Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Flora Tassone | -- | 7889 | 2023-09-27 13:55:19 | | | |
| 2 | Camila Xu | Meta information modification | 7889 | 2023-09-28 03:40:57 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Tassone, F.; Protic, D.; Allen, E.G.; Archibald, A.D.; Baud, A.; Brown, T.W.; Budimirovic, D.B.; Cohen, J.; Dufour, B.; Eiges, R.; et al. The Molecular Basis of Fragile X-Premutation-Associated Conditions. Encyclopedia. Available online: https://encyclopedia.pub/entry/49717 (accessed on 21 July 2026).
Tassone F, Protic D, Allen EG, Archibald AD, Baud A, Brown TW, et al. The Molecular Basis of Fragile X-Premutation-Associated Conditions. Encyclopedia. Available at: https://encyclopedia.pub/entry/49717. Accessed July 21, 2026.
Tassone, Flora, Dragana Protic, Emily Graves Allen, Alison D. Archibald, Anna Baud, Ted W. Brown, Dejan B. Budimirovic, Jonathan Cohen, Brett Dufour, Rachel Eiges, et al. "The Molecular Basis of Fragile X-Premutation-Associated Conditions" Encyclopedia, https://encyclopedia.pub/entry/49717 (accessed July 21, 2026).
Tassone, F., Protic, D., Allen, E.G., Archibald, A.D., Baud, A., Brown, T.W., Budimirovic, D.B., Cohen, J., Dufour, B., Eiges, R., Elvassore, N., Gabis, L.V., Grudzien, S.J., Hall, D.A., Hessl, D., Hogan, A., Hunter, J.E., Jin, P., Jiraanont, P., ...Hagerman, R.J.. (2023, September 27). The Molecular Basis of Fragile X-Premutation-Associated Conditions. In Encyclopedia. https://encyclopedia.pub/entry/49717
Tassone, Flora, et al. "The Molecular Basis of Fragile X-Premutation-Associated Conditions." Encyclopedia. Web. 27 September, 2023.
Copy Citation
The premutation of the fragile X messenger ribonucleoprotein 1 (FMR1) gene is characterized by an expansion of the CGG trinucleotide repeats (55 to 200 CGGs) in the 5’ untranslated region and increased levels of FMR1 mRNA. Molecular mechanisms leading to fragile X-premutation-associated conditions (FXPAC) include cotranscriptional R-loop formations, FMR1 mRNA toxicity through both RNA gelation into nuclear foci and sequestration of various CGG-repeat-binding proteins, and the repeat-associated non-AUG (RAN)-initiated translation of potentially toxic proteins.
FMR1 premutation
FXPAC
FXTAS
FXAND
FXPOI
1. Introduction
The discovery and sequencing of the fragile X messenger ribonucleoprotein 1 (FMR1) gene [1] have led to new molecular testing to facilitate the diagnosis of those with fragile X syndrome (FXS) with >200 CGG repeats, and the methylation of the promoter and the repeats located within the 5′UTR of the gene. Carriers of the premutation (PM) were found to have 55 to 200 CGG repeats, did not have methylation, could pass on the full mutation to their offspring, and were presumed to be unaffected because FMR1 protein (FMRP) levels were usually normal. Males with the PM were called “non-penetrant and transmitting males” because they were thought to be unaffected and passed on the PM to their daughters without the repeat expanding to the FM range. The PM term reflected the lack of clinical involvement, and this concept was soon to crumble.
