These complexes, if not inhibited, would induce the apoptotic signaling cascade, demonstrating that clusterin has antiapoptotic activity. Furthermore, when the psCLU isoform is found in the cytoplasm, it can stabilize the Ku70-Bax complex, thus preventing the arrival of Bax into the mitochondria [9]. In the cytoplasm, it can also bind to cytotoxic substances and promote their proteasomal degradation, which further contributes to its antiapoptotic activity [16].
CLU expression can be induced by various factors, such as oxidative stress, chemotherapeutic agents, ionizing and ultraviolet radiation, estrogen, or androgen deprivation in hormone-sensitive tumors, or her2-neu receptor blockade [17][18]. In response to proapoptotic stimuli of various origins, CLU expression increases to protect and enable cell survival. There are two forms of CLU with opposite functions: the sCLU secretory form has a cytoprotective role, whereas the nCLU form can promote cell death [19][20]. Each form of CLU may be subject to its own regulatory pathway.
The regulation of CLU depends not only on the form of the protein, but also on the cell type. In addition to being found in Sertoli cells [21], CLU can be found in a wide variety of cells, such as motor neurons [22], dermal fibroblasts [23] and epithelial cells [24]. CLU expression in these varies considerably and this is thought to be due to both tissue type and different regulatory pathways acting in physiological or pathological situations [2][25]. Various factors, such as TGF-β, NGF (Nerve Growth Factor), EGF (epidermal growth factor), cytokines such as TNF-α and IL-1, IL-2, and IL-6, are involved in the regulation of CLU synthesis [2]. The Wnt signaling pathway and transcription factors such as TCF1, Jak/STAT1 (Signal Transducers and Activators of Transcription 1) and IGF1 (Insulin-Like Growth Factor 1) also play a role in CLU expression [26][27]. In addition, ATM (Ataxia Telangiectasia-Mutated) has been found to act as a sensor of DNA damage, being necessary for the induction of the IGF1-sCLU axis after ionizing radiation [28]. On the other hand, Ha-Ras and c-myc oncogenes can downregulate CLU expression [10]. CLU regulation is mediated by several elements present in the CLU promoter region, such as AP-1 (Activator Protein-1), AP-2 (Activator Protein-2), SP-1 (Stimulatory Element-1), CLE (Clusterin Element), heat shock element (HSE)-like sequences, TCF (T-Cell Factor), NF-κB (Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells) and STAT1 binding sites [29][30]. CLU can also be epigenetically regulated by DNA methylation and histone acetylation due to the presence of CpG island-rich domains [31][32].
2. Colorectal Cancer
Colorectal cancer (CRC) is the third most frequently diagnosed cancer (10%) in both sexes, behind only breast cancer and lung cancer [33]. It ranks second in terms of overall mortality, with a 65% survival rate [34][35]. Unfortunately, approximately 25% of patients present late for consultations, leading to diagnosis at advanced or metastatic stages and, therefore, to delayed treatment [36][37]. In 2019 alone, 60% of newly diagnosed cases were advanced disease, of which 22% had distant metastases [38].
Similar to many other types of cancer, CRC risk is influenced by a range of health behaviors and lifestyle factors such as moderate to heavy alcohol consumption, smoking, a diet high in fat and low in vegetables, obesity, and sedentary lifestyle and non-modifiable factors such as age, ethnicity, and genetic predisposition [39]. In fact, it is estimated that approximately 15% to 30% of CRC cases have a hereditary component in first- and second-degree relatives [40], with a higher risk observed in individuals who have first- and second-degree relatives affected by CRC. In addition, inflammatory bowel diseases, such as Crohn’s disease and ulcerative colitis, increase the risk of developing CRC, especially when the inflammation is chronic and long-lasting [41] (Figure 2).
