Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Xiaofei Huang | -- | 3240 | 2023-10-06 07:09:33 | | | |
| 2 | Lindsay Dong | -1 word(s) | 3239 | 2023-10-06 15:05:51 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Huang, X.; Xie, M.; Lu, X.; Mei, F.; Song, W.; Liu, Y.; Chen, L. The Roles of Periodontal Bacteria in Atherosclerosis. Encyclopedia. Available online: https://encyclopedia.pub/entry/49858 (accessed on 25 July 2026).
Huang X, Xie M, Lu X, Mei F, Song W, Liu Y, et al. The Roles of Periodontal Bacteria in Atherosclerosis. Encyclopedia. Available at: https://encyclopedia.pub/entry/49858. Accessed July 25, 2026.
Huang, Xiaofei, Mengru Xie, Xiaofeng Lu, Feng Mei, Wencheng Song, Yang Liu, Lili Chen. "The Roles of Periodontal Bacteria in Atherosclerosis" Encyclopedia, https://encyclopedia.pub/entry/49858 (accessed July 25, 2026).
Huang, X., Xie, M., Lu, X., Mei, F., Song, W., Liu, Y., & Chen, L. (2023, October 06). The Roles of Periodontal Bacteria in Atherosclerosis. In Encyclopedia. https://encyclopedia.pub/entry/49858
Huang, Xiaofei, et al. "The Roles of Periodontal Bacteria in Atherosclerosis." Encyclopedia. Web. 06 October, 2023.
Copy Citation
Atherosclerosis (AS) is an inflammatory vascular disease that constitutes a major underlying cause of cardiovascular diseases (CVD) and stroke. Infection is a contributing risk factor for AS. Epidemiological evidence has implicated individuals afflicted by periodontitis displaying an increased susceptibility to AS and CVD.
atherosclerosis
CVD
periodontal pathogens
Porphyromonas gingivalis
Aggregatibacter actinomycetemcomitans
Fusobacterium nucleatum
plaque
1. Introduction
Atherosclerotic cardiovascular disease (CVD) is a major public health problem of all humankind. It is the primary contributor to death and disability and accounts for 1/3 of the deaths in the world [1][2]. Atherosclerosis (AS) is one of the most common causes of CVD. Stenosis, obstruction or rupture of blood vessels can lead to ischemic CVDs such as myocardial infarction, stroke, and limb ischemia [3]. AS is considered to be a chronic inflammatory disease of the arterial wall caused by a variety of stimulating factors, characterized by the formation, progression, and instability of atherosclerotic plaques. It often involves medium and large arteries. The development of AS is a long-term and slow accumulation process. It usually begins with the injury of the vascular endothelial barrier and is followed by cholesterol-rich lipoprotein accumulating subcutaneously. Vascular smooth muscle cells (VSMCs) migrate from vascular media to subendothelium, proliferate and synthesize extracellular matrix (ECM), resulting in intimal thickening, which is called diffuse intimal thickening (DIT). Subsequently, the resident VSMCs and blood monocyte-derived macrophages recruited in the subendothelial space uncontrolled uptake modified lipoproteins by scavenger receptors, transforming into lipid-rich cells called “foam cells” and leading to the formation and enlargement of AS plaques. With the death of cells and the disfunction of efferocytosis, the arterial plaque gradually becomes unstable, exhibits necrosis and calcification, or even ruptures and detaches to form a thrombus [4].
Traditional risk factors for AS include lifestyle factors, primarily smoking, dyslipidemia, hypertension, and altered glucose metabolism [5]. Studies in recent decades have revealed that infection plays an important role in AS. Beginning with Fabricant and colleagues, who induced AS in chickens by Marek’s disease virus infection and prevented atherosclerotic changes by vaccination [6], microbial infections such as herpes simplex virus [7], Chlamydia pneumoniae [8], Porphyromonas gingivalis (Pg) [9], Helicobacter pylori [10], influenza A virus [11], hepatitis C virus [12], cytomegalovirus [13], and HIV [14] have all been identified as risk factors for AS.
As one of the four major human bacterial reservoirs, more than 700 bacterial species exist in the oral cavity [15][16]. It is worth noting that these bacteria maintain an ecological balance within a healthy periodontium. However, in the presence of periodontal disease, microbial dysbiosis emerges, leading to a shift from Gram-positive anaerobic bacteria to Gram-negative anaerobic bacteria. Consequently, certain bacteria opportunistically acquire pathogenic capabilities, further exacerbating the pathogenesis of the disease [17][18]. Local or systemic infections of oral origin are prevalent in the human population; for example, periodontitis is the sixth most prevalent disease on a worldwide scale, with a global prevalence of 45–50% [19]. It not only contributes to the destruction of local tissues but is also related to the development of a variety of systemic diseases such as AS. The American Heart Association (AHA) acknowledges the correlation between periodontal disease (PD) and atherosclerotic cardiovascular disease, irrespective of known confounding factors [20]. Epidemiologic evidence shows that the incidence of AS in patients with periodontitis is 1.27 times higher than that in patients without periodontitis [21].
Oral bacteria can cause temporary bacteremia during some therapy like periodontal treatment, tooth extraction or during daily oral hygiene practices such as chewing, brushing, and flossing, especially in subjects with existing periodontitis and dental pulp infections. Periodontal pathogens can reach distant organs through the blood. Researchers have detected DNA of periodontitis pathogen from atherosclerotic plaques [22][23], providing direct evidence for the link between periodontitis and AS. The main periodontal pathogens detected in the plaques include Pg, Aggregatibacter actinomycetemcomitans (Aa), Fusobacterium nucleatum (Fn), Prevotella intermedia (Pi), Tannerella forsythia (Tf), Treponema denticola (Td), and Campylobacter rectus (Cr). In addition, several benign species associated with dental plaque on the tooth surface were detected in the plaques [24].
2. Porphyromonas gingivalis
Pg is a dark, lytic, nonmotile, Gram-negative obligate anaerobes that derive energy from the fermentation of amino acids, which facilitates its survival in the subgingival sulcus and periodontal pockets. Pg is a main pathogen of periodontitis, and it forms the “red complex” with Tf and Td, which is responsible for the severe clinical manifestation of periodontal disease. Pg is one of the most common bacteria found in the subgingival biofilm of patients with periodontitis [25][26]. A retrospective study conducted in Germany involving 7804 adults diagnosed with periodontitis reported a detection rate of 68.2% for Pg in the biofilm of periodontal pockets [27]. Pg has a variety of virulence factors, such as lipopolysaccharide (LPS) on the bacterial outer membrane, which can activate the pathogen-related pattern recognition receptor signaling pathway, cause inflammatory response and the secretion of cytokines. Gingipains are trypsin-like cysteine proteinases generated by Pg that can cleave laminin, fibronectin, and collagen, activate complement pathways, and induce dysregulation of coagulation and fibrinolytic pathways.
Endothelial Barrier Disruption
Endothelial dysfunction is an early cardiovascular response to stimuli and is considered an “alarm” of AS. After entrance to the blood, Pg adheres to the cell surface of endothelial cells (ECs) via a variety of adhesins (including fimA and HagB) to interact with E-selectin, vascular cell adhesion molecule 1 (VCAM-1), intercellular cell adhesion molecule 1 (ICAM-1), and other molecules on the cell surface [28][29][30]. It can also be internalized into ECs by lipid rafts on the cell membrane.