Even before the discovery of the FMR1 gene, four women, who had sons with FXS, attending a National Fragile X Foundation (NFXF) conference luncheon in 1987, surprised the others at the table, including scientists, as they all spoke about early menopause in their 30 s. In a subsequent survey, 104 female carriers were divided into those that had an IQ less than 85 vs. greater than or equal to 85. Thirteen percent of carriers with an IQ of 85 or above were found to have an early menopause versus 0% of those with an IQ < 85 and 5% of the normal controls. Although this finding did not quite reach statistical significance, it suggested that carriers with an average or greater IQ (who later turned out to have the PM) had an increased prevalence of early menopause [2]. Subsequent studies have confirmed the presence of fragile X-associated primary ovarian insufficiency (FXPOI) in PM carriers, which is associated with a bell-shaped relationship with the CGG repeat number; those with repeats between 85 and 100 have the highest risk and earliest onset of FXPOI [3][4][5]. Drs. Flora Tassone and Paul Hagerman discovered elevated levels of the FMR1 mRNA in PM carriers compared to controls, the opposite of what was expected. The blood of carriers had between two and eight times the normal values of the FMR1 mRNA, with a positive association with the CGG repeat number in the PM range [6]. The same year, at the NFXF meeting in Los Angeles in 2000, the Hagerman team presented case summaries from five aging male carriers with a history of tremor, ataxia, and atrophy using magnetic resonance imaging (MRI), and these cases were published in 2001 [7]. The researchers thought that this was a rare finding; however, when the family audience, which included over 100 carriers, were asked if they knew of relatives with similar problems, about 50% raised their hands, leading to a multitude of studies documenting the phenotype of what was later known as the fragile X-associated tremor/ataxia syndrome (FXTAS). The name of FXTAS and the original diagnostic criteria were established with the description of over 40 cases, as reported in Jacquemont et al. [8]. The awareness of FXTAS was dramatically improved with another paper published in JAMA documenting the prevalence of tremor and ataxia in carriers utilizing all the families identified in California at that time [9]. They found that the incidence of FXTAS increased with age in male carriers; 17% in their 50 s had tremor and balance problems, but this number gradually increased with age, such that 75% had tremor and ataxia in their 80 s. The researchers also found that females had fewer motor symptoms than males [9].
FXTAS is now well recognized as a neurodegenerative disorder with tremor, ataxia, neuropathy, and parkinsonian features, as well as cognitive changes beginning with memory problems and executive function deficits [10][11][12][13][14][15][16]. Additionally, MRI findings of white-matter disease, usually in the middle cerebellar peduncles (MCP sign) and periventricular areas, in addition to the splenium of the corpus callosum [17], have been documented. Neuropathological studies have demonstrated the presence of intranuclear inclusions in both neurons and astrocytes [18], and more recently, the enhanced activation and frequent death of astrocytes [14], iron overload [19], frequent microbleeds [20], parkinsonian features, including loss of dopamine cells, and occasional Lewy-body inclusions [21]. Eventually, 50% of males with FXTAS develop dementia [22], but this is far less common in females with FXTAS [23].
The pathophysiology of FXTAS involves multiple mechanisms, including RNA toxicity [24][25][26], clogging of the proteasome [27], RAN translation [28][29], and mitochondrial dysfunction [30][31][32] (for more details, see Section 2, ‘The molecular basis of FXPAC’). Recent papers have shown that males progress more rapidly in motor symptoms than females, presumably because of the protective effects in addition to the presence of the normal second X chromosome [33]. Therefore, the phenotype of FXTAS appears to be somewhat different in females, but emotional problems, such as anxiety and even pain symptoms, are more common in females than in males, and these problems progress faster in females [23][33][34].
The expanded phenotype beyond FXPOI and FXTAS in female carriers dates to the study by Coffey and colleagues [35] who studied 128 non-FXTAS adult female carriers and 18 women with FXTAS compared to age-matched controls [35]. The authors found multiple medical conditions, including neuropathy, hypertension, autoimmune thyroid disease, chronic muscle pain, intermittent tremor, and fibromyalgia, that were significantly increased in carriers compared to controls, and many of these issues were seen in carriers without FXTAS. These findings have led to further studies of problems that occur in carriers before the onset of FXTAS and of disorders that can occur even in childhood in a subgroup of carriers. Although most carriers have normal intellectual abilities and are without neuropsychological issues, studies have shown that a subgroup of carriers have psychiatric problems in childhood, including anxiety [36], attention deficit hyperactivity disorder (ADHD) [37][38], social deficits [39], and even autism spectrum disorder (ASD) [37][40][41][42]. For carriers who experience seizures, there is a higher incidence of ASD or intellectual disabilities (ID) compared to carriers without seizures [43], and 20% of carriers with ID and ASD have a second genetic hit, as detected with whole-exome sequencing (WES) or microarray studies [44].