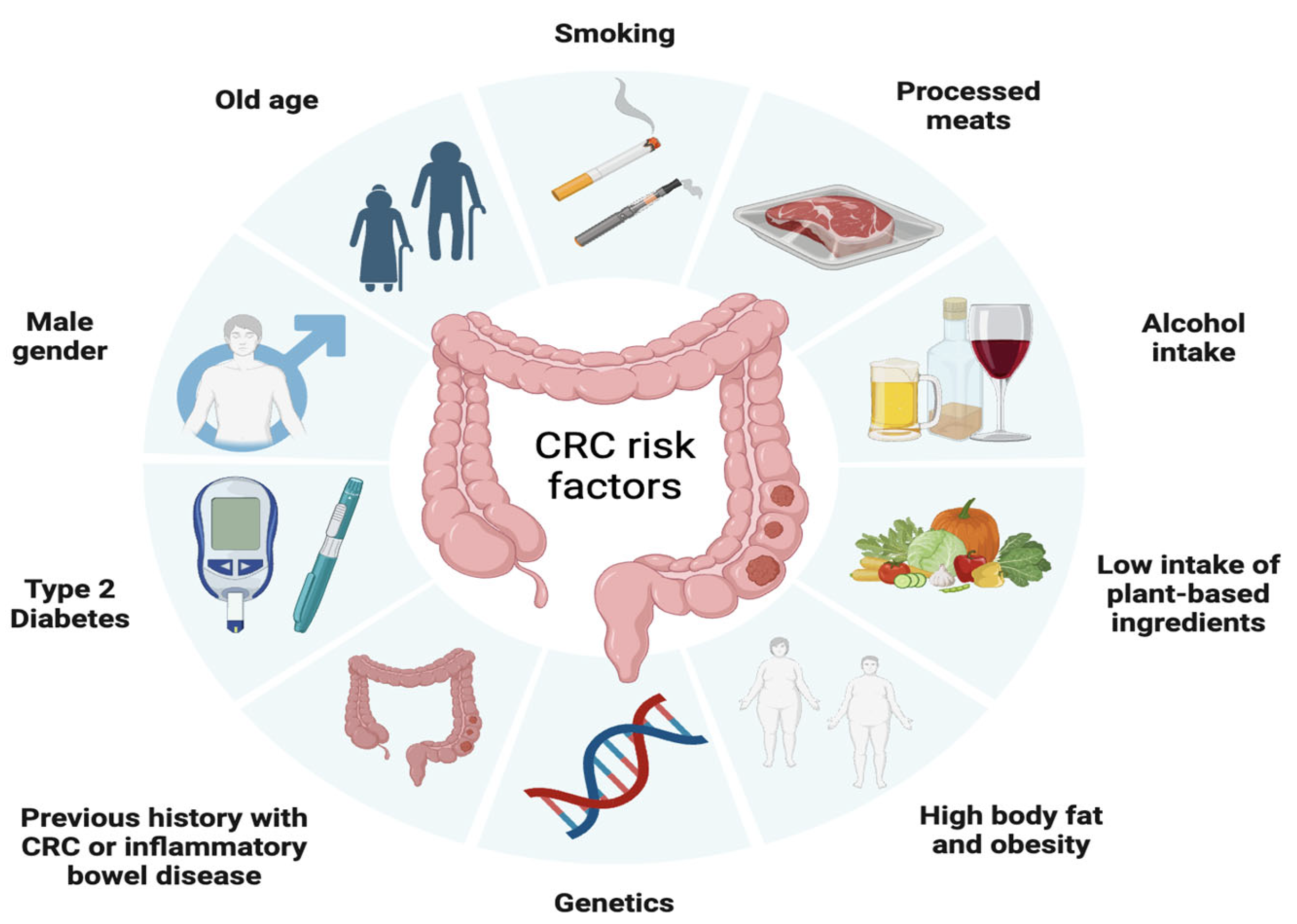
Figure 2. Factors associated with the development of colorectal cancer including modifiable factors (moderate to heavy alcohol consumption, smoking, a diet high in fat and low in vegetables, obesity, and sedentary lifestyle), non-modifiable factors (age, ethnicity, inflammatory bowel disease) and hereditary component.
The progression of colorectal cancer follows a multi-step process, starting from normal epithelial cells and advancing through distinct stages, from the formation of a premalignant lesion, commonly known as an adenoma, into a malignant lesion referred to as carcinoma. Carcinomas are characterized by invasive growth into surrounding tissues and eventually, if left unchecked, can spread systemically.
Advancements in understanding the molecular genetics and epigenetics of colorectal cancer has led to the identification of well-established alterations related to CRC such as chromosomal instability, microsatellite instability (MSI-H) and the CpG island methylator (CGI) phenotype stand out [42].
Chromosomal instability, present in 84% of cases, is related to the activation of oncogenes such as PIK3CA, K-RAS, APC, SMAD4 and TP53 [43]. Mutations in the APC oncogene occur in 80% of CRC cases [44] and lead to WNT signaling pathway activation, which promotes cells proliferation and differentiation [45]. While transformation from adenoma to carcinoma is usually caused by mutations in TP53, K-RAS and DCC 18q genes [46], progression to metastasis is normally associated with the accumulation of genetic changes in APC-KRAS-TP53 (Adenomatous Polyposis Coli–Kirsten rat sarcoma viral oncogene- the tumor suppressor p53), according to the Vogelstein model [47]. In addition, approximately 10% of CRC cases are caused by serrated neoplasia, characterized by mutations in the BRAF and K-RAS genes, which activates the MAPKinase signaling pathway, which has a main role in the regulation of gene expression, cellular growth, and survival [44][48][49].
Microsatellite instability (MSI-H) is present in 15% to 20% of CRC cases and is due to hypermethylation of the hMSH2 and hMLH1 promoters [50], as well as to mutations in mismatch repair (MMR) genes. On the other hand, the CpG-rich island (CGI) methylating phenotype involves methylation of the 5’ ends of half of all genes with short, CpG dinucleotide-rich sequences [51].
CRC is classified into different stages depending on the extent of the tumor and the presence of metastases (Figure 3) [52]. Stage 0 (carcinoma in situ) implies that the cancer cells are found only in the innermost layer of the lining of the colon or rectum, without invading nearby tissues or spreading to lymph nodes or other parts of the body. In stage I, cancer has grown through the innermost layer of the lining, but has not spread to lymph nodes or distant organs. In stage II, cancer has grown through the lining of the colon or rectum, but has not spread to lymph nodes or distant organs. In stage III, the cancer has invaded nearby lymph nodes, but has not reached distant organs. In stage IV, the cancer has spread to distant organs. Prognosis and treatment vary at each stage, and may include surgery, radiation therapy, chemotherapy, and targeted therapy or immunotherapy [53].