Pg can suppress the proliferation of vascular ECs and induce cell apoptosis, thus destroying the endothelial barrier [31]. It is evidenced that gingipains can cleave neural cadherin and vascular endothelial cadherin, and degrade integrin β1, making ECs disconnect from the ECM, and come to anoikis [32][33]. Systemic inflammation induced by Pg can also promote the endothelial–mesenchymal transition (EndMT) of ECs, thereby promoting the fibrosis of arterial plaques and destroying the permeability and integrity of the vessel wall [34]. Pg-activated ECs secrete angiotensin II and pro-inflammatory cytokines such as interleukin (IL)-6, monocyte chemoattractant protein-1 (MCP-1), and granulocyte-macrophage colony-stimulating factor (GM-CSF), amplifying vascular inflammation and arterial hypertension [35].
Vascular oxidative stress is one of the pathological mechanisms of many cardiovascular diseases, including AS, hypercholesterolemia, hypertension, and diabetes mellitus [36]. Excessive or sustained reactive oxygen species (ROS) are the main characteristic of oxidative stress. Pg can promote ROS production in multiple ways. It is identified that Pg can motivate DNA methyltransferase 1 (DNMT-1) to methylate basic helix-loop-helix ARNT like 1 (BMAL1) promoter via activating TLRs-NF-κB signal axis, followed by the restrain of BMAL1 expression and release of circadian locomotor output cycles kaput (CLOCK). In turn, CLOCK phosphorylates P65 and further enhances the NF-κB signal, which aggravates oxidative stress and inflammatory response in human aortic ECs, thereby aggravating vascular endothelial injury and promoting the progress of AS [22]. Pg is also able to reduce the antioxidant mechanism and accelerate the oxidative damage of ECs through the NOS/BH4/Nrf2/GSK-3β pathway [37].
Monocyte Adherence and Aggregation
Monocyte recruitment to the endothelium is a crucial step in AS. Endothelium injury causes the subsequent chemotaxis and aggregation of monocytes to the subendothelium. Pg upregulates the expression of MCP-1, ICAM-1, VCAM-1, and E- selectin in ECs, and the expression of C-C chemokine receptor 2 (CCR2) and integrinαMβ2 in monocytes, promotes the adhesion and aggregation of monocytes to the endothelium [38][39][40][41][42]. The NF-κB pathway plays a vital role in this process. Restraint of the NF-κB pathway can abrogate ICAM-1 expression in ECs [43]. NOD1, an intracellular pattern recognition reporter, is overexpressed in Pg-infected ECs. After NOD1 recognizes Pg, the expression of ICAM-1 and VCAM-1 in ECs is up-regulated through the NF-κB signaling pathway [39]. Gas6 inhibits Pg-LPS-induced monocyte–endothelial cell interaction in vitro through the Akt/NF-κB pathway [44]. Macrophage migration inhibitory factor (MIF) secreted is augmented in Pg-infected ECs, which binds to CD74 and CXCR4 on the surface of ECs to form a receptor–ligand complex and activates ECs to express more ICAM-1 [45]. Exposure to Pg induces the increased expression of these adhesion molecules and attracts a large number of monocytes to accumulate in the subendothelium of the artery, which results in extensive secretion of inflammatory factors and exacerbates vascular and systemic inflammation [34][46].
Foam Cell Formation
The formation of foam cells is a hallmark of AS. Macrophages serve as one of the primary sources of foam cells in plaque. Pg and its components, including outer membrane vesicles (OMVs), can boost the binding and phagocytosis of macrophages to low-density lipoprotein (LDL), and macrophage-mediated modification of LDL [47]. Pg can increase the expression of CD36, a scavenger receptor that mediates cholesterol uptake through the c-Jun-AP-1 pathway [48] or ERK/NF-κB [49], as well as lysosomal integral membrane protein 2 (LIMP2) involved in cholesterol transport [50][51], so as to intensify the lipid accumulation of macrophages. Pg-infected macrophages up-regulate fatty acid binding protein 4 (FABP4), an intracellular transport protein for fatty acids, presenting more intake of fatty acids [52]. Moreover, a notable positive association was observed between serum Pg antibody and FABP4 level in clinical periodontitis patients, suggesting that Pg can promote AS and other systemic diseases by affecting FABP4 [52].
Calcification and Angiogenesis in Plaque
In the advanced stages of AS, the presence of calcium deposition within plaques, known as calcification, is frequently observed. Calcified plaques contribute to luminal narrowing and impede blood flow. However, it is worth noting that there is a prevailing viewpoint suggesting that calcified plaques exhibit greater stability and reduced susceptibility to rupture compared to non-calcified plaques. VSMCs also constitute a large portion of the plaque. Pg promotes phenotypic transformation, apoptosis, and matrix vesicle release of VSMCs, and consequently intensifies inorganic phosphate-induced vascular calcification [53]. Pg boosted VSMCs proliferation and intimal hyperplasia, and the expression of vascular cell proliferative phenotypic markers S100 calcium-binding protein A9 (S100A9) and embryonic isoform of smooth muscle myosin heavy chain (SMemb) was observed higher on the surface of VSMCs of Pg-infected mice and in aneurysm specimens from Pg-infected patients [54][55].
In response to the oxygen and nutrient demands, new blood vessels gradually form within the growing plaque, while they are often structurally abnormal and fragile. Angiogenesis may cause leakage of blood cells and inflammatory cells into the plaque, or even rupture and lead to intraplaque hemorrhage. Microarray analysis revealed that gingipains influence the focal adhesion activation, ECM receptor interactions, and the actin cytoskeleton pathway of Pg-mediated VSMCs, suggesting an impact on VSMC motility, phenotype transition, and angiogenesis processes [56]. Pg and its gingipains have been demonstrated to facilitate the upregulation of the high angiopoietin 2 (Angpt2)/Angpt1 expression ratio in VSMCs, manifesting their potential involvement in promoting vascular neogenesis, SMC proliferation, and pro-inflammatory phenotypic changes. Conversely, fimbriae and LPS lack the ability to elicit similar effects [57].
Plaque Destabilization
There is currently a dearth of research regarding the association between Pg and plaque destabilization in AS. A study conducted in 2017 demonstrated that Pg can facilitate the imbalance between Th17 and Treg cells, and encourage intra-plaque inflammation by modulating T cell differentiation during the progression of AS. This contributed to an enlarged AS lesion area, accompanied by an escalation in macrophage content and a reduction in VSMC area, thereby fostering plaque instability [58]. Pg also triggered macrophages to secret MMP9, thereby inducing the fragmentation of vascular type IV collagen, which weakened the structural support of the plaque and worsen its destabilized [59]. Be, the heightened vascular inflammation, also impairs plaque stability [60].
3. Aggregatibacter actinomycetemcomitans
Aa, a Gram-negative facultent-anaerobic coccobacillus, is the predominant bacterium isolated from caries in adolescents and adults with stage Ⅲ or Ⅳ periodontitis [61] and can lead to premature tooth loss. Aa is also one of the major bacteria in the subgingival biofilm of patients with periodontitis [26]. Ramin Akhi et al. observed that the levels of salivary IgA antibodies to MAA-LDL (p = 0.034) and Aa-HSP60 (p = 0.045) increased with an elevated number of teeth with probing depths of 4–5 mm, which may suggest the cross-activation of the humoral immune may potentially mediate the association between PD and systemic disorders [62]. Pili is an important structure for Aa adhesion to the host. Isolated Aa pili contained a low molecular mass protein (about 6.5 kDa), called Flp, and a small amount of a 54-kda protein, called Fup [63]. Examination of the binding of Aa to hydroxyapatite surfaces coated with saliva exhibited a highly adhesive interaction that seemed to rely on the formation of glycoconjugates [64]. In an oral colonization model infected with the Flp mutant of Aa, the absence of soft tissue or plaque colonization, as well as the absence of bone loss, in the Flp mutant of Aa, provides compelling evidence supporting the critical role of Flp in Aa’s virulence [65].