Chen et al. [45] have demonstrated that PM neurons die more easily in culture, leading to the concept that they may be more vulnerable to environmental toxins, as seen in the cellular studies of Song et al. [46], who studied the effects of several toxins. In the clinical realm, it can be seen that exposure to isoflurane in general anesthesia can lead to the onset of FXTAS after surgery in elderly carriers [47]. In addition, toxic substances, such as illicit drugs, opioids, and excessive alcohol consumption, can lead to the more rapid progression of FXTAS [48][49][50][51][52]. Furthermore, research suggests that lifestyle changes to avoid toxins, environmental exposures, adverse experiences, and illnesses, such as diabetes, vitamin deficiencies, or hypothyroidism, may be helpful to slow down the progression of FXTAS [53].
It is likely that the pathophysiological changes in carriers, including mitochondrial dysfunction [30][31][54] and calcium dysregulation [55], can occur well before FXTAS and lead to GABA deficits [56], chronic pain [34], chronic fatigue [57][58], increased stress [59], mental health problems, and sensitivity to environmental stimuli [60]. In addition, several medical problems occur more frequently in carriers of the PM compared to the general population, such as autoimmune diseases [61], hypertension [62], insomnia [57], migraines [63], and connective tissue problems [64], which can rarely present as sudden coronary artery dissection (SCAD) [65] and cardiac arrhythmias [66]. Recognition of these findings will likely lead to further research and treatment endeavors [53].
Mental health impact has been documented particularly in female carriers compared to controls over the last two decades, including anxiety, depression, obsessive–compulsive behavior, ADHD inattentive type, and the broad autism phenotype [67][68][69] (reviewed in [60]). Roberts et al. [70] have reported that psychiatric symptoms can become more common with age in adulthood. Women have expressed that their physicians do not take their concerns seriously and basically blame these psychological problems on the stress of raising a child with FXS, even though these problems can be seen in carriers without children or without children with FXS [71][72]. Although many scientists doubted that psychological/psychiatric problems could be related to the PM, the work of Marsha Mailick and colleagues has validated some of these findings [73]. They studied the Marshfield cohort of over 20,000 patients and conducted FMR1 genotyping on the sample, but the patients and clinicians were naive to the results of the DNA testing. This research found elevated rates of agoraphobia, social anxiety or social phobia, and panic disorder, but not higher rates of major depression episodes in the medical records database in the male and female carriers compared to male and female noncarriers. This study demonstrated a higher prevalence of anxiety conditions in an unbiased group of people with the PM from the general population, as smaller studies have previously shown. A strong argument for the association between the PM status and psychological/psychiatric problems in female carriers was provided by the finding of highly significant (nonlinear) negative correlations between the size of CGG repeats and a great majority of SCL-90-R subscale scores and all the global indices [74].
The psychological difficulties can be severe and can occur in up to 50% of adult carriers. The name, fragile X-associated neuropsychiatric disorders (FXAND), was coined as an umbrella term to encompass the problems that are increased in carriers compared to controls, and are listed in the DSM5 [60]. Johnson et al. [75] have objected to the term FXAND because there are milder mental health impacts that do not meet the criteria for a disorder, so they proposed the term fragile X-PM-associated conditions (FXPAC), avoiding the use of the term “disorder”. Thus, the various physical and mental conditions mentioned above, and any of the problems associated with the PM, can appropriately be called FXPAC so that the more specific and detrimental PM issues, such as FXAND, FXPOI, and FXTAS, can fall under this category.
2. The Molecular Basis of FXPAC
The PM alleles are characterized by increased levels of FMR1 mRNA, which correlate with the length of the repeat tract, in both male and female carriers of a PM allele [6][76][77]. Although the elevated mRNA levels result from an increase in transcriptional gene activity [78], a CGG-repeat-length-dependent decrease in the expression of the FMR1 protein, FMRP, likely results from the impaired scanning of ribosomal preinitiation complexes through CGG-repeat tracts [6][76][79][80]. The increased expression of the FMR1 mRNA (up to six-to-eight-fold of that seen in normal alleles) leads to transcriptionally activated cellular stress pathways, RNA-mediated toxicity triggering CGG-binding-protein sequestration, and repeat-associated non-AUG-initiated (RAN) translation, which are the current basic and central molecular mechanisms proposed to explain the pathogenesis of FXTAS.