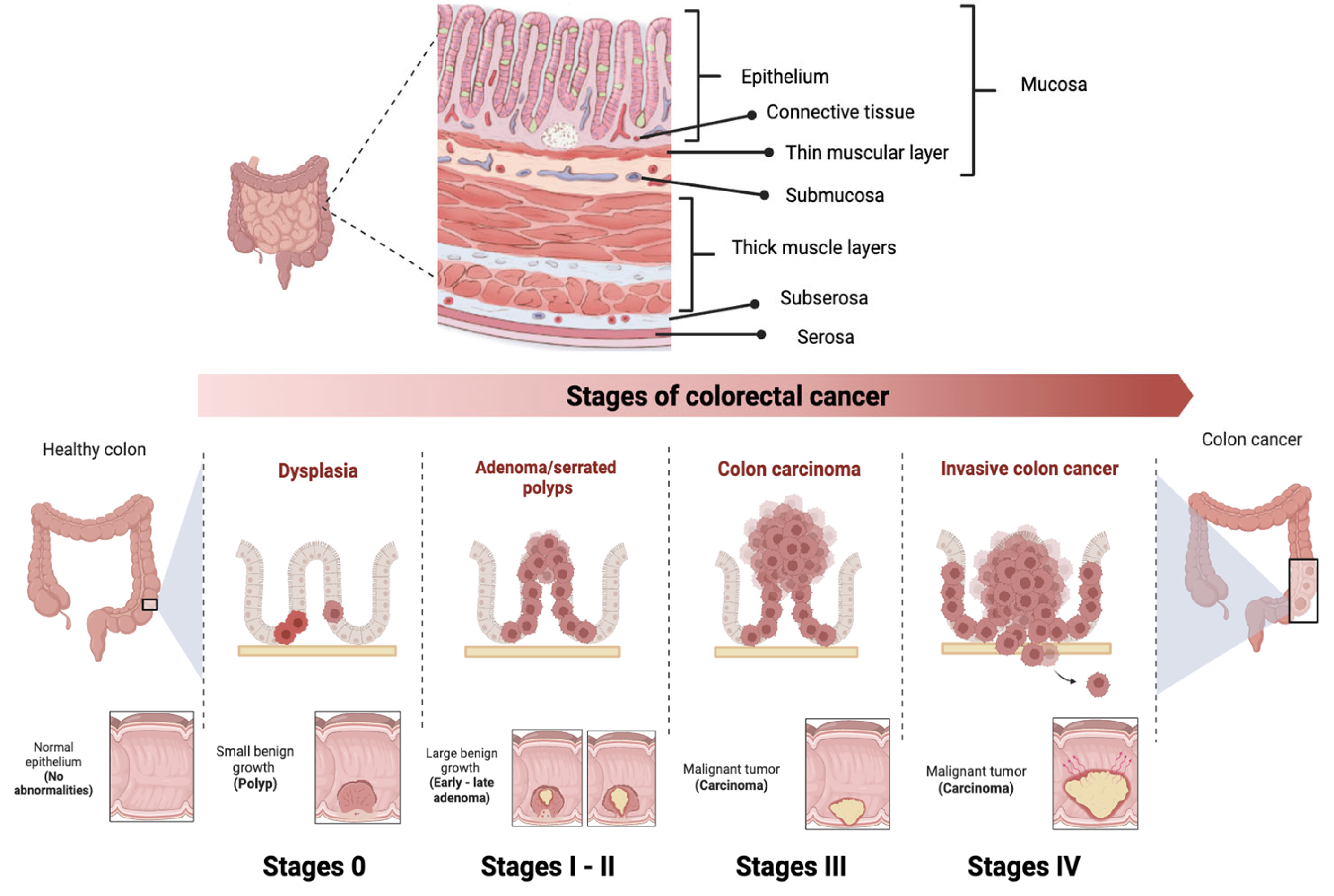
Figure 3. Stages of colorectal cancer depending on the extent of the tumor and the presence of metastases. The TNM staging system is used to evaluate cancer and determine its stage. It focuses on Tumor (T), describing the depth of growth of the primary tumor into the intestinal lining, with categories ranging from T0 (no cancer) to T4b (invasion of other organs or structures). The results of T, along with the assessment of Lymph Nodes (N) and Metastases (M), are combined to assign a stage to the cancer, ranging from 0 to IV.
Since the 5-year survival rate is 91% for stage I, decreasing to 72% for locally advanced stage disease and dropping further to 14% for stage IV [38], there is a current need for the improvement of CRC prevention and screening programs that allow for early detection. Nowadays, colonoscopy is the first option for the patient at is at higher risk of CRC due to genetic syndromes, personal or family history of colorectal cancer or the presence of precancerous polyps. Despite being a relatively expensive invasive procedure, with limitations due to the intestinal preparation required and the complications of the procedure itself [54], colonoscopy represent the most decisive test for detecting CRC because it allows the entire colon to be analyzed, biopsies of suspicious lesions to be obtained and the polyp to be removed in the same session. with a 5% miss rate.
If the patient is not at high risk, the first option is the test based on the detection of blood in stool. Despite its low sensitivity, this test was the first one used for population screening because it is the most economical and the least invasive so far available, reason why it is currently being replaced by methods such as the fecal immunohistochemical test [54]. This test was firstly approved in 2014 by the US Food and Drug Administration (FDA) and recommended for screening tests in asymptomatic patients aged 50–85 years [55].