Aa is known to generate two types of toxins, LtxA and cytolethal-distending toxins (Cdts). LtxA binds with lymphocyte function-associated antigen-1 (LFA-1; CD11a/CD18), the receptor for LtxA on leukocytes [66][67][68], to induce macrophages pyroptosis and activation of inflammasome to release inflammatory cytokines and induce secondary immune response [69]. Aa has a significant impact on inflammation as well as the aggregation and adhesion of monocytes by up-regulating ICAM-1 and VCAM-1 on ECs [70] and macrophages [71][72]. LtxA can also arrest the G2/M phase of the cell cycle in microvascular ECs, thus hindering cell proliferation and driving cell apoptosis [70]. LPS of Aa can induce TNF-α and IL-1β production, and restrain the expression of scavenger receptor class B type-I (SR-BI) and ABCA1 in macrophages, followed by augmented cholesterol accumulation [73]. Infection with Aa elevates serum and intramural levels of TH17 cell-related factors, such as IL-1β, IL-17, IL-6, TGF-β, and IL-1β, indicating a potential induction of TH17 activation and the promotion of vascular inflammation [74]. This evidence illustrates a certain correlation between Aa and CVD. Cdts, a heterotrimeric AB2 toxin, can be internalized into cells and induce cell-cycle retardation and apoptosis in lymphocytes and other cell types [75].
4. Fusobacterium nucleatum
Fn is a species of bacteria that belongs to the genus Fusobacterium. It is a Gram-negative anaerobic bacterium. Fn acts as a bridge bacterium, facilitating the adherence of other bacteria to form complex microbial communities. It exhibits various virulence factors that contribute to its pathogenicity, including adhesins such as adhesin FadA and Fap2 [76], outer membrane proteins like radial proteins D [76], hemagglutinins, secreted toxins like butyric acid [77], LPS, and some proteases.
Recent studies have elucidated that Fn can enhance EC permeability and reduce the abundance of EC adhesion molecule-1, leading to endothelial dysfunction [78]. Fn has been shown to impair ECs proliferation and induce apoptosis [79][80]. Fn and its GroEL Fn are capable to upregulate the expression of chemotactic factors, including MCP1 and IL-8, as well as cell adhesion molecules including ICAM-1, VCAM-1, and E-selectin in ECs [81]. Fn also disrupts lipid metabolism and transport processes. Fn fosters hepatic glycolysis and lipid synthesis through the PI3K/Akt/mTOR signaling pathway, thus uplifting plasma lipid concentrations and exacerbating AS in mice [82]. Fn-infected macrophages exhibit an activation of the PI3K-AKT/MAPK/NF-κB signaling pathway, propelling the inflammatory responses and cholesterol uptake, concurrently reducing lipid excretion, leading to lipid deposition [83].
5. Prevotella intermedia
Pi, a Gram-negative bacterium, is a dominant bacterium of periodontitis and is predominant in adult patients with periodontitis [84][85]. Two genotypes, I and II, have been identified for Pi [86]. In 1992, Harou N N Shan et al. identified a new genetic group in the Pi strain, which was significantly different from genotype I in terms of DNA–DNA hybridization characteristics and peptidase and lipase activities. The new species was named Prevotella nigrescens (Pn) [87]. Pi and Pn can simultaneously exist in oral mucosa, the tongue, and tonsils, as well as in subgingival plaque in deep periodontal pockets [88][89]. Some studies have proposed that Pn strains are associated with healthy sites, whereas Pi strains are isolated from deeper sites of periodontal lesions and are thought to connect with periodontal breakdown [90][91][92]. In addition to periodontitis, Pi is also found to be abundant in colorectal cancer [93]. Moreover, Pi has been linked to subclinical hypothyroidism [94], infectious endocarditis [95], and other related conditions.
Currently, research on the impact of Pi on macrophages is limited. The pro-inflammatory effect of Pi on macrophages may aggravate the progression of AS. LPS is the major virulence factor of Pi. Similar to the LPS of Pg, the LPS of Pi differs greatly in the structure from LPS of Escherichia coli. Pi-LPS contains fewer and longer fatty acids than E. coli-type lipid A [96]. Pi-derived LPS can attract the production of macrophage inflammatory mediators such as nitric oxide (NO), IL-1β, and IL-6 through TLR4 signaling pathway [97]. In addition, a novel non-endotoxin protein, prevotella glycoprotein, was isolated from Pi, which is composed of carbohydrates and protein and is free of fatty acids [98]. PCG raises IL-8 production by human monocyte THP-1 cells and motivates human and mouse monocytes through CD14 and TLR2 but not TLR4-dependent pathways [99].
6. Tannerella forsythia
Tf is an anaerobic Gram-negative member of the Cytophaga-Bacteroides family. It was first isolated by the Forsyth Institute in the 1970s from subjects with advanced progressive periodontitis, and originally described as Fusiform Bacteroides [85]. According to the 16S rRNA phylogenetic analysis, it was reclassified to Tf [100][101]. Tf is one of the members of the “red complex”, and participates in the development of gingivitis and periodontitis. Multiple research studies have successfully detected Tf in the subgingival biofilm of patients using various techniques [26]. Several potential virulence factors have been discovered in T. forsythia, including trypsin-like [102] and PrtH proteases [103], NanH [104], a leucine-rich repeat protein BspA [105], alpha-D-glucosidase, N-acetyl-beta-glucosaminidase [106], components of the bacterial S-layer, and methylglyoxal [107].
The mechanisms underlying the involvement of Tf in the promotion of AS remain inadequately understood. Existing research findings indicate that Tf and its components, including LPS and OMVs, can enhance the secretion of pro-inflammatory mediators by macrophages, including IL-1β, IL-6, TNF-α, and IL-8 [108][109][110], while S-layer-deficient Tf mutants yield a remarkably higher secretion level [109]. Similarly, infection of mice with Tf mutant strains lacking an intact S-layer glycan core has been shown to provoke robust Th17 cell responses and researchers considered that the surface glycosylation of Tf may contribute to its persistence within the host by restraining Th17 responses [111]. Tf and BspA can also induce THP-1 to form foam cells [112]. This evidence manifests that Tf may evade recognition by the innate immune system, elicit a chronic inflammatory response, and catalyze foam cell formation in AS plaque.
7. Treponema denticola
Td is a Gram-negative bacterium from the Spirochetes family. As a partner of the “red-complex” organisms, Td is commonly found in the oral cavity, especially in subgingival plaque. Td possesses several virulence factors, such as the major outer sheath protein (MSP), ortholog of oligopeptide transporter unit (OppA), factor H-like protein-1 binding proteins, coaggregation, dentilisin, lipooligosaccharide [113], peptidoglycan, and cystalysin, which assist Td in adhesion, locomotion, immune escape and destruction of host cells [114]. Td can activate human ECs by inducing IL-8 and MCP-1 expression [115], which facilitates the chemotaxis and aggregation of monocytes to the subendothelium.
8. Campylobacter rectus
Cr is a Gram-negative anaerobic bacterium. It was initially named and identified in 1981 as Wolinella recta, and was reclassified as Campylobacter in 1991 based on phylogenetic analysis [116]. It is common in the oral cavity and gastrointestinal tract and mainly participates in oral and periodontal infections, but it is also detected in cases of severe infection outside the gastrointestinal tract [117].
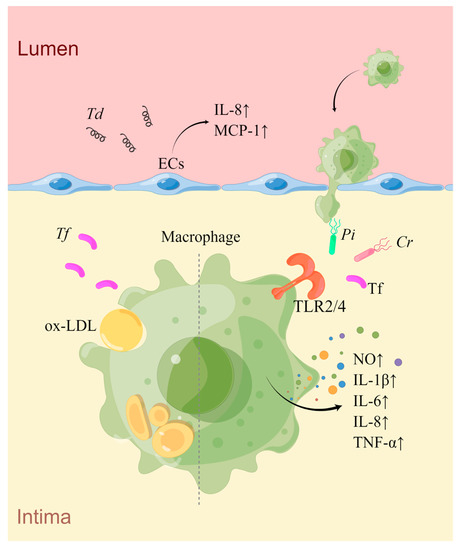
Similar to other oral bacteria, Cr possesses potent TLR4 stimulating activity, effectively triggering the macrophage TLR4 signaling pathway and inducing IL-6 secretion [118]. Currently, there is a lack of evidence of how Cr affects AS, and further research is needed (Figure 1).
Figure 1. An overview of the mechanisms of Pi, Tf, Td, and Cr in AS. Td infection induces the secretion of IL-8 and MCP1 by ECs, promoting the adhesion and aggregation of monocytes towards the sub-endothelial space. Pi, Cr, and Tf can activate macrophages via the TLR2/4 signaling pathway, leading to increased production of inflammatory mediators. Additionally, Tf can also promote macrophage-derived foam cell formation induced by ox-LDL (drawn by Figdraw).
References
- Murray, C.J.; Vos, T.; Lozano, R.; Naghavi, M.; Flaxman, A.D.; Michaud, C.; Ezzati, M.; Shibuya, K.; Salomon, J.A.; Abdalla, S.; et al. Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990–2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2197–2223.
- Herrington, W.; Lacey, B.; Sherliker, P.; Armitage, J.; Lewington, S. Epidemiology of Atherosclerosis and the Potential to Reduce the Global Burden of Atherothrombotic Disease. Circ. Res. 2016, 118, 535–546.
- Frostegard, J. Immunity, atherosclerosis and cardiovascular disease. BMC Med. 2013, 11, 117.
- Silvestre-Roig, C.; de Winther, M.P.; Weber, C.; Daemen, M.J.; Lutgens, E.; Soehnlein, O. Atherosclerotic plaque destabilization: Mechanisms, models, and therapeutic strategies. Circ. Res. 2014, 114, 214–226.
- Sanz, M.; Del Castillo, A.M.; Jepsen, S.; Gonzalez-Juanatey, J.R.; D’Aiuto, F.; Bouchard, P.; Chapple, I.; Dietrich, T.; Gotsman, I.; Graziani, F.; et al. Periodontitis and Cardiovascular Diseases. Consensus Report. Glob. Heart 2020, 15, 1.
- Fabricant, C.G.; Fabricant, J.; Minick, C.R.; Litrenta, M.M. Herpesvirus-induced atherosclerosis in chickens. Fed. Proc. 1983, 42, 2476–2479.
- Ibrahim, A.I.; Obeid, M.T.; Jouma, M.J.; Moasis, G.A.; Al-Richane, W.L.; Kindermann, I.; Boehm, M.; Roemer, K.; Mueller-Lantzsch, N.; Gartner, B.C. Detection of herpes simplex virus, cytomegalovirus and Epstein-Barr virus DNA in atherosclerotic plaques and in unaffected bypass grafts. J. Clin. Virol. 2005, 32, 29–32.
- Campbell, L.A.; Kuo, C.C. Chlamydia pneumoniae—An infectious risk factor for atherosclerosis? Nat. Rev. Microbiol. 2004, 2, 23–32.
- Seymour, G.J.; Ford, P.J.; Cullinan, M.P.; Leishman, S.; Yamazaki, K. Relationship between periodontal infections and systemic disease. Clin. Microbiol. Infect. 2007, 13 (Suppl. S4), 3–10.
- Franceschi, F.; Sepulveda, A.R.; Gasbarrini, A.; Pola, P.; Silveri, N.G.; Gasbarrini, G.; Graham, D.Y.; Genta, R.M. Cross-reactivity of anti-CagA antibodies with vascular wall antigens: Possible pathogenic link between Helicobacter pylori infection and atherosclerosis. Circulation 2002, 106, 430–434.
- Gurevich, V.S.; Pleskov, V.M.; Levaia, M.V.; Bannikov, A.I.; Mitrofanova, L.B.; Urazgil’deeva, S.A. Influenza virus infection in progressing atherosclerosis. Kardiologiia 2002, 42, 21–24.
- Hsu, Y.H.; Muo, C.H.; Liu, C.Y.; Tsai, W.C.; Hsu, C.C.; Sung, F.C.; Kao, C.H. Hepatitis C virus infection increases the risk of developing peripheral arterial disease: A 9-year population-based cohort study. J. Hepatol. 2015, 62, 519–525.
- Adam, E.; Melnick, J.L.; Probtsfield, J.L.; Petrie, B.L.; Burek, J.; Bailey, K.R.; McCollum, C.H.; DeBakey, M.E. High levels of cytomegalovirus antibody in patients requiring vascular surgery for atherosclerosis. Lancet 1987, 2, 291–293.
- Hsue, P.Y.; Waters, D.D. HIV infection and coronary heart disease: Mechanisms and management. Nat. Rev. Cardiol. 2019, 16, 745–759.
- Yamashita, Y.; Takeshita, T. The oral microbiome and human health. J. Oral Sci. 2017, 59, 201–206.
- Parahitiyawa, N.B.; Scully, C.; Leung, W.K.; Yam, W.C.; Jin, L.J.; Samaranayake, L.P. Exploring the oral bacterial flora: Current status and future directions. Oral Dis. 2010, 16, 136–145.
- Darveau, R.P.; Tanner, A.; Page, R.C. The microbial challenge in periodontitis. Periodontol. 2000 1997, 14, 12–32.
- Bui, F.Q.; Almeida-da-Silva, C.L.C.; Huynh, B.; Trinh, A.; Liu, J.; Woodward, J.; Asadi, H.; Ojcius, D.M. Association between periodontal pathogens and systemic disease. Biomed. J. 2019, 42, 27–35.
- Kassebaum, N.J.; Bernabe, E.; Dahiya, M.; Bhandari, B.; Murray, C.J.; Marcenes, W. Global burden of severe periodontitis in 1990–2010: A systematic review and meta-regression. J. Dent. Res. 2014, 93, 1045–1053.
- Lockhart, P.B.; Bolger, A.F.; Papapanou, P.N.; Osinbowale, O.; Trevisan, M.; Levison, M.E.; Taubert, K.A.; Newburger, J.W.; Gornik, H.L.; Gewitz, M.H.; et al. Periodontal disease and atherosclerotic vascular disease: Does the evidence support an independent association?: A scientific statement from the American Heart Association. Circulation 2012, 125, 2520–2544.
- Zeng, X.T.; Leng, W.D.; Lam, Y.Y.; Yan, B.P.; Wei, X.M.; Weng, H.; Kwong, J.S. Periodontal disease and carotid atherosclerosis: A meta-analysis of 17,330 participants. Int. J. Cardiol. 2016, 203, 1044–1051.
- Xie, M.; Tang, Q.; Nie, J.; Zhang, C.; Zhou, X.; Yu, S.; Sun, J.; Cheng, X.; Dong, N.; Hu, Y.; et al. BMAL1-Downregulation Aggravates Porphyromonas Gingivalis-Induced Atherosclerosis by Encouraging Oxidative Stress. Circ. Res. 2020, 126, e15–e29.
- Kannosh, I.; Staletovic, D.; Toljic, B.; Radunovic, M.; Pucar, A.; Matic Petrovic, S.; Grubisa, I.; Lazarevic, M.; Brkic, Z.; Knezevic Vukcevic, J.; et al. The presence of periopathogenic bacteria in subgingival and atherosclerotic plaques—An age related comparative analysis. J. Infect. Dev. Ctries. 2018, 12, 1088–1095.
- Fernandes, C.P.; Oliveira, F.A.F.; Silva, P.G.d.B.; Alves, A.P.N.N.; Mota, M.R.L.; Montenegro, R.C.; Burbano, R.M.R.; Seabra, A.D.; Lobo Filho, J.G.; Lima, D.L.F.; et al. Molecular analysis of oral bacteria in dental biofilm and atherosclerotic plaques of patients with vascular disease. Int. J. Cardiol. 2014, 174, 710–712.
- Datta, H.K.; Ng, W.F.; Walker, J.A.; Tuck, S.P.; Varanasi, S.S. The cell biology of bone metabolism. J. Clin. Pathol. 2008, 61, 577–587.
- Sanz, M.; Lau, L.; Herrera, D.; Morillo, J.M.; Silva, A. Methods of detection of Actinobacillus actinomycetemcomitans, Porphyromonas gingivalis and Tannerella forsythensis in periodontal microbiology, with special emphasis on advanced molecular techniques: A review. J. Clin. Periodontol. 2004, 31, 1034–1047.
- Jepsen, K.; Falk, W.; Brune, F.; Fimmers, R.; Jepsen, S.; Bekeredjian-Ding, I. Prevalence and antibiotic susceptibility trends of periodontal pathogens in the subgingival microbiota of German periodontitis patients: A retrospective surveillance study. J. Clin. Periodontol. 2021, 48, 1216–1227.
- Komatsu, T.; Nagano, K.; Sugiura, S.; Hagiwara, M.; Tanigawa, N.; Abiko, Y.; Yoshimura, F.; Furuichi, Y.; Matsushita, K. E-selectin mediates Porphyromonas gingivalis adherence to human endothelial cells. Infect. Immun. 2012, 80, 2570–2576.
- Song, H.; Belanger, M.; Whitlock, J.; Kozarov, E.; Progulske-Fox, A. Hemagglutinin B is involved in the adherence of Porphyromonas gingivalis to human coronary artery endothelial cells. Infect. Immun. 2005, 73, 7267–7273.
- Walter, C.; Zahlten, J.; Schmeck, B.; Schaudinn, C.; Hippenstiel, S.; Frisch, E.; Hocke, A.C.; Pischon, N.; Kuramitsu, H.K.; Bernimoulin, J.P.; et al. Porphyromonas gingivalis strain-dependent activation of human endothelial cells. Infect. Immun. 2004, 72, 5910–5918.
- Xie, M.; Tang, Q.; Yu, S.; Sun, J.; Mei, F.; Zhao, J.; Chen, L. Porphyromonas gingivalis disrupts vascular endothelial homeostasis in a TLR-NF-kappaB axis dependent manner. Int. J. Oral Sci. 2020, 12, 28.
- Sheets, S.M.; Potempa, J.; Travis, J.; Casiano, C.A.; Fletcher, H.M. Gingipains from Porphyromonas gingivalis W83 induce cell adhesion molecule cleavage and apoptosis in endothelial cells. Infect. Immun. 2005, 73, 1543–1552.
- Aoudjit, F.; Vuori, K. Matrix attachment regulates Fas-induced apoptosis in endothelial cells: A role for c-flip and implications for anoikis. J. Cell. Biol. 2001, 152, 633–643.
- Suh, J.S.; Kim, S.; Bostrom, K.I.; Wang, C.Y.; Kim, R.H.; Park, N.H. Periodontitis-induced systemic inflammation exacerbates atherosclerosis partly via endothelial-mesenchymal transition in mice. Int. J. Oral Sci. 2019, 11, 21.
- Viafara-Garcia, S.M.; Morantes, S.J.; Chacon-Quintero, Y.; Castillo, D.M.; Lafaurie, G.I.; Buitrago, D.M. Repeated Porphyromonas gingivalis W83 exposure leads to release pro-inflammatory cytokynes and angiotensin II in coronary artery endothelial cells. Sci. Rep. 2019, 9, 19379.
- Forstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735.
- Sampath, C.; Okoro, E.U.; Gipson, M.J.; Chukkapalli, S.S.; Farmer-Dixon, C.M.; Gangula, P.R. Porphyromonas gingivalis infection alters Nrf2-phase II enzymes and nitric oxide in primary human aortic endothelial cells. J. Periodontol. 2021, 92, 54–65.
- Hashizume, T.; Kurita-Ochiai, T.; Yamamoto, M. Porphyromonas gingivalis stimulates monocyte adhesion to human umbilical vein endothelial cells. FEMS Immunol. Med. Microbiol. 2011, 62, 57–65.
- Wan, M.; Liu, J.; Ouyang, X. Nucleotide-binding oligomerization domain 1 regulates Porphyromonas gingivalis-induced vascular cell adhesion molecule 1 and intercellular adhesion molecule 1 expression in endothelial cells through NF-kappaB pathway. J. Periodontal Res. 2015, 50, 189–196.
- Takahashi, Y.; Davey, M.; Yumoto, H.; Gibson, F.C., 3rd; Genco, C.A. Fimbria-dependent activation of pro-inflammatory molecules in Porphyromonas gingivalis infected human aortic endothelial cells. Cell. Microbiol. 2006, 8, 738–757.
- Li, Q.; Liu, J.; Liu, W.; Chu, Y.; Zhong, J.; Xie, Y.; Lou, X.; Ouyang, X. LOX-1 Regulates -Induced Monocyte Migration and Adhesion to Human Umbilical Vein Endothelial Cells. Front. Cell. Dev. Biol. 2020, 8, 596.
- Xu, W.; Pan, Y.; Xu, Q.; Wu, Y.; Pan, J.; Hou, J.; Lin, L.; Tang, X.; Li, C.; Liu, J.; et al. Porphyromonas gingivalis ATCC 33277 promotes intercellular adhesion molecule-1 expression in endothelial cells and monocyte-endothelial cell adhesion through macrophage migration inhibitory factor. BMC Microbiol. 2018, 18, 16.
- Zhang, D.; Zheng, H.; Zhao, J.; Lin, L.; Li, C.; Liu, J.; Pan, Y. Porphorymonas gingivalis induces intracellular adhesion molecule-1 expression in endothelial cells through the nuclear factor-kappaB pathway, but not through the p38 MAPK pathway. J. Periodontal Res. 2011, 46, 31–38.
- Wang, X.; Liu, Y.; Zhang, S.; Ouyang, X.; Wang, Y.; Jiang, Y.; An, N. Crosstalk between Akt and NF-kappaB pathway mediates inhibitory effect of gas6 on monocytes-endothelial cells interactions stimulated by P. gingivalis-LPS. J. Cell. Mol. Med. 2020, 24, 7979–7990.
- Wu, Y.; Xu, W.; Hou, J.; Liu, Y.; Li, R.; Liu, J.; Li, C.; Tang, X.; Lin, L.; Pan, Y.; et al. Porphyromonas gingivalis-Induced MIF Regulates Intercellular Adhesion Molecule-1 Expression in EA.hy926 Cells and Monocyte-Endothelial Cell Adhesion Through the Receptors CD74 and CXCR4. Inflammation 2019, 42, 874–883.
- Hayashi, C.; Gudino, C.V.; Gibson, F.C., 3rd; Genco, C.A. Review: Pathogen-induced inflammation at sites distant from oral infection: Bacterial persistence and induction of cell-specific innate immune inflammatory pathways. Mol. Oral Microbiol. 2010, 25, 305–316.
- Qi, M.; Miyakawa, H.; Kuramitsu, H.K. Porphyromonas gingivalis induces murine macrophage foam cell formation. Microb. Pathog. 2003, 35, 259–267.
- Li, X.Y.; Wang, C.; Xiang, X.R.; Chen, F.C.; Yang, C.M.; Wu, J. Porphyromonas gingivalis lipopolysaccharide increases lipid accumulation by affecting CD36 and ATP-binding cassette transporter A1 in macrophages. Oncol. Rep. 2013, 30, 1329–1336.
- Liang, D.Y.; Liu, F.; Chen, J.X.; He, X.L.; Zhou, Y.L.; Ge, B.X.; Luo, L.J. Porphyromonas gingivalis infected macrophages upregulate CD36 expression via ERK/NF-kappaB pathway. Cell. Signal. 2016, 28, 1292–1303.
- Yang, Y.; He, X.; Xia, S.; Liu, F.; Luo, L. Porphyromonas gingivalis facilitated the foam cell formation via lysosomal integral membrane protein 2 (LIMP2). J. Periodontal Res. 2021, 56, 265–274.
- Ruan, Q.; Guan, P.; Qi, W.; Li, J.; Xi, M.; Xiao, L.; Zhong, S.; Ma, D.; Ni, J. Porphyromonas gingivalis regulates atherosclerosis through an immune pathway. Front. Immunol. 2023, 14, 1103592.
- Kim, D.J.; Rho, J.H.; Woo, B.H.; Joo, J.Y.; Lee, J.Y.; Song, J.M.; Lee, J.H.; Park, H.R. Periodontal Pathogens Modulate Lipid Flux via Fatty Acid Binding Protein 4. J. Dent. Res. 2019, 98, 1511–1520.
- Park, H.J.; Kim, Y.; Kim, M.K.; Park, H.R.; Kim, H.J.; Bae, S.K.; Bae, M.K. Infection of Porphyromonas gingivalis Increases Phosphate-Induced Calcification of Vascular Smooth Muscle Cells. Cells 2020, 9, 2694.
- Hokamura, K.; Inaba, H.; Nakano, K.; Nomura, R.; Yoshioka, H.; Taniguchi, K.; Ooshima, T.; Wada, K.; Amano, A.; Umemura, K. Molecular analysis of aortic intimal hyperplasia caused by Porphyromonas gingivalis infection in mice with endothelial damage. J. Periodontal Res. 2010, 45, 337–344.
- Inaba, H.; Hokamura, K.; Nakano, K.; Nomura, R.; Katayama, K.; Nakajima, A.; Yoshioka, H.; Taniguchi, K.; Kamisaki, Y.; Ooshima, T.; et al. Upregulation of S100 calcium-binding protein A9 is required for induction of smooth muscle cell proliferation by a periodontal pathogen. Febs Lett. 2009, 583, 128–134.
- Zhang, B.; Sirsjo, A.; Khalaf, H.; Bengtsson, T. Transcriptional profiling of human smooth muscle cells infected with gingipain and fimbriae mutants of Porphyromonas gingivalis. Sci. Rep. 2016, 6, 21911.
- Zhang, B.; Khalaf, H.; Sirsjo, A.; Bengtsson, T. Gingipains from the Periodontal Pathogen Porphyromonas gingivalis Play a Significant Role in Regulation of Angiopoietin 1 and Angiopoietin 2 in Human Aortic Smooth Muscle Cells. Infect. Immun. 2015, 83, 4256–4265.
- Yang, J.; Wu, J.; Zhang, R.; Yao, M.; Liu, Y.; Miao, L.; Sun, W. Porphyromonas gingivalis oral infection promote T helper 17/Treg imbalance in the development of atherosclerosis. J. Dent. Sci. 2017, 12, 60–69.
- Mubarokah, S.N.; Susilawati, I.D.A.; Sumarno, S.; Muliartha, I.K.G.; Sargowo, D. Porphyromonas gingivalis Induced Fragmentation of Type IV Collagen Through Macrophage-Activated MMP-9: (In Vitro Study of Collagenolytic Mechanism in Pathogenesis of Atherosclerotic Plaque Rupture). Indones. Biomed. J. 2009, 1, 88.
- Shah, P.K. Inflammation and plaque vulnerability. Cardiovasc. Drugs Ther. 2009, 23, 31–40.
- Herbert, B.A.; Novince, C.M.; Kirkwood, K.L. Aggregatibacter actinomycetemcomitans, a potent immunoregulator of the periodontal host defense system and alveolar bone homeostasis. Mol. Oral Microbiol. 2016, 31, 207–227.
- Akhi, R.; Nissinen, A.E.; Wang, C.; Kyrklund, M.; Paju, S.; Mantyla, P.; Buhlin, K.; Sinisalo, J.; Pussinen, P.J.; Horkko, S. Salivary IgA antibody to malondialdehyde-acetaldehyde associates with mild periodontal pocket depth. Oral Dis. 2022, 28, 2285–2293.
- Ishihara, K.; Honma, K.; Miura, T.; Kato, T.; Okuda, K. Cloning and sequence analysis of the fimbriae associated protein (fap) gene from Actinobacillus actinomycetemcomitans. Microb. Pathog. 1997, 23, 63–69.
- Fine, D.H.; Furgang, D.; Kaplan, J.; Charlesworth, J.; Figurski, D.H. Tenacious adhesion of Actinobacillus actinomycetemcomitans strain CU1000 to salivary-coated hydroxyapatite. Arch. Oral Biol. 1999, 44, 1063–1076.
- Schreiner, H.C.; Sinatra, K.; Kaplan, J.B.; Furgang, D.; Kachlany, S.C.; Planet, P.J.; Perez, B.A.; Figurski, D.H.; Fine, D.H. Tight-adherence genes of Actinobacillus actinomycetemcomitans are required for virulence in a rat model. Proc. Natl. Acad. Sci. USA 2003, 100, 7295–7300.
- Kieba, I.R.; Fong, K.P.; Tang, H.Y.; Hoffman, K.E.; Speicher, D.W.; Klickstein, L.B.; Lally, E.T. Aggregatibacter actinomycetemcomitans leukotoxin requires beta-sheets 1 and 2 of the human CD11a beta-propeller for cytotoxicity. Cell. Microbiol. 2007, 9, 2689–2699.
- Dileepan, T.; Kachlany, S.C.; Balashova, N.V.; Patel, J.; Maheswaran, S.K. Human CD18 is the functional receptor for Aggregatibacter actinomycetemcomitans leukotoxin. Infect. Immun. 2007, 75, 4851–4856.
- Lally, E.T.; Kieba, I.R.; Sato, A.; Green, C.L.; Rosenbloom, J.; Korostoff, J.; Wang, J.F.; Shenker, B.J.; Ortlepp, S.; Robinson, M.K.; et al. RTX toxins recognize a beta2 integrin on the surface of human target cells. J. Biol. Chem. 1997, 272, 30463–30469.
- Shenker, B.J.; Ojcius, D.M.; Walker, L.P.; Zekavat, A.; Scuron, M.D.; Boesze-Battaglia, K. Aggregatibacter actinomycetemcomitans cytolethal distending toxin activates the NLRP3 inflammasome in human macrophages, leading to the release of proinflammatory cytokines. Infect. Immun. 2015, 83, 1487–1496.
- Dietmann, A.; Millonig, A.; Combes, V.; Couraud, P.O.; Kachlany, S.C.; Grau, G.E. Effects of Aggregatibacter actinomycetemcomitans leukotoxin on endothelial cells. Microb. Pathog. 2013, 61–62, 43–50.
- Tsutsumi, T.; Nakashima, K.; Isoda, T.; Yokota, M.; Nishihara, T. Involvement of adhesion molecule in in vitro plaque-like formation of macrophages stimulated with Aggregatibacter actinomycetemcomitans lipopolysaccharide. J. Periodontal Res. 2010, 45, 550–556.
- Oksaharju, A.; Lappalainen, J.; Tuomainen, A.M.; Pussinen, P.J.; Puolakkainen, M.; Kovanen, P.T.; Lindstedt, K.A. Pro-atherogenic lung and oral pathogens induce an inflammatory response in human and mouse mast cells. J. Cell. Mol. Med. 2009, 13, 103–113.
- Lakio, L.; Lehto, M.; Tuomainen, A.M.; Jauhiainen, M.; Malle, E.; Asikainen, S.; Pussinen, P.J. Pro-atherogenic properties of lipopolysaccharide from the periodontal pathogen Actinobacillus actinomycetemcomitans. J. Endotoxin Res. 2006, 12, 57–64.
- Jia, R.; Hashizume-Takizawa, T.; Du, Y.; Yamamoto, M.; Kurita-Ochiai, T. Aggregatibacter actinomycetemcomitans induces Th17 cells in atherosclerotic lesions. Pathog. Dis. 2015, 73, ftu027.
- Boesze-Battaglia, K.; Dhingra, A.; Walker, L.M.; Zekavat, A.; Shenker, B.J. Internalization and Intoxication of Human Macrophages by the Active Subunit of the Aggregatibacter actinomycetemcomitans Cytolethal Distending Toxin Is Dependent Upon Cellugyrin (Synaptogyrin-2). Front. Immunol. 2020, 11, 1262.
- Kaplan, C.W.; Ma, X.; Paranjpe, A.; Jewett, A.; Lux, R.; Kinder-Haake, S.; Shi, W. Fusobacterium nucleatum outer membrane proteins Fap2 and RadD induce cell death in human lymphocytes. Infect. Immun. 2010, 78, 4773–4778.
- Lee, P.; Tan, K.S. Fusobacterium nucleatum activates the immune response through retinoic acid-inducible gene I. J. Dent. Res. 2014, 93, 162–168.
- Farrugia, C.; Stafford, G.P.; Gains, A.F.; Cutts, A.R.; Murdoch, C. Fusobacterium nucleatum mediates endothelial damage and increased permeability following single species and polymicrobial infection. J. Periodontol. 2022, 93, 1421–1433.
- Mendes, R.T.; Nguyen, D.; Stephens, D.; Pamuk, F.; Fernandes, D.; Van Dyke, T.E.; Kantarci, A. Endothelial Cell Response to Fusobacterium nucleatum. Infect. Immun. 2016, 84, 2141–2148.
- Wang, Q.; Zhao, L.; Xu, C.; Zhou, J.; Wu, Y. Fusobacterium nucleatum stimulates monocyte adhesion to and transmigration through endothelial cells. Arch. Oral Biol. 2019, 100, 86–92.
- Lee, H.R.; Jun, H.K.; Kim, H.D.; Lee, S.H.; Choi, B.K. Fusobacterium nucleatum GroEL induces risk factors of atherosclerosis in human microvascular endothelial cells and ApoE(-/-) mice. Mol. Oral Microbiol. 2012, 27, 109–123.
- Zhou, L.J.; Lin, W.Z.; Meng, X.Q.; Zhu, H.; Liu, T.; Du, L.J.; Bai, X.B.; Chen, B.Y.; Liu, Y.; Xu, Y.; et al. Periodontitis exacerbates atherosclerosis through Fusobacterium nucleatum-promoted hepatic glycolysis and lipogenesis. Cardiovasc. Res. 2023, 119, 1706–1717.
- Shen, S.; Sun, T.; Ding, X.; Gu, X.; Wang, Y.; Ma, X.; Li, Z.; Gao, H.; Ge, S.; Feng, Q. The exoprotein Gbp of Fusobacterium nucleatum promotes THP-1 cell lipid deposition by binding to CypA and activating PI3K-AKT/MAPK/NF-kappaB pathways. J. Adv. Res. 2023.
- Slots, J.; Bragd, L.; Wikstrom, M.; Dahlen, G. The occurrence of Actinobacillus actinomycetemcomitans, Bacteroides gingivalis and Bacteroides intermedius in destructive periodontal disease in adults. J. Clin. Periodontol. 1986, 13, 570–577.
- Tanner, A.C.; Haffer, C.; Bratthall, G.T.; Visconti, R.A.; Socransky, S.S. A study of the bacteria associated with advancing periodontitis in man. J. Clin. Periodontol. 1979, 6, 278–307.
- Lie, M.A.; van der Weijden, G.A.; Timmerman, M.F.; Loos, B.G.; van Steenbergen, T.J.M.; van der Velden, U. Occurrence of Prevotella intermedia and Prevotella nigrescens in relation to gingivitis and gingival health. J. Clin. Periodontol. 2001, 28, 189–193.
- Shah, H.N.; Gharbia, S.E. Biochemical and chemical studies on strains designated Prevotella intermedia and proposal of a new pigmented species, Prevotella nigrescens sp. nov. Int. J. Syst. Bacteriol. 1992, 42, 542–546.
- Van der Velden, U.; Van Winkelhoff, A.J.; Abbas, F.; De Graaff, J. The habitat of periodontopathic micro-organisms. J. Clin. Periodontol. 1986, 13, 243–248.
- Van Winkelhoff, A.J.; Van der Velden, U.; Winkel, E.G.; de Graaff, J. Black-pigmented Bacteroides and motile organisms on oral mucosal surfaces in individuals with and without periodontal breakdown. J. Periodontal Res. 1986, 21, 434–439.
- Gharbia, S.E.; Haapasalo, M.; Shah, H.N.; Kotiranta, A.; Lounatmaa, K.; Pearce, M.A.; Devine, D.A. Characterization of Prevotella intermedia and Prevotella nigrescens isolates from periodontic and endodontic infections. J. Periodontol. 1994, 65, 56–61.
- Dahlen, G.; Wikstrom, M.; Renvert, S.; Gmur, R.; Guggenheim, B. Biochemical and serological characterization of Bacteroides intermedius strains isolated from the deep periodontal pocket. J. Clin. Microbiol. 1990, 28, 2269–2274.
- Matto, J.; Saarela, M.; von Troil-Linden, B.; Kononen, E.; Jousimies-Somer, H.; Torkko, H.; Alaluusua, S.; Asikainen, S. Distribution and genetic analysis of oral Prevotella intermedia and Prevotella nigrescens. Oral Microbiol. Immunol. 1996, 11, 96–102.
- Lo, C.H.; Wu, D.C.; Jao, S.W.; Wu, C.C.; Lin, C.Y.; Chuang, C.H.; Lin, Y.B.; Chen, C.H.; Chen, Y.T.; Chen, J.H.; et al. Enrichment of Prevotella intermedia in human colorectal cancer and its additive effects with Fusobacterium nucleatum on the malignant transformation of colorectal adenomas. J. Biomed. Sci. 2022, 29, 88.
- Dong, T.; Xu, S.; Chen, Z.Y.; Liang, Y.J.; Meng, X.Q.; Niu, C.G.; Yuan, K.Y.; Li, P.L.; Duan, S.Z.; Huang, Z.W. Prevotella intermedia Aggravates Subclinical Hypothyroidism. J. Dent. Res. 2023, 102, 814–824.
- Boukobza, M.; Raffoul, R.; Duval, X.; Laissy, J.P. First report of prosthetic aortic valve Infective Endocarditis due to Prevotella Intermedia. Ann. Cardiol. Angeiol. 2022, 71, 240–242.
- Hashimoto, M.; Asai, Y.; Tamai, R.; Jinno, T.; Umatani, K.; Ogawa, T. Chemical structure and immunobiological activity of lipid A from Prevotella intermedia ATCC 25611 lipopolysaccharide. Febs Lett. 2003, 543, 98–102.
- Choi, E.Y.; Jin, J.Y.; Lee, J.Y.; Choi, J.I.; Choi, I.S.; Kim, S.J. Melatonin inhibits Prevotella intermedia lipopolysaccharide-induced production of nitric oxide and interleukin-6 in murine macrophages by suppressing NF-kappaB and STAT1 activity. J. Pineal Res. 2011, 50, 197–206.
- Iki, K.; Kawahara, K.; Sawamura, S.; Arakaki, R.; Sakuta, T.; Sugiyama, A.; Tamura, H.; Sueda, T.; Hamada, S.; Takada, H. A novel component different from endotoxin extracted from Prevotella intermedia ATCC 25611 activates lymphoid cells from C3H/HeJ mice and gingival fibroblasts from humans. Infect. Immun. 1997, 65, 4531–4538.
- Sugawara, S.; Yang, S.; Iki, K.; Hatakeyama, J.; Tamai, R.; Takeuchi, O.; Akashi, S.; Espevik, T.; Akira, S.; Takada, H. Monocytic cell activation by Nonendotoxic glycoprotein from Prevotella intermedia ATCC 25611 is mediated by toll-like receptor 2. Infect. Immun. 2001, 69, 4951–4957.
- Sakamoto, M.; Suzuki, M.; Umeda, M.; Ishikawa, I.; Benno, Y. Reclassification of Bacteroides forsythus (Tanner et al. 1986) as Tannerella forsythensis corrig., gen. nov., comb. nov. Int. J. Syst. Evol. Microbiol. 2002, 52, 841–849.
- Maiden, M.F.; Cohee, P.; Tanner, A.C. Proposal to conserve the adjectival form of the specific epithet in the reclassification of Bacteroides forsythus Tanner et al. 1986 to the genus Tannerella Sakamoto et al. 2002 as Tannerella forsythia corrig., gen. nov., comb. nov. Request for an Opinion. Int. J. Syst. Evol. Microbiol. 2003, 53, 2111–2112.
- Song, Q.; Zhang, X.; Li, N.; Shen, J.; Cheng, J. A propeptide-independent protease from Tannerella sp.6_1_58FAA_CT1 displays trypsin-like specificity. J. Basic. Microbiol. 2017, 57, 50–56.
- Hamlet, S.M.; Taiyeb-Ali, T.B.; Cullinan, M.P.; Westerman, B.; Palmer, J.E.; Seymour, G.J. Tannerella forsythensis prtH genotype and association with periodontal status. J. Periodontol. 2007, 78, 344–350.
- Thompson, H.; Homer, K.A.; Rao, S.; Booth, V.; Hosie, A.H. An orthologue of Bacteroides fragilis NanH is the principal sialidase in Tannerella forsythia. J. Bacteriol. 2009, 191, 3623–3628.
- Sharma, A.; Inagaki, S.; Honma, K.; Sfintescu, C.; Baker, P.J.; Evans, R.T. Tannerella forsythia-induced alveolar bone loss in mice involves leucine-rich-repeat BspA protein. J. Dent. Res. 2005, 84, 462–467.
- Hughes, C.V.; Malki, G.; Loo, C.Y.; Tanner, A.C.; Ganeshkumar, N. Cloning and expression of alpha-D-glucosidase and N-acetyl-beta-glucosaminidase from the periodontal pathogen, Tannerella forsythensis (Bacteroides forsythus). Oral Microbiol. Immunol. 2003, 18, 309–312.
- Retamal, I.N.; Hernandez, R.; Gonzalez-Rivas, C.; Caceres, M.; Arancibia, R.; Romero, A.; Martinez, C.; Tobar, N.; Martinez, J.; Smith, P.C. Methylglyoxal and methylglyoxal-modified collagen as inducers of cellular injury in gingival connective tissue cells. J. Periodontal Res. 2016, 51, 812–821.
- Bodet, C.; Chandad, F.; Grenier, D. Inflammatory responses of a macrophage/epithelial cell co-culture model to mono and mixed infections with Porphyromonas gingivalis, Treponema denticola, and Tannerella forsythia. Microbes Infect. 2006, 8, 27–35.
- Sekot, G.; Posch, G.; Messner, P.; Matejka, M.; Rausch-Fan, X.; Andrukhov, O.; Schaffer, C. Potential of the Tannerella forsythia S-layer to delay the immune response. J. Dent. Res. 2011, 90, 109–114.
- Cecil, J.D.; O’Brien-Simpson, N.M.; Lenzo, J.C.; Holden, J.A.; Singleton, W.; Perez-Gonzalez, A.; Mansell, A.; Reynolds, E.C. Outer Membrane Vesicles Prime and Activate Macrophage Inflammasomes and Cytokine Secretion In Vitro and In Vivo. Front. Immunol. 2017, 8, 1017.
- Settem, R.P.; Honma, K.; Nakajima, T.; Phansopa, C.; Roy, S.; Stafford, G.P.; Sharma, A. A bacterial glycan core linked to surface (S)-layer proteins modulates host immunity through Th17 suppression. Mucosal Immunol. 2013, 6, 415–426.
- Lee, H.R.; Jun, H.K.; Choi, B.K. Tannerella forsythia BspA increases the risk factors for atherosclerosis in ApoE(-/-) mice. Oral Dis. 2014, 20, 803–808.
- Tanabe, S.; Bodet, C.; Grenier, D. Treponema denticola lipooligosaccharide activates gingival fibroblasts and upregulates inflammatory mediator production. J. Cell. Physiol. 2008, 216, 727–731.
- Ishihara, K. Virulence factors of Treponema denticola. Periodontol. 2000 2010, 54, 117–135.
- Okuda, T.; Kimizuka, R.; Miyamoto, M.; Kato, T.; Yamada, S.; Okuda, K.; Ishihara, K. Treponema denticola induces interleukin-8 and macrophage chemoattractant protein 1 production in human umbilical vein epithelial cells. Microbes Infect. 2007, 9, 907–913.
- Vandamme, P.; Falsen, E.; Rossau, R.; Hoste, B.; Segers, P.; Tytgat, R.; De Ley, J. Revision of Campylobacter, Helicobacter, and Wolinella taxonomy: Emendation of generic descriptions and proposal of Arcobacter gen. nov. Int. J. Syst. Bacteriol. 1991, 41, 88–103.
- Lam, J.Y.; Wu, A.K.; Ngai, D.C.; Teng, J.L.; Wong, E.S.; Lau, S.K.; Lee, R.A.; Woo, P.C. Three cases of severe invasive infections caused by Campylobacter rectus and first report of fatal C. rectus infection. J. Clin. Microbiol. 2011, 49, 1687–1691.
- Marchesan, J.; Jiao, Y.; Schaff, R.A.; Hao, J.; Morelli, T.; Kinney, J.S.; Gerow, E.; Sheridan, R.; Rodrigues, V.; Paster, B.J.; et al. TLR4, NOD1 and NOD2 mediate immune recognition of putative newly identified periodontal pathogens. Mol. Oral Microbiol. 2016, 31, 243–258.
More
Information
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
580
Revisions:
2 times
(View History)
Update Date:
06 Oct 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No