2.1. Molecular Basis of the FMR1 Locus
The PM alleles in females are unstable and prone to expansion on intergenerational transmission, with expansion into alleles harboring greater than 200 CGG repeats, leading to FXS. Generally, one or two AGG interruptions are observed within the repeat tract of normal and intermediate FMR1 alleles (6–44 CGG and 45–54 CGG repeats, respectively), while one or none are observed in PM alleles, and they are known to influence the stability of the repeats during parental transmission. Specifically, the presence of AGG interruptions decrease the intergenerational instability of the CGG repeats, thus decreasing the risk of expansion to a full-mutation allele [81][82]. In addition to AGG interruptions, other factors that increase the risk of expansion to full-mutation alleles during maternal transmission include the maternal CGG repeat number and age [81][82]. Interestingly, no association was found to correlate with either the transcriptional or translational activity of the gene [78][80][83][84].
As observed in other trinucleotide disorders, a bidirectional transcription at the FMR1 locus has been demonstrated and specific alternative splicings of the antisense FMR1 (ASFMR1) gene have been identified [85]. The ASFMR1 gene is expressed in all tissues, with high expression observed in the brain, spans approximately 59 kb of genomic DNA, and contains 13 exons and 45 ASFMR1 isoforms that are identified, 19 of which are expressed only in the PM [86]. Some of these isoforms are, as for the FMR1 gene, highly expressed in the PM as compared to the controls [87]. Although the ASFMR1 has been suggested to play a critical role in the pathogenesis of FXTAS [88][89], further studies are warranted to shed light on the contribution of the ASFMR1 in the clinical phenotypes of FXTAS.
Recently, it has been demonstrated that alleles in the PM range can be somatically unstable in both male and female carriers of a PM allele [86][90]. As observed with intergenerational instability, it was demonstrated that the extent of somatic instability directly correlates with the number of CGG repeats and, inversely, with the number of AGG interruptions. Increased levels of somatic expansion are observed over time in blood (PBMCs) derived from female carriers of a PM allele [86], and are mainly due to unmethylated FMR1 alleles and, therefore, limited to the active X chromosome. Recent evidence suggests that DNA repair factors FAN1 and MSH3 are both also modifiers of the expansion risk in females with specific genotypes associated with increased somatic instability [86] (these genes have also been implicated in other repeat expansion disorders (Genetic Modifiers of Huntington’s Disease Consortium)), suggesting a common expansion mechanism. Genetic factors that affect the somatic expansion risk may contribute to the variable penetrance for FXPAC that is seen. The extent of somatic instability in female PM carriers has shown a significant correlation with a diagnosis of ADHD [91], and may also affect the risk of various PM conditions in both males and females.
Allelic instability, observed in individuals with FMR1 mutations, leads to both intra- and intertissue mosaicism (PBMCs, fibroblasts, and brain tissues), and may account for some of the variability observed in the clinical phenotype of individual carriers of the PM [90]. During the International Premutation Conference, new data were presented about allelic instability within the FMR1 gene, confirming its occurrence between and within different tissues derived from the same individuals. Unstable alleles were exhibited among the majority of both female and male PM carriers. In addition, diverse allele profiles were displayed between PBMCs and fibroblasts from the same individuals among PM males, in accordance with previous studies [90][92][93][94][95]. Allelic instability affirms the complexity of FMR1 mutations and may relate to diverse phenotypes, including cognitive abilities and behavioral features observed in both FXS and PM disorders [96], specifically in female carriers of a PM allele with ADHD and depression [91]. The activation ratio (AR) is a clinically relevant parameter for females with both full-mutation [97] and PM conditions [86], as it reflects the fraction of normal alleles present on the active X chromosome [98]. The X-inactivation process is widely recognized as a factor that can influence the symptoms and severity of many diseases [99]. In FXS, although the size of the CGG repeat in the promoter region of the FMR1 gene is a significant factor, it is not sufficient to entirely determine the functionality of the gene. Hence, factors such as the AR and methylation status of the gene in females carrying an FMR1 mutation may also contribute to the regulation of FMRP levels. Therefore, to accurately interpret phenotypic characteristics in individuals with both FXS and FXPAC, it is necessary to assess methylation-status analyses [100][101][102][103].