Conventional treatments used to treat CRC include surgery, chemotherapy, and radiotherapy, alone or in combination, depending on the location [53]:
-
Surgery
Total excision through surgery is the option used to treat localized CRC if the tumor location is easily accessible [56]. Since complete elimination of all cancer cells is not always possible so approximately 66% of stage II and III CRC patients must undergo additional treatments, where adjuvant chemotherapy and/or radiotherapy are included, respectively [57] and, in addition, 54% of patients often relapse even after undergoing adjuvant treatment [58]. This evidences the need for alternative treatments and more effects to treat CRC patients [56].
-
Radiotherapy
Neoadjuvant radiation therapy is another important clinical option available to treat CRC, especially for those at an intermediate or advanced stage for whom surgery is not feasible or chemotherapy cannot be tolerated well. However, the use of radiotherapy is quite limited as it has low sensitivity for CRC and high toxicity in surrounding healthy tissues [59]. This neoadjuvant treatment can be given as a short course followed by surgery or as chemoradiotherapy with 5-fluorouracil or capecitabine, where possible [60].
-
Chemotherapy
The first chemotherapeutic used against CRC was 5-fluorouracil (5-FU) [61]. Its combination with leucovorin became the standard for metastatic colon and rectal cancer (mCRCC) allowing a median overall survival of 8 to 9 months. Years later, the approval of oxaliplatin and the demonstration that it induces cell death by immunogenic mechanisms, allowed the establishment of the current standard chemotherapy of 5-FU and oxilaplatin, commonly known as FOLFOX, which has demonstrated a has superior efficacy with a median survival of 18 to 20 months [62][63].
Recently, the Chinese Clinical Oncological Society has recommended the application of neoadjuvant chemoradiotherapy in patients with CRC, specifically with sigmoid colon cancer, and with a locally advanced stage (T4b) since the response of such treatment can be increased from 26.3% to 38.1%. However, whether MSI-H patients can benefit from this approach, remains as a controversial issue [64]. Postoperative adjuvant chemotherapy is recommended for all stage III CRC [65].
-
Targeted treatments
Monoclonal Antibodies
After 20 years of translational and clinical research, the epidermal growth factor receptor (EGFR) family and its intracellular signaling pathways constitute one of the foundations of molecular targeted therapy for CRC [66]. EGFR serve as cell-surface protein receptors for the peptide ligands of the epidermal growth factor (EGF) family with enzymatic tyrosine kinase activity [66]. Ligand binding induces receptor conformational change, activation, and subsequent phosphorylation of intracellular tyrosine residues, which leads to the activation of different intracellular pathways including the RAS-RAF-MEK-MAPK and PTEN-I3K-AKT-mTOR pathways, which play a main role the regulation of cell proliferation, survival, dissemination, and angiogenesis [67]. Given the role of these processes in cancer development and tumor progression, targeting the EGFR has become a key strategy in the treatment of metastatic colorectal cancer (mCRC) [68][69].
For its part, the RAS family comprises four genes (KRAS4A, KRAS4B, HRAS and NRAS) which are among the most frequently altered oncogenes in human cancer, encode for four proteins with pivotal roles in cells signaling. KRAS4A (K-RAS), the predominant splice variant, and part of the RAS/MAPK pathway. Upon stimulation by upstream receptors such as EGFR, KRAS switches from inactive to active state, and promotes RAF recruitment to the cell membrane, leading to RAF dimerization and activation of downstream effector pathways. KRAS mutations, which are present in approximately 40% of CRC cases, determine constitutive activation of RAS, and promote tumorigenesis as well as modulation of the tumor microenvironment by inducing immune escape and cancer progression [70], reason why RAS mutations are generally associated with poor prognosis and low response to conventional CRC therapies [71]. On the other hand, BRAF mutations, which are seen in 10% to 15% of CRC cases [72], usually lead to constitutive activation of the mitogen-activated protein kinase (MAPK) signaling pathway, conferring high clinical aggressiveness, resistance to anti-EGFR monoclonal antibody therapy, and poor survival [73].
The first successful step towards personalized cancer medicine has been the definition of different treatment options and sequences, which are based on tumor molecular stratification [74]. Since the validation of KRAS and BRAF mutations as predictive biomarkers to anti-EGFR monoclonal antibodies in mCRC, regulatory agencies, such as the FDA and the European Medicines Agency (EMA), established the need to assess K-RAS and BRAF mutation status for patient stratification and management.
Bevacizumab (Avastin; Genentech, South San Francisco, CA, USA), a human antiendothelial growth factor receptor 2 (VEGF) monoclonal antibody, anti-VEGF-A, was the first antigenic drug to be successfully added to the therapeutic armamentarium for CRC. Given its very modest action as a single agent the FDA approves its use in combination with fluoropyrimidine [75]. Nowadays, bevacizumab is widely used for the treatment of patients with mCRC in combination with oxaliplatin-based chemotherapy, due to evidence of improved patient survival [76], either as first-line or second-line therapeutic options [77][78].
Other available drugs approved as second-line treatment for mCRC, Ziv-aflibercept (Zaltrap; Regeneron Pharmaceuticals, Tarrytown, NY, USA) a fully humanized recombinant fusion protein that blocks VEGF-A with higher binding affinity than Bevacizumad [79] and Ramucirumab (Cyramza; Eli Lilly, Indianapolis, IN, USA) a fully humanized IgG1completely humanized monoclonal antibody (mAb) targeting the extracellular domain of VEGF, both of which have demonstrated efficacy for second-line use against CRC in combination with leucovorin and ironotecan (FOLFIRI) or irinotecan alone.
References
- Hogg, S.D.; Embery, G. The isolation and partial characterization of a sulphated glycoprotein from human whole saliva which aggregates strains of Streptococcus sanguis but not Streptococcus mutans. Arch. Oral Biol. 1979, 24, 791–797.
- Trougakos, I.P.; Gonos, E.S. Clusterin/apolipoprotein J in human aging and cancer. Int. J. Biochem. Cell Biol. 2002, 34, 1430–1448.
- Pucci, S.; Bettuzzi, S. Chapter 3: The shifting balance between CLU forms during tumor progression. Adv. Cancer Res. 2009, 104, 25–32.
- Yu, J.-T.; Tan, L. The role of clusterin in Alzheimer’s disease: Pathways, pathogenesis, and therapy. Mol. Neurobiol. 2012, 45, 314–326.
- Poon, S.; Easterbrook-Smith, S.B.; Rybchyn, M.S.; Carver, J.A.; Wilson, M.R. Clusterin is an ATP-independent chaperone with very broad substrate specificity that stabilizes stressed proteins in a folding-competent state. Biochemistry 2000, 39, 15953–15960.
- Zoubeidi, A.; Martin, G. Small heat shock proteins in cancer therapy and prognosis. Int. J. Biochem. Cell Biol. 2012, 44, 1646–1656.
- Herring, S.K.; Moon, H.-L.; Rawal, P.; Chhibber, A.; Zhao, L. Brain clusterin protein isoforms and mitochondrial localization. eLife 2019, 8, e48255.
- Foster, E.M.; Dangla-Valls, A.; Lovestone, S.; Ribe, E.M.; Buckley, N.J. Clusterin in Alzheimer’s Disease: Mechanisms, Genetics, and Lessons from Other Pathologies. Front. Neurosci. 2019, 13, 164.
- Trougakos, I.P.; Gonos, E.S. Regulation of clusterin/apolipoprotein J, a functional homologue to the small heat shock proteins, by oxidative stress in aging and age-related diseases. Free. Radic. Res. 2006, 40, 1324–1334.
- Bettuzzi, S.; Scorcioni, F.; Astancolle, S.; Davalli, P.; Scaltriti, M.; Corti, A. Clusterin (SGP-2) transient overexpression decreases proliferation rate of SV40-immortalized human prostate epithelial cells by slowing down cell cycle progression. Oncogene 2002, 21, 4328–4334.
- Pucci, S.; Bonanno, E.; Pichiorri, F.; Angeloni, C.; Spagnoli, L.G. Modulation of different clusterin isoforms in human colon tumorigenesis. Oncogene 2004, 23, 2298–2304.
- Caccamo, A.E.; Scaltriti, M.; Caporali, A.; D’Arca, D.; Corti, A.; Corvetta, D.; Sala, A.; Bettuzzi, S. Ca2+ depletion induces nuclear clusterin, a novel effector of apoptosis in immortalized human prostate cells. Cell Death Differ. 2005, 12, 101–104.
- Caccamo, A.E.; Scaltriti, M.; Caporali, A.; D’Arca, D.; Scorcioni, F.; Astancolle, S.; Mangiola, M.; Bettuzzi, S. Cell detachment and apoptosis induction of immortalized human prostate epithelial cells are associated with early accumulation of a 45 kDa nuclear isoform of clusterin. Biochem. J. 2004, 382, 157–168.
- Leskov, K.S.; Klokov, D.Y.; Li, J.; Kinsella, T.J.; Boothman, D.A. Synthesis and functional analyses of nuclear clusterin, a cell death protein. J. Biol. Chem. 2003, 278, 11590–11600.
- Li, N.; Zoubeidi, A.; Beraldi, E.; Gleave, M.E. GRP78 regulates clusterin stability, retrotranslocation and mitochondrial localization under ER stress in prostate cancer. Oncogene 2013, 32, 1933–1942.
- Nizard, P.; Tetley, S.; Le Dréan, Y.; Watrin, T.; Le Goff, P.; Wilson, M.R.; Michel, D. Stress-Induced Retrotranslocation of Clusterin/ApoJ into the Cytosol. Traffic 2007, 8, 554–565.
- Trougakos, I.P. The Molecular Chaperone Apolipoprotein J/Clusterin as a Sensor of Oxidative Stress: Implications in Therapeutic Approaches—A Mini-Review. Gerontology 2013, 59, 514–523.
- July, L.V.; Akbari, M.; Zellweger, T.; Jones, E.C.; Goldenberg, S.L.; Gleave, M.E. Clusterin expression is significantly enhanced in prostate cancer cells following androgen withdrawal therapy. Prostate 2002, 50, 179–188.
- Scaltriti, M.; Bettuzzi, S.; Sharrard, R.M.; Caporali, A.; Caccamo, A.E.; Maitland, N.J. Clusterin overexpression in both malignant and nonmalignant prostate epithelial cells induces cell cycle arrest and apoptosis. Br. J. Cancer 2004, 91, 1842–1850.
- Scaltriti, M.; Santamaria, A.; Paciucci, R.; Bettuzzi, S. Intracellular clusterin induces G2-M phase arrest and cell death in PC-3 prostate cancer cells1. Cancer Res. 2004, 64, 6174–6182.
- Collard, M.W.; Griswold, M.D. Biosynthesis, and molecular cloning of sulfated glycoprotein 2 secreted by rat Sertoli cells. Biochemistry 1987, 26, 3297–3303.
- Danik, M.; Chabot, J.G.; Hassan-Gonzalez, D.; Suh, M.; Quirion, R. Localization of sulfated glycoprotein-2/clusterin mRNA in the rat brain by in situ hybridization. J. Comp. Neurol. 1993, 334, 209–227.
- Scaltriti, M.; Brausi, M.; Amorosi, A.; Caporali, A.; D’Arca, D.; Astancolle, S.; Corti, A.; Bettuzzi, S. Clusterin (SGP-2, ApoJ) expression is downregulated in low- and high-grade human prostate cancer. Int. J. Cancer 2004, 108, 23–30.
- Aronow, B.J.; Lund, S.D.; Brown, T.L.; Harmony, J.A.; Witte, D.P. Apolipoprotein J expression at fluid-tissue interfaces: Potential role in barrier cytoprotection. Proc. Natl. Acad. Sci. USA 1993, 90, 725–729.
- Rizzi, F.; Coletta, M.; Bettuzzi, S. Clusterin (CLU): From one gene and two transcripts to many proteins. Adv. Cancer Res. 2009, 104, 9–23.
- Park, S.; Mathis, K.W.; Lee, I.K. The physiological roles of apolipoprotein J/clusterin in metabolic and cardiovascular diseases. Rev. Endocr. Metab. Disord. 2014, 15, 45–53.
- Zoubeidi, A.; Chi, K.; Gleave, M. Targeting the cytoprotective chaperone, clusterin, for treatment of advanced cancer. Clin. Cancer Res. 2010, 16, 1088–1093.
- Goetz, E.M.; Shankar, B.; Zou, Y.; Morales, J.C.; Luo, X.; Araki, S.; Bachoo, R.; Mayo, L.D.; Boothman, D.A. ATM dependent IGF-1 induction regulates secretory clusterin expression after DNA damage and in genetic instability. Oncogene. Nat. Publ. Group 2011, 30, 3745–3754.
- Prochnow, H.; Gollan, R.; Rohne, P.; Hassemer, M.; Koch-Brandt, C.; Baiersdörfer, M. Non-Secreted Clusterin Isoforms Are Translated in Rare Amounts from Distinct Human mRNA Variants and Do Not Affect BaxMediated Apoptosis or the NF-κB Signaling Pathway. PLoS ONE 2013, 8, e75303.
- Michel, D.; Chatelain, G.; North, S.; Brun, G. Stress-induced transcription of the clusterin/apoJ gene. Biochem. J. 1997, 50, 45–50.
- Serrano, A.; Redondo, M.; Tellez, T.; Castro-Vega, I.; Roldan, M.J.; Mendez, R.; Rueda, A.; Jiménez, E. Regulation of clusterin expression in human cancer via DNA methylation. Tumour Biol. 2009, 30, 286–291.
- Deb, M.; Sengupta, D.; Rath, S.K.; Kar, S.; Parbin, S.; Shilpi, A.; Pradhan, N.; Bhutia, S.K.; Roy, S.; Patra, S.K. Clusterin gene is predominantly regulated by histone modifications in human colon cancer and ectopic expression of the nuclear isoform induces cell death. Biochim. Biophys. Acta 2015, 1852, 1630–1645.
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249.
- Miller, K.D.; Nogueira, L.; Devasia, T.; Mariotto, A.B.; Yabroff, K.R.; Jemal, A.; Kramer, J.; Siegel, R.L. Cancer treatment and survivorship statistics, 2022. CA Cancer J. Clin. 2022, 72, 409–436.
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33.
- Zarcos-Pedrinaci, I.; Fernández-López, A.; Téllez, T.; Rivas-Ruiz, F.; Rueda, A.; Morales Suarez-Varela, M.M.; Briones, E.; Baré, M.; Escobar, A.; Sarasqueta, C.; et al. CARESS-CCR Study Group. Factors that influence treatment delay in patients with colorectal cancer. Oncotarget 2017, 8, 36728–36742.
- Padilla-Ruiz, M.; Morales-Suárez-Varela, M.; Rivas-Ruiz, F.; Alcaide, J.; Varela-Moreno, E.; Zarcos-Pedrinaci, I.; Téllez, T.; Fernández-de Larrea-Baz, N.; Baré, M.; Bilbao, A.; et al. On Behalf of Caress–Ccr Study Group. Influence of Diagnostic Delay on Survival Rates for Patients with Colorectal Cancer. Int. J. Environ. Res. Public Health 2022, 19, 3626.
- Siegel, R.L.; Wagle, N.S.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 233–254.
- Sninsky, J.A.; Shore, B.M.; Lupu, G.V.; Crockett, S.D. Risk Factors for Colorectal Polyps and Cancer. Gastrointest. Endosc. Clin. N. Am. 2022, 32, 195–213.
- Li, J.; Ma, X.; Chakravarti, D.; Shalapour, S.; DePinho, R.A. Genetic and biological hallmarks of colorectal cancer. Genes Dev. 2021, 35, 787–820.
- Shah, S.C.; Itzkowitz, S.H. Colorectal Cancer in Inflammatory Bowel Disease: Mechanisms and Management. Gastroenterology 2022, 162, 715–730.e3.
- Ciardiello, F.; Ciardiello, D.; Martini, G.; Napolitano, S.; Tabernero, J.; Cervantes, A. Clinical management of metastatic colorectal cancer in the era of precision medicine. CA Cancer J. Clin. 2022, 72, 372–401.
- Sullivan, B.A.; Noujaim, M.; Roper, J. Cause, Epidemiology, and Histology of Polyps and Pathways to Colorectal Cancer. Gastrointest. Endosc. Clin. N. Am. 2022, 32, 177–194.
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767.
- Silva, A.L.; Dawson, S.N.; Arends, M.J.; Guttula, K.; Hall, N.; Cameron, E.A.; Huang, T.H.; Brenton, J.D.; Tavaré, S.; Bienz, M.; et al. Boosting Wnt activity during colorectal cancer progression through selective hypermethylation of Wnt signaling antagonists. BMC Cancer 2014, 14, 891.
- Jass, J.R. Classification of colorectal cancer based on correlation of clinical, morphological, and molecular features. Histopathology 2007, 50, 113–130.
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480.
- Bettington, M.; Walker, N.; Clouston, A.; Brown, I.; Leggett, B.; Whitehall, V. The serrated pathway to colorectal carcinoma: Current concepts and challenges. Histopathology 2013, 62, 367–386.
- Chang, K.; Willis, J.A.; Reumers, J.; Taggart, M.W.; San Lucas, F.A.; Thirumurthi, S.; Kanth, P.; Delker, D.A.; Hagedorn, C.H.; Lynch, P.M.; et al. Colorectal premalignancy is associated with consensus molecular subtypes 1 and 2. Ann. Oncol. 2018, 29, 2061–2067.
- Toyota, M.; Ahuja, N.; Ohe-Toyota, M.; Herman, J.G.; Baylin, S.B.; Issa, J.P.J. CpG island methylator phenotype in colorectal cancer. Proc. Natl. Acad. Sci. USA 1999, 96, 8681–8686.
- Bird, A.P. CpG-rich islands and the function of DNA methylation. Nature 1986, 321, 209–213.
- Delattre, J.F.; Selcen Oguz Erdogan, A.; Cohen, R.; Shi, Q.; Emile, J.F.; Taieb, J.; Tabernero, J.; André, T.; Meyerhardt, J.A.; Nagtegaal, I.D.; et al. A comprehensive overview of tumor deposits in colorectal cancer: Towards a next TNM classification. Cancer Treat. Rev. 2022, 103, 102325.
- Van Cutsem, E.; Cervantes, A.; Adam, R.; Sobrero, A.; Van Krieken, J.H.; Aderka, D.; Aranda Aguilar, E.; Bardelli, A.; Benson, A.; Bodoky, G.; et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann. Oncol. 2016, 27, 1386–1422.
- Kormi, S.M.A.; Ardehkhani, S.; Kerachian, M.A. New insights into colorectal cancer screening and early detection tests. Color. Cancer 2017, 6, 63–68.
- Hwang, T.J.; Lehmann, L.S.; Kesselheim, A.S. Precision medicine and the FDA’s draft guidance on laboratory-developed tests. Nat. Biotechnol. 2015, 33, 449–451.
- Johdi, N.A.; Sukor, N.F. Colorectal Cancer Immunotherapy: Options and Strategies. Front. Immunol. 2020, 11, 1624.
- Park, S.C.; Sohn, D.K.; Kim, M.J.; Chang, H.J.; Chang, H.J.; Han, K.S.; Hyun, J.H.; Joo, J.; Oh, J.H. Phase II Clinical Trial to Evaluate the Efficacy of Transanal Endoscopic Total Mesorectal Excision for Rectal Cancer. Dis. Colon Rectum 2018, 61, 554–560.
- Ogura, A.; Konishi, T.; Cunningham, C.; Garcia-Aguilar, J.; Iversen, H.; Toda, S.; Lee, I.K.; Lee, H.X.; Uehara, K.; Lee, P.; et al. Neoadjuvant (Chemo)radiotherapy With Total Mesorectal Excision Only Is Not Sufficient to Prevent Lateral Local Recurrence in Enlarged Nodes: Results of the Multicenter Lateral Node Study of Patients with Low cT3/4 Rectal Cancer. J. Clin. Oncol. 2019, 37, 33–43.
- Kulka, U.; Schaffer, M.; Siefert, A.; Schaffer, P.M.; Olsner, A.; Kasseb, K.; Hofstetter, A.; Duhmke, E.; Jori, G. Photofrin as a radiosensitizer in an in vitro cell survival assay. Biochem. Biography. Jt. Res. 2003, 311, 98–103.
- Kuipers, E.J.; Grady, W.M.; Lieberman, D.; Seufferlein, T.; Sung, J.J.; Boelens, P.G.; van de Velde, C.J.; Watanabe, T. Colorectal cancer. Nat. Rev. Dis. Primers 2015, 1, 15065.
- Duschinsky, R.; Pleven, E.; Heidelberger, C. The Synthesis of 5-Fluoropyrimidines. J. Am. Chem. Soc. 1957, 79, 4559–4560.
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic Cell Death in Cancer Therapy. Annu. Rev. Immunol. 2013, 31, 51–72.
- Grothey, A.; Sargent, D.; Goldberg, R.M.; Schmoll, H.J. Survival of Patients with Advanced Colorectal Cancer Improves with the Availability of Fluorouracil-Leucovorin, Irinotecan, and Oxaliplatin in the Course of Treatment. J. Clin. Oncol. 2004, 22, 1209–1214.
- Zhou, C.; Jiang, T.; Xiao, Y.; Wang, Q.; Zeng, Z.; Cai, P.; Zhao, Y.; Zhao, Z.; Wu, D.; Lin, H.; et al. Good Tumor Response to Chemoradioimmunotherapy in dMMR/MSI-H Advanced Colorectal Cancer: A Case Series. Front. Immunol. 2021, 12, 784336.
- Brenner, H.; Kloor, M.; Pox, C.P. Colorectal cancer. Lancet 2014, 383, 1490–1502.
- Ciardiello, F.; Tortora, G. EGFR Antagonists in Cancer Treatment. N. Engl. J. Med. 2008, 358, 1160–1174.
- Zhu, G.; Pei, L.; Xia, H.; Tang, Q.; Bi, F. Role of oncogenic KRAS in the prognosis, diagnosis and treatment of colorectal cancer. Mol. Cancer 2021, 20, 143.
- Chan, D.L.H.; Segelov, E.; Wong, R.S.; Smith, A.; Herbertson, R.A.; Li, B.T.; Tebbutt, N.; Price, T.; Pavlakis, N. Epidermal growth factor receptor (EGFR) inhibitors for metastatic colorectal cancer. Cochrane Database Syst. Rev. 2017, 6, CD007047.
- Halder, S.; Basu, S.; Lall, S.P.; Ganti, A.K.; Batra, S.K.; Seshacharyulu, P. Targeting the EGFR signaling pathway in cancer therapy: What’s new in 2023? Expert Opin. Ther. Targets 2023, 27, 305–324.
- Dias Carvalho, P.; Guimarães, C.F.; Cardoso, A.P.; Mendonça, S.; Costa, Â.M.; Oliveira, M.J.; Velho, S. KRAS Oncogenic Signaling Extends beyond Cancer Cells to Orchestrate the Microenvironment. Cancer Res. 2018, 78, 7–14.
- Nassar, A.H.; Adib, E.; Kwiatkowski, D.J. Distribution of KRAS G12C Somatic Mutations across Race, Sex, and Cancer Type. N. Engl. J. Med. 2021, 384, 185–187.
- Ciombor, K.K.; Strickler, J.H.; Bekaii-Saab, T.S.; Yaeger, R. BRAF-Mutated Advanced Colorectal Cancer: A Rapidly Changing Therapeutic Landscape. J. Clin. Oncol. 2022, 40, 2706–2715.
- Cohen, R.; Liu, H.; Fiskum, J.; Adams, R.; Chibaudel, B.; Maughan, T.S.; Van Cutsem, E.; Venook, A.; Douillard, J.Y.; Heinemann, V.; et al. BRAF V600E Mutation in First-Line Metastatic Colorectal Cancer: An Analysis of Individual Patient Data from the ARCAD Database. JNCI J. Natl. Cancer Inst. 2021, 113, 1386–1395.
- Saito, T.; Niida, A.; Uchi, R.; Hirata, H.; Komatsu, H.; Sakimura, S.; Hayashi, S.; Nambara, S.; Kuroda, Y.; Ito, S.; et al. A temporal shift of the evolutionary principle shaping intratumor heterogeneity in colorectal cancer. Nat. Commun. 2018, 9, 2884.
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus Irinotecan, Fluorouracil, and Leucovorin for Metastatic Colorectal Cancer. N. Engl. J. Med. 2004, 350, 2335–2342.
- Cassidy, J.; Clarke, S.; Díaz-Rubio, E.; Scheithauer, W.; Figer, A.; Wong, R.; Koski, S.; Rittweger, K.; Gilberg, F.; Saltz, L. XELOX vs. FOLFOX-4 as first-line therapy for metastatic colorectal cancer: NO16966 updated results. Br. J. Cancer 2011, 105, 58–64.
- Kemeny, N.E.; Jarnagin, W.R.; Capanu, M.; Fong, Y.; Gewirtz, A.N.; DeMatteo, R.P.; D’Angelica, M.I. Randomized Phase II Trial of Adjuvant Hepatic Arterial Arterial Infusion and Systemic Chemotherapy with or without Bevacizumab in Patients with Resected Hepatic Metastases from Colorectal Cancer. J. Clin. Oncol. 2011, 29, 884–889.
- Giantonio, B.J.; Catalano, P.J.; Meropol, N.J.; O’Dwyer, P.J.; Mitchell, E.P.; Alberts, S.R.; Schwartz, M.A.; Benson, A.B., 3rd. Eastern Cooperative Oncology Group Study E3200. Bevacizumab in Combination with Oxaliplatin, Fluorouracil, and Leucovorin (FOLFOX4) for Previously Treated Metastatic Colorectal Cancer: Results from the Eastern Cooperative Oncology Group Study E3200. J. Clin. Oncol. 2007, 25, 1539–1544.
- Tang, P.A.; Cohen, S.J.; Kollmannsberger, C.; Bjarnason, G.; Virik, K.; MacKenzie, M.J.; Lourenco, L.; Wang, L.; Chen, A.; Moore, M.J. Phase II Clinical and Pharmacokinetic Study of Aflibercept in Patients with Previously Treated Metastatic Colorectal Cancer. Clin. Cancer Res. 2012, 18, 6023–6031.
More
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
799
Revisions:
2 times
(View History)
Update Date:
12 Oct 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No