Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | YAJIN ZHAO | -- | 2810 | 2023-10-10 15:52:40 | | | |
| 2 | Peter Tang | Meta information modification | 2810 | 2023-10-11 03:26:18 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Zhao, Y.; Main, K.; Aujla, T.; Keshavjee, S.; Liu, M. Necroptosis in Solid Organ Transplantation. Encyclopedia. Available online: https://encyclopedia.pub/entry/50067 (accessed on 24 June 2026).
Zhao Y, Main K, Aujla T, Keshavjee S, Liu M. Necroptosis in Solid Organ Transplantation. Encyclopedia. Available at: https://encyclopedia.pub/entry/50067. Accessed June 24, 2026.
Zhao, Yajin, Kimberly Main, Tanroop Aujla, Shaf Keshavjee, Mingyao Liu. "Necroptosis in Solid Organ Transplantation" Encyclopedia, https://encyclopedia.pub/entry/50067 (accessed June 24, 2026).
Zhao, Y., Main, K., Aujla, T., Keshavjee, S., & Liu, M. (2023, October 10). Necroptosis in Solid Organ Transplantation. In Encyclopedia. https://encyclopedia.pub/entry/50067
Zhao, Yajin, et al. "Necroptosis in Solid Organ Transplantation." Encyclopedia. Web. 10 October, 2023.
Copy Citation
Necroptosis is a type of programmed cell death involved in many diseases and has been studied in the setting of all major solid organ transplants, including the kidney, heart, liver, and lung. It is determined by the underlying donor organ conditions (e.g., age, alcohol consumption, fatty liver, hemorrhage shock, donation after circulatory death, etc.), preservation conditions and reperfusion, and allograft rejection. The specific molecular mechanisms of necroptosis have been uncovered in the organ transplantation setting, and potential targeting drugs have been identified.
programmed cell death
donor organ condition
ischemia–reperfusion injury
allograft rejection
inflammation
1. Introduction
Over the past several decades, transplantation has been performed for all the major solid organs and has become the most effective therapy for patients with end-stage organ failure [1]. However, the current practice continues to face major challenges, including shortages in organ donation and the quality of donated organs. The latter is determined by the donor conditions (e.g., age, sex, smoking history, obesity, and organ damage before donation), the types of donations (e.g., donation after brain death (DBD) vs. donation after circulatory death (DCD)), organ preservation conditions (e.g., temperature, time, static storage vs. machine perfusion), and reperfusion conditions (e.g., anesthesia, surgery, post-operative care). Graft quality is linked to post-operative primary graft dysfunction (PGD), which is a major cause of early morbidity and mortality after organ transplantation [2]. It also contributes to acute and chronic rejection post-transplantation. Moreover, the increased incidence and severity of infection (viral, bacterial, or fungal), the transfer of cancer from donor to recipient, and carcinogenesis are also challenges for transplant recipients. Among these pathological processes, acute and chronic inflammation and diverse types of cell death are two major underlying mechanisms for graft injury. Thus, therapeutics targeting these processes may prevent or reduce organ injury, leading to improved post-transplant outcomes.
Ischemia–reperfusion (IR) injury is an inevitable consequence of donor organ preservation and transplantation. IR injury is also recognized as one of the primary causes of PGD [3][4][5]. Cellular damage resulting from IR is an important risk factor not only for PGD but also for acute and chronic rejection [6]. It consists of complex pathophysiological processes, including endothelial and epithelial cell dysfunction, acute inflammation, and the activation of innate and adaptive immune responses. IR-induced cell death can be induced by a loss of energy supply, the elaboration of inflammatory mediators and toxic molecules, and the activation of programmed cell death (PCD). Recently, multiple different types of inflammation-related PCD (e.g., necroptosis, pyroptosis, ferroptosis, autophagy-associated cell death) have been reported in IR injury in lung transplantation [7]. Among these types of PCD, necroptosis is particularly interesting, as it has been investigated in all major solid organ transplantations with clinical relevance.
Necroptosis is defined as cell death mediated through a pathway that depends on the Receptor-Interacting Serine/Threonine-Protein Kinase 1/3 (RIPK1/3) complex (also called the necrosome), which can be induced by a class of death receptors (DRs), including TNF receptor 1 (TNFR1), TNF-related apoptosis-inducing ligand-receptors (TRAIL-Rs), Fas, and toll-like receptors (TLRs) [8][9][10][11][12]. The release of inflammatory cytokines, pathogen-associated molecular patterns, and damage-associated molecular patterns (DAMPs) in the donor and recipient graft can activate these DRs. Upon activation, the necrosome then recruits mixed lineage kinase domain-like protein (MLKL), which can be phosphorylated by RIPK3 [13] and subsequently translocated to lipid rafts in the plasma membrane, ultimately resulting in cell membrane rupture and cell death [10][11][14]. Necroptotic cells further release DAMPs, such as high mobility group box 1 (HMGB1), and cytokines to propagate the inflammatory response to surrounding cells [15]. Necroptosis can also be initiated by a range of intrinsic factors, such as reactive oxygen species (ROS) and intracellular Ca2+ overload [16], which, through p53 and cyclophilin D (CypD), exerts control over mitochondria permeability transition pore (mPTP) opening to trigger regulated necrosis (Figure 1) [17].
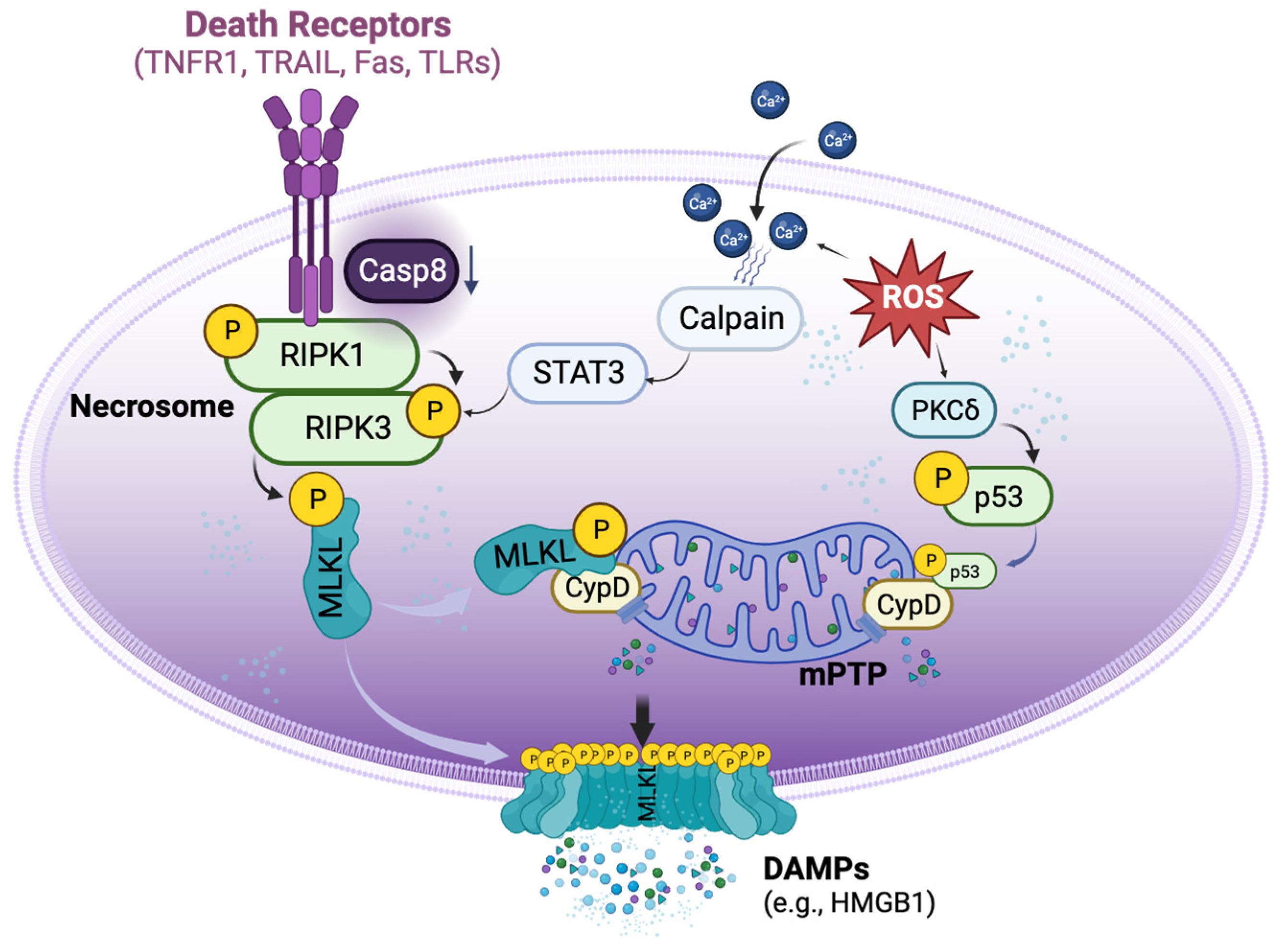
Figure 1. Basic signalling pathways of necroptosis. Necroptosis can be triggered by the activation of death receptors, which induce the phosphorylation of receptor-interacting protein kinase 1 (RIPK1) and RIPK3 to form the necrosome when the activity of caspase 8 is reduced. The necrosome then phosphorylates mixed lineage kinase domain-like protein (MLKL), causing it to translocate to the cell membrane and form pore-like structures that disrupt the integrity of the plasma membrane. These lead to the release of damage-associated molecular patterns (DAMPs), such as high mobility group box 1 (HMGB1). Aside from the activation of death receptors, necroptosis can also be triggered by reactive oxygen species (ROS). ROS can induce the translocation of p53 into the mitochondria, where it forms a complex with cyclophilin D (CypD), leading to the opening of the mitochondrial permeability transition pore (mPTP) and ultimately inducing regulated necrosis. Additionally, ROS can increase the accumulation of cytosolic Ca2+ and activate proteases such as calpain, which in turn activate necrosome formation.
2. Necroptosis in Kidney Transplantation
The first evidence of necroptosis in organ transplantation was derived from rodent kidney studies. In 2013, a research group led by Dr. Zhu-Xu Zhang and Dr. Anthony Jevnikar at the University of Western Ontario, in collaboration with Dr. Linkermann (an expert in necroptosis research), demonstrated that Ripk3−/− mice are resistant to renal IR injury and exhibited better renal function, less necrosis, and lower levels of HMGB-1 (a chromatin protein that can be secreted by immune cells or released by dead cells) in kidney tissue. Ripk3−/− kidney allografts also had reduced histological injury scores, neutrophil infiltration, fibrosis, tubulitis, and vascular injury. Moreover, animals receiving Ripk3−/− kidney allografts achieved greater rejection-free survival [18]. As RIPK1/3 are the most important mediators of necroptosis, this study built the foundation for further investigations on the role of necroptosis in solid organ transplantation.
Cold ischemia (CI) is commonly induced to prolong graft viability following donor organ harvest until the commencement of transplantation. However, prolonged CI is a risk factor for post-transplant acute kidney injury. Jain et al. used inbred mice to study the effects of CI on kidney transplantation. After 1 h of CI, the right kidney was transplanted to a recipient, and 7 days later, the native left kidney was removed and the function of the right renal graft at postoperative day 8 was assessed [19]. After transplantation, the acute tubular necrosis score was significantly higher in CI-preserved than directly transplanted renal grafts. This was accompanied by the increased expression of DRs including TNFR1 and TLR4 in the CI-preserved kidney. Additionally, the expression of RIPK1, RIPK3, and phosphorylated MLKL (pMLKL) was significantly higher in CI-preserved kidneys after transplantation [19]. These results indicate that donor kidney preservation conditions may influence the pathogenesis of necroptosis and post-transplant renal function.
Hypothermic machine perfusion (HMP) is a method used to optimize donor kidney quality during the cold preservation period. Unlike simple CI, HMP provides continuous perfusion and oxygenation, which can better sustain organ function and reduce preservation-related injury during transplantation. In rabbit kidneys, 25 min of warm ischemic exposure to simulate clinical DCD conditions was followed by either CI static preservation or HMP at 4–8 °C for 4 h. One day later, renal function was assessed, and HMP significantly inhibited the markers of inflammation, apoptosis, and necroptosis in donor kidneys compared to those preserved statically [20].
3. Necroptosis in Heart Transplantation
Zhang and Jevnikar’s group have also extensively studied necroptosis in heart transplantation. To investigate the effects of necroptosis on post-transplant rejection, B6 Ripk3−/− donor hearts were heterotopically transplanted into fully major histocompatibility complex (MHC) mismatched recipient BALB/c mice. Twelve days after transplantation, Ripk3−/− heart grafts showed significantly lower levels of endothelial damage, lymphocyte infiltration, necrosis, and HMGB1 compared to those from wild-type mice. Moreover, a brief immunosuppressive treatment with sirolimus markedly prolonged Ripk3−/− cardiac allograft survival [21]. They further studied the role of RIPK3 in chronic cardiac allograft rejection and demonstrated that RIPK3 deficiency protected donor cardiac grafts from CD4+ T cell-mediated chronic rejection and improved graft survival. Using a cell culture model, they also demonstrated that CD4+ T cells can induce RIPK3-dependent necroptosis in microvascular endothelial cells (MVEC) by releasing TNFα and inducing direct cell–cell contact cytotoxicity. Furthermore, Fas ligand and granzyme B may contribute to activated alloreactive CD4+ T cell-induced necroptosis in MVECs [22].
This group further reported that the mPTP opening is an important hallmark of necroptosis in cardiac allografts [23]. The opening of mPTP is largely regulated by CypD, which has been shown to regulate both apoptotic and necrotic cell death [24]. CypD inhibition or deficiency protected MVECs from necroptosis in cell culture. Moreover, CypD-deficient cardiac grafts showed prolonged survival in mice [23]. To further elucidate the role of necroptosis in IR injury during cardiac transplant, a cold hypoxia and warm reoxygenation cell culture model was used [25]. In MVECs, after exposure to the simulated IR conditions, apoptosis-inducing factor (AIF) translocated to the nucleus, which was prevented by the RIPK1 inhibitor, necrostatin-1 stable (Nec-1s), or CypD deficiency. Overall, CypD deficiency in donor cardiac grafts significantly mitigated graft IR injury and prolonged cardiac allograft survival [25].
Another hallmark of IR injury-induced cell death in transplantation is the cellular leakage of RNA, which can interact with TLR3 on neighbouring cells and propagate inflammation to aggravate graft injury [26]. The interaction between TLR3 and RNA leads to caspase-dependent apoptosis, RIPK-dependent necroptosis, and CypD-regulated mitochondrial damage in mouse MVECs. Moreover, TLR3 deficiency protects cardiac grafts from apoptotic and necroptotic cell death and tissue damage post-transplant in a Tlr3−/− heterotopic heart transplant model [26].
Tuuminen et al. performed an intra-abdominal heterotopic heart transplantation with fully MHC-mismatched rats, and they reported that 4 h of CI followed by 6 h of reperfusion increased the mRNA and protein expression of RIPK1/3 in rat cardiac allografts. The pre-treatment of donors and recipients with a single dose of simvastatin (an HMG-CoA reductase inhibitor) 2 h prior to allograft transplant reduced these changes and had a beneficial effect on IR injury [27].
4. Necroptosis in Liver Transplantation
IR injury is also an important contributor to graft dysfunction in liver transplantation [5]. When C57BL6 mice were subjected to warm hepatic IR (90 min ischemia/240 min reperfusion), pre-treatment with RIPK1 inhibitor necrostatin-1 (Nec-1) before ischemic onset did not attenuate IR-induced leukocyte migration, perfusion failure, and hepatocellular injury. Western blot analysis showed baseline RIPK1 expression in livers from sham-operated mice, which was reduced in IR groups. Interestingly, caspase 3 activity was significantly elevated after IR. The authors proposed that the activation of caspase 3, which can cleave RIPK1, may negatively regulate hepatic necroptosis under these specific experimental conditions [28].
Steatotic livers are prone to more severe IR injury, which has limited their usability for transplantation. In a fatty liver mouse model induced by a Western diet, steatotic livers had increased levels of RIPK1, RIPK3, and MLKL. These mice had more severe liver injury and necrosis after IR than mice on a control diet. When Mlkl−/− mice were used, although the development of hepatic steatosis was not affected, these mice had decreased hepatic neutrophil infiltration and inflammation, irrespective of diet. Moreover, Ripk3−/− or Ripk3 kinase-dead knock-in mice were protected against IR injury 24 h after reperfusion, irrespective of diet [29]. Alcohol consumption increased the levels of liver MLKL and RIPK3 in mice and corresponded with more severe liver IR injury [30].
To investigate the possible age-dependent effects of necroptosis, young (8 weeks) and aged (100 weeks) mice were subjected to liver IR (90 min ischemia/6 h reperfusion). In the aged group, there were significant increases in liver necroptosis post-IR injury [31]. Interestingly, Nec-1 was effective at reducing hepatocyte necroptosis only in aged mice, with no significant reduction in younger mice. Furthermore, IR induced endoplasmic reticulum (ER) stress in the livers of both young and aged mice, especially aged mice. The administration of an ER stress antagonist, 4-phenylbutyrate, alleviated liver IR injury in both young and aged mice. ER stress inhibition reduced hepatocyte necroptosis primarily in aged mice [31]. These studies demonstrated that pre-existing morbidities (fatty liver, aging, alcohol consumption, etc.) constitute a risk factor for necroptosis, which can be further exacerbated by IR injury during organ preservation and transplantation.
Early allograft dysfunction (EAD) following liver transplantation is a major threat to the clinical outcome of recipients [32]. In a rat isograft liver transplant model, donor livers were preserved statically for 22 h of CI followed by transplantation. One day later, significantly higher pMLKL was observed in livers with IR injury. When human samples were studied, the pMLKL score was significantly higher in the grafts of patients who developed EAD after transplantation compared to non-EAD grafts. The pMLKL score at 1 h after reperfusion and the ratio of the pMLKL score between 1 h of reperfusion and at the end of preservation were highly predictive for EAD. Human liver grafts with a high pMLKL index had significantly elevated serum levels of aminotransferases and lactate dehydrogenase 24 h after transplantation, signifying lytic cell death. Thus, the pMLKL index can serve as a reliable prognostic tool for the progression of EAD [33].
5. Necroptosis in Lung Transplantation
Interestingly, the first report on transplant-related necroptosis in the lung was from a study that investigated a phenomenon commonly referred to as kidney–lung crosstalk. In a rat allogeneic kidney transplantation model, RIPK1 and RIPK3 expression were significantly enhanced in the lung. Nec-1 given to recipients with ischemic renal grafts improved lung morphology, evidenced by decreased hemorrhage and leukocyte infiltration. The acute immune rejection of renal allograft exacerbated lung injury with enhanced RIPK1 expression. The treatment of animals with cyclosporine A, an immunosuppressive agent, significantly reduced lung injury, and when Nec-1 was combined with cyclosporine A, there was an additive benefit for lung protection [34]. Cyclosporine A is not only an immunosuppressive drug, but also an effective inhibitor of mPTP opening-related regulated necrosis [35]. It was further proposed that osteopontin, a multifunctional glycophosphoprotein that may mediate systemic inflammation, is involved in necroptosis and lung injury after the transplantation of ischemic renal allografts [36].
To determine the underlying mechanisms of IR injury in the lung transplant setting, Kim et al. used a CI and warm reperfusion cell culture model that mimicked the IR process of lung transplantation [37][38]. Human lung epithelial cells were stored at 4 °C in 50% oxygen to simulate donor lung preservation, and the cells were then returned to a serum-containing culture medium at body temperature (37 °C) to simulate warm reperfusion. Reperfusion induced mPTP opening and regulated necrosis after prolonged CI. This is mediated by the translocation of p53 into mitochondria to form a complex with CypD. Protein kinase C delta (PKCδ) plays an important role in signal transduction related to oxidative stress. The selective inhibition of PKCδ by small interference RNA (siRNA) or dV1-1, a peptide PKCδ inhibitor, reduced IR-induced inflammation, ER stress, and cell death. dV1-1 also reduced PKCδ and p53 translocation to mitochondria [37][39]. dV1-1 and its nano formula also prevented lung IR injury in a left-lung transplantation model and in a warm pulmonary IR injury model in rats [37][39].
Kim et al. further found that the RIPK1 inhibitor, Nec-1, reduced IR-induced necroptosis using the aforementioned cell culture model [38]. Necroptosis is usually triggered by the activation of DRs [40]; however, in this study, blocking DRs did not affect IR-induced necroptosis [38]. Interestingly, N-acetyl-Leu-Leu-norleucinal (ALLN), a protease calpain inhibitor, reduced RIPK1/RIPK3 expression and pMLKL via the signal transducer and activator of transcription 3 (STAT3) pathway. Blocking this calpain–STAT3–RIPK axis reduced ER stress and mitochondrial calcium dysregulation. In human lung transplant samples, mRNA levels of RIPK1, MLKL, and STAT3 were significantly increased at 2 h of reperfusion or post-transplant. Additionally, the levels of pRIPK1, pMLKL, and pSTAT3 are higher in human lung tissue samples from patients who developed PGD than those who did not [38]. The administration of Nec-1 to both donor lungs (preserved at 4 °C for 18 h) and recipients in a rat lung transplant model significantly improved pulmonary gas exchange, reduced lung edema, and inhibited necrosis [41]. Nec-1 has also been shown to significantly alleviate IR-induced lung injury, cytokine release, and the necroptosis of epithelial cells after left lung hilum clamping in mice [42]. Similarly, necrosulfonamide, an MLKL inhibitor, attenuated IR injury in a rat left hilum clamping and reperfusion model [43].
Wang et al. used a mouse left-lung transplant model to determine the donor lung conditions that may promote the development of PGD. In a single-hit model, donor lungs from inbred C57BL/6 mice were CI-preserved at 0 °C for 1 h, 72 h, or 96 h before engraftment. Multi-hit models were established by inducing 24 h of hemorrhage shock and/or 3 h of brain death before 24 h of CI preservation. Extending CI to 96 h led to increased necroptosis activation in lung grafts 24 h post-transplantation. Animals in the multi-hit group showed increased lung injury, cellular infiltration, and the activation of both necroptotic and apoptotic pathways. Nec-1 treatment significantly decreased necroptosis activation in both single- and multi-hit models of IR injury [44].
The initial recruitment of neutrophils to the reperfused lung is a critical step in IR injury hallmark post-transplantation. Using a mouse left-lung transplant model, Li et al. measured lipid peroxidation products with lipidomics and found that oxidized phosphatidylcholine species were rapidly increased after reperfusion [45], which is a signal associated with necroptosis [46]. The administration of Nec-1 to transplant recipients or using donor lungs from Ripk3−/− mice significantly improved graft function by reducing neutrophil extravasation and aggregation, leading to better subpleural vessel integrity. Graft levels of oxidized phosphatidylcholine species were not elevated in RIPK3-deficient lungs [45].
References
- WHO; Transplantation Society (TTS); Organizatión Nacional de Transplantes (ONT). Third WHO Global Consultation on Organ Donation and Transplantation: Striving to achieve self-sufficiency, March 23–25, 2010, Madrid, Spain. Transplantation 2011, 91 (Suppl. 11), S27–S28.
- Lee, J.C.; Christie, J.D. Primary graft dysfunction. Proc. Am. Thorac. Soc. 2009, 6, 39–46.
- Al-Adhami, A.; Singh, S.S.A.; Das De, S.; Singh, R.; Panjrath, G.; Shah, A.; Dalzell, J.R.; Schroder, J.; Al-Attar, N. Primary Graft Dysfunction after Heart Transplantation—Unravelling the Enigma. Curr. Probl. Cardiol. 2022, 47, 100941.
- Gelman, A.E.; Fisher, A.J.; Huang, H.J.; Baz, M.A.; Shaver, C.M.; Egan, T.M.; Mulligan, M.S. Report of the ISHLT Working Group on Primary Lung Graft Dysfunction Part III: Mechanisms: A 2016 Consensus Group Statement of the International Society for Heart and Lung Transplantation. J. Heart Lung Transplant. 2017, 36, 1114–1120.
- Hirao, H.; Nakamura, K.; Kupiec-Weglinski, J.W. Liver ischaemia–reperfusion injury: A new understanding of the role of innate immunity. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 239–256.
- Zhao, H.; Alam, A.; Soo, A.P.; George, A.J.T.; Ma, D. Ischemia-Reperfusion Injury Reduces Long Term Renal Graft Survival: Mechanism and Beyond. EBioMedicine 2018, 28, 31–42.
- Capuzzimati, M.; Hough, O.; Liu, M. Cell death and ischemia-reperfusion injury in lung transplantation. J. Heart Lung Transplant. 2022, 41, 1003–1013.
- He, S.; Wang, L.; Miao, L.; Wang, T.; Du, F.; Zhao, L.; Wang, X. Receptor Interacting Protein Kinase-3 Determines Cellular Necrotic Response to TNF-α. Cell 2009, 137, 1100–1111.
- Zhang, D.-W.; Shao, J.; Lin, J.; Zhang, N.; Lu, B.-J.; Lin, S.-C.; Dong, M.-Q.; Han, J. RIP3, an Energy Metabolism Regulator That Switches TNF-Induced Cell Death from Apoptosis to Necrosis. Science 2009, 325, 332–336.
- Zhao, J.; Jitkaew, S.; Cai, Z.; Choksi, S.; Li, Q.; Luo, J.; Liu, Z.-G. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 5322–5327.
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.-F.; Wang, F.-S.; Wang, X. Mixed Lineage Kinase Domain-like Protein MLKL Causes Necrotic Membrane Disruption upon Phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146.
- Vanden Berghe, T.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147.
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed Lineage Kinase Domain-like Protein Mediates Necrosis Signaling Downstream of RIP3 Kinase. Cell 2012, 148, 213–227.
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.-C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.-G.; Liu, Z.-G. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 2014, 16, 55–65.
- Todd, J.L.; Palmer, S.M. Danger signals in regulating the immune response to solid organ transplantation. J. Clin. Investig. 2017, 127, 2464–2472.
- Pasparakis, M.; Vandenabeele, P. Necroptosis and its role in inflammation. Nature 2015, 517, 311–320.
- Vaseva, A.V.; Marchenko, N.D.; Ji, K.; Tsirka, S.E.; Holzmann, S.; Moll, U.M. p53 Opens the Mitochondrial Permeability Transition Pore to Trigger Necrosis. Cell 2012, 149, 1536–1548.
- Lau, A.; Wang, S.; Jiang, J.; Haig, A.; Pavlosky, A.; Linkermann, A.; Zhang, Z.-X.; Jevnikar, A.M. RIPK3-Mediated Necroptosis Promotes Donor Kidney Inflammatory Injury and Reduces Allograft Survival. Am. J. Transplant. 2013, 13, 2805–2818.
- Jain, S.; Plenter, R.B.; Nydam, T.; Jani, A. Injury Pathways That Lead to AKI in a Mouse Kidney Transplant Model. Transplantation 2020, 104, 1832–1841.
- Yang, Z.; Zhong, Z.; Li, M.; Xiong, Y.; Wang, Y.; Peng, G.; Ye, Q. Hypothermic machine perfusion increases A20 expression which protects renal cells against ischemia/reperfusion injury by suppressing inflammation, apoptosis and necroptosis. Int. J. Mol. Med. 2016, 38, 161–171.
- Pavlosky, A.; Lau, A.; Su, Y.; Lian, D.; Huang, X.; Yin, Z.; Haig, A.; Jevnikar, A.M.; Zhang, Z.-X. RIPK3-Mediated Necroptosis Regulates Cardiac Allograft Rejection. Am. J. Transplant. 2014, 14, 1778–1790.
- Kwok, C.; Pavlosky, A.; Lian, D.; Jiang, J.; Huang, X.; Yin, Z.; Liu, W.; Haig, A.; Jevnikar, A.M.; Zhang, Z.-X. Necroptosis Is Involved in CD4+ T Cell-Mediated Microvascular Endothelial Cell Death and Chronic Cardiac Allograft Rejection. Transplantation 2017, 101, 2026–2037.
- Gan, I.; Jiang, J.; Lian, D.; Huang, X.; Fuhrmann, B.; Liu, W.; Haig, A.; Jevnikar, A.M.; Zhang, Z.-X. Mitochondrial permeability regulates cardiac endothelial cell necroptosis and cardiac allograft rejection. Am. J. Transplant. 2019, 19, 686–698.
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W., II; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662.
- Qamar, A.; Zhao, J.; Xu, L.; McLeod, P.; Huang, X.; Jiang, J.; Liu, W.; Haig, A.; Zhang, Z.-X. Cyclophilin D Regulates the Nuclear Translocation of AIF, Cardiac Endothelial Cell Necroptosis and Murine Cardiac Transplant Injury. Int. J. Mol. Sci. 2021, 22, 11038.
- Zhao, J.; Huang, X.; Mcleod, P.; Jiang, J.; Liu, W.; Haig, A.; Jevnikar, A.M.; Jiang, Z.; Zhang, Z.-X. Toll-like receptor 3 is an endogenous sensor of cell death and a potential target for induction of long-term cardiac transplant survival. Am. J. Transplant. 2021, 21, 3268–3279.
- Tuuminen, R.; Holmström, E.; Raissadati, A.; Saharinen, P.; Rouvinen, E.; Krebs, R.; Lemström, K.B. Simvastatin pretreatment reduces caspase-9 and RIPK1 protein activity in rat cardiac allograft ischemia-reperfusion. Transpl. Immunol. 2016, 37, 40–45.
- Rosentreter, D.; Funken, D.; Reifart, J.; Mende, K.; Rentsch, M.; Khandoga, A. RIP1-Dependent Programmed Necrosis is Negatively Regulated by Caspases During Hepatic Ischemia-Reperfusion. Shock 2015, 44, 72–76.
- Ni, H.-M.; Chao, X.; Kaseff, J.; Deng, F.; Wang, S.; Shi, Y.-H.; Li, T.; Ding, W.-X.; Jaeschke, H. Receptor-Interacting Serine/Threonine-Protein Kinase 3 (RIPK3)–Mixed Lineage Kinase Domain-Like Protein (MLKL)–Mediated Necroptosis Contributes to Ischemia-Reperfusion Injury of Steatotic Livers. Am. J. Pathol. 2019, 189, 1363–1374.
- Chen, H.; McKeen, T.; Chao, X.; Chen, A.; Deng, F.; Jaeschke, H.; Ding, W.-X.; Ni, H.-M. The role of MLKL in Hepatic Ischemia-Reperfusion Injury of Alcoholic Steatotic Livers. Int. J. Biol. Sci. 2022, 18, 1096–1106.
- Zhong, W.; Wang, X.; Rao, Z.; Pan, X.; Sun, Y.; Jiang, T.; Wang, P.; Zhou, H.; Wang, X. Aging aggravated liver ischemia and reperfusion injury by promoting hepatocyte necroptosis in an endoplasmic reticulum stress-dependent manner. Ann. Transl. Med. 2020, 8, 869.
- Olthoff, K.M.; Kulik, L.; Samstein, B.; Kaminski, M.; Abecassis, M.; Emond, J.; Shaked, A.; Christie, J.D. Validation of a current definition of early allograft dysfunction in liver transplant recipients and analysis of risk factors. Liver Transplant. 2010, 16, 943–949.
- Shi, S.; Bonaccorsi-Riani, E.; Schurink, I.; Bosch, T.v.D.; Doukas, M.; Lila, K.A.; Roest, H.P.; Xhema, D.; Gianello, P.; de Jonge, J.; et al. Liver Ischemia and Reperfusion Induce Periportal Expression of Necroptosis Executor pMLKL Which Is Associated with Early Allograft Dysfunction After Transplantation. Front. Immunol. 2022, 13, 890353.
- Zhao, H.; Ning, J.; Lemaire, A.; Koumpa, F.-S.; Sun, J.J.; Fung, A.; Gu, J.; Yi, B.; Lu, K.; Ma, D. Necroptosis and parthanatos are involved in remote lung injury after receiving ischemic renal allografts in rats. Kidney Int. 2015, 87, 738–748.
- Piot, C.; Croisille, P.; Staat, P.; Thibault, H.; Rioufol, G.; Mewton, N.; Elbelghiti, R.; Cung, T.T.; Bonnefoy, E.; Angoulvant, D.; et al. Effect of Cyclosporine on Reperfusion Injury in Acute Myocardial Infarction. N. Engl. J. Med. 2008, 359, 473–481.
- Zhao, H.; Chen, Q.; Huang, H.; Suen, K.C.; Alam, A.; Cui, J.; Ciechanowicz, S.; Ning, J.; Lu, K.; Takata, M.; et al. Osteopontin mediates necroptosis in lung injury after transplantation of ischaemic renal allografts in rats. Br. J. Anaesth. 2019, 123, 519–530.
- Kim, H.; Zhao, J.; Zhang, Q.; Wang, Y.; Lee, D.; Bai, X.; Turrell, L.; Chen, M.; Gao, W.; Keshavjee, S.; et al. δV1-1 Reduces Pulmonary Ischemia Reperfusion-Induced Lung Injury by Inhibiting Necrosis and Mitochondrial Localization of PKCδ and p53. Am. J. Transplant. 2016, 16, 83–98.
- Kim, H.; Zamel, R.; Bai, X.-H.; Lu, C.; Keshavjee, S.; Keshavjee, S.; Liu, M. Ischemia-reperfusion induces death receptor-independent necroptosis via calpain-STAT3 activation in a lung transplant setting. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 315, L595–L608.
- Lee, D.; Zhao, J.; Yang, H.; Xu, S.; Kim, H.; Pacheco, S.; Keshavjee, S.; Liu, M. Effective delivery of a rationally designed intracellular peptide drug with gold nanoparticle–peptide hybrids. Nanoscale 2015, 7, 12356–12360.
- Linkermann, A.; Green, D.R. Necroptosis. N. Engl. J. Med. 2014, 370, 455–465.
- Kanou, T.; Ohsumi, A.; Kim, H.; Chen, M.; Bai, X.; Guan, Z.; Hwang, D.; Cypel, M.; Keshavjee, S.; Liu, M. Inhibition of regulated necrosis attenuates receptor-interacting protein kinase 1–mediated ischemia-reperfusion injury after lung transplantation. J. Heart Lung Transplant. 2018, 37, 1261–1270.
- Dong, L.; Liang, F.; Lou, Z.; Li, Y.; Li, J.; Chen, Y.; Ding, J.; Jiang, B.; Wu, C.; Yu, H.; et al. Necrostatin-1 Alleviates Lung Ischemia-Reperfusion Injury via Inhibiting Necroptosis and Apoptosis of Lung Epithelial Cells. Cells 2022, 11, 3139.
- Ueda, S.; Chen-Yoshikawa, T.F.; Tanaka, S.; Yamada, Y.; Nakajima, D.; Ohsumi, A.; Date, H. Protective effect of necrosulfonamide on rat pulmonary ischemia-reperfusion injury via inhibition of necroptosis. J. Thorac. Cardiovasc. Surg. 2022, 163, e113–e122.
- Wang, X.; O’brien, M.E.; Yu, J.; Xu, C.; Zhang, Q.; Lu, S.; Liang, L.; An, X.; McDyer, J.F.; Mallampalli, R.K. Prolonged Cold Ischemia Induces Necroptotic Cell Death in Ischemia–Reperfusion Injury and Contributes to Primary Graft Dysfunction after Lung Transplantation. Am. J. Respir. Cell Mol. Biol. 2019, 61, 244–256.
- Li, W.; Terada, Y.; Tyurina, Y.Y.; Tyurin, V.A.; Bery, A.I.; Gauthier, J.M.; Higashikubo, R.; Tong, A.Y.; Zhou, D.; Nunez-Santana, F.; et al. Necroptosis triggers spatially restricted neutrophil-mediated vascular damage during lung ischemia reperfusion injury. Proc. Natl. Acad. Sci. USA 2022, 119, e2111537119.
- Wiernicki, B.; Dubois, H.; Tyurina, Y.Y.; Hassannia, B.; Bayir, H.; Kagan, V.E.; Vandenabeele, P.; Wullaert, A.; Berghe, T.V. Excessive phospholipid peroxidation distinguishes ferroptosis from other cell death modes including pyroptosis. Cell Death Dis. 2020, 11, 922.
More
Information
Subjects:
Biophysics
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
663
Revisions:
2 times
(View History)
Update Date:
11 Oct 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No