The extent of phenotypic variation based on the AR is demonstrated by the observation that approximately 30% to 50% of females carrying a full mutation and exhibiting normal intelligence have the mutation primarily on their inactive X chromosome [103]. Moreover, studies have indicated that female PM carriers with a higher AR exhibit a significantly lower FXTAS incidence [35][104][105]. On the other hand, individuals with a normal allele that is predominantly methylated, and therefore inactive, may be at a higher risk of developing FXTAS. Additionally, several studies have suggested that lower AR values could be linked to cognitive and behavioral challenges in female PM carriers [97][106][107][108][109][110], potentially affecting the risk, severity, and age of onset of FXPAC. Despite numerous studies investigating the role and impact of the AR in PM carriers, there are discrepancies among their findings that may be partially attributable to technical variability, as previously reported [86][111][112][113][114], or to differences in the methods employed to calculate the AR (as discussed in Protic et al., this special issue [115]). At the International Premutation Conference, novel data were presented demonstrating a noteworthy correlation between clinical measures and the AR. As anticipated, the study revealed that higher ARs were linked to reduced FMR1 transcript levels for any given repeat length, and associated with enhanced performance, verbal, and full-scale IQ scores, as well as lower levels of depression, and a smaller number of medical conditions. Based on this evidence, it is advisable to evaluate the methylation status, including the AR in females with both PM and full-mutation alleles of the FMR1 gene, to better understand their clinical phenotypes.
2.2. Molecular Mechanisms Leading to FXTAS Pathology—RNA Toxicity and RAN Translation at CGG Repeats: Mechanistic Insights and Their Contribution to Disease Pathology
There are currently three nonexclusive models for how CGG repeats elicit pathogenesis in FXTAS (Figure 1).
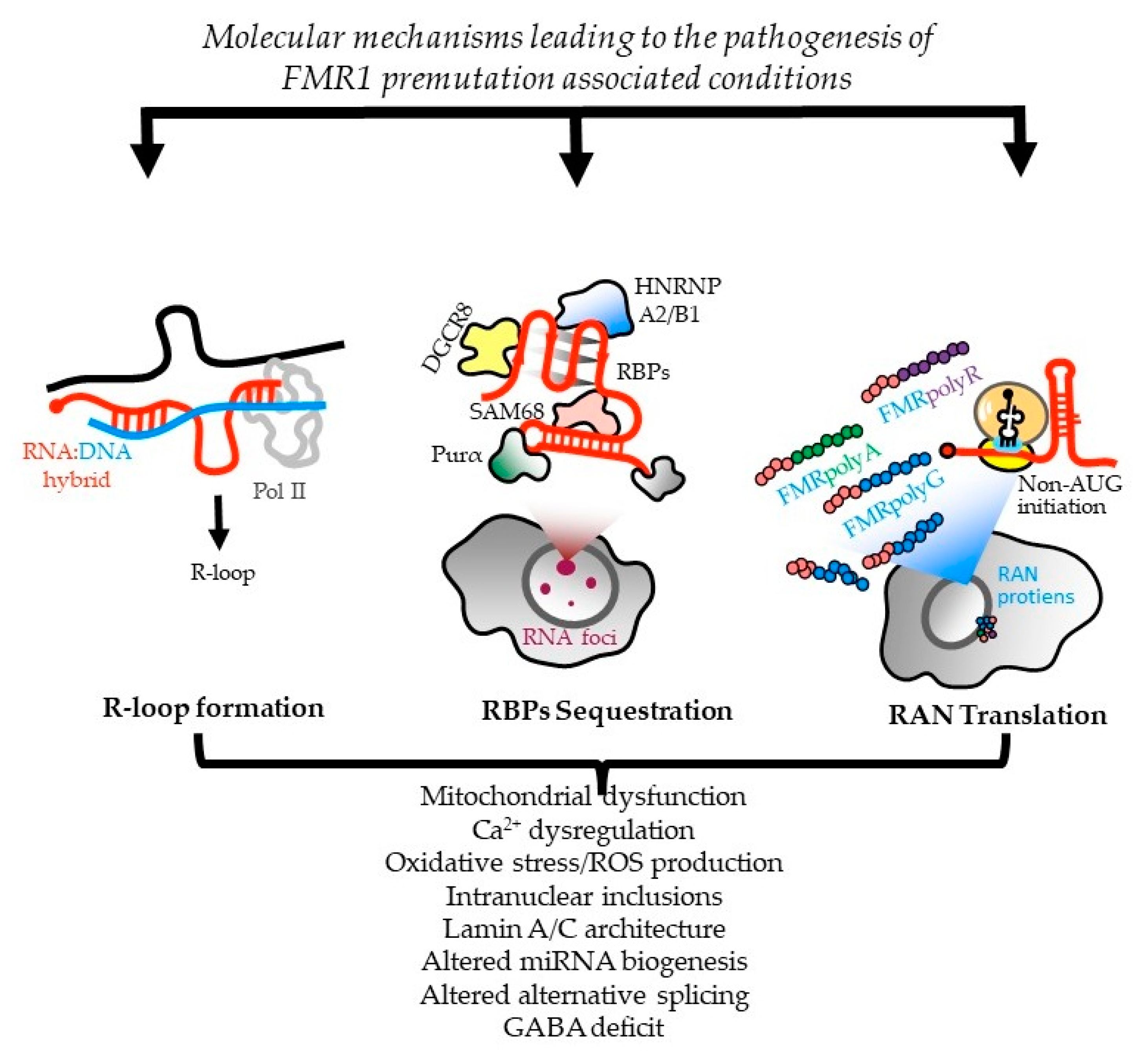
Figure 1. Molecular mechanisms leading to FMR1-PM-associated conditions. Three nonexclusive models are proposed for how CGG repeats contribute to the pathogenesis of PM conditions, including FXTAS. First, the cotranscriptional R-loop formation, which compromises genomic stability and triggers a DNA-damage response that can activate inflammatory cascades [116][117]. Second, CGG-repeat RNAs can elicit a gain-of-function toxicity through RNA gelation into nuclear foci and sequestration of various rCGG-repeat-binding proteins, leading to their functional depletion [25][26]. Third, repeat-associated non-AUG (RAN)-initiated translation generates potentially toxic proteins that accumulate within intranuclear neuronal inclusions in FXTAS patients. The relative contribution from each mechanism to downstream sequelae, such as mitochondrial dysfunction and neuronal death, and their potential synergies in disease pathogenesis, are areas of ongoing research in the field. Adapted from [118].
In one, CGG-repeat RNAs elicit a gain-of-function toxicity through both RNA gelation into nuclear foci and sequestration of various rCGG-repeat-binding proteins [25][26]. Mass-spectrometric and immunohistochemical analyses have identified over 20 proteins in the frontal cortex inclusions of FXTAS patients, including RNA-binding proteins (RBPs), HNRNP A2/B1 (heterogeneous nuclear ribonucleoprotein A1), and MBNL1 (muscleblind-like protein 1), as well as some neurofilament proteins, such as lamin A/C and α-internexin. These proteins are involved in various neurological disorders [119]. Pur α and HNRNP A2/B1 bind directly to rCGG repeats in inclusions, and their overexpression in a Drosophila model expressing PM CGG repeat expansions suppressed neurodegeneration phenotypes [25][26]. Sequestration of other proteins, such as CUGBP1 (CUGBP Elav-like family member 1), SAM68 (Src-associated substrate during mitosis of 68-kDa), Rm62 (ATP-dependent RNA helicase p62), and DGCR8 (DiGeorge syndrome critical region 8), leads to altered mRNA splicing and transport, as well as dysregulated microRNAs, supporting a toxic RNA gain-of-function mechanism mediated by the expanded CGG repeats in FMR1 [24][26][120][121][122].
HNRNPA2/B1 is present in intranuclear inclusions of FXTAS patients and it binds directly to rCGG repeats. Its overexpression, along with its two homologs in Drosophila, suppresses the neurodegenerative eye phenotype caused by the rCGG repeat [26]. HNRNP A2/B1 also mediates the indirect interaction between CGG repeats and CUGBP1 involved in myotonic dystrophy type 1 (DM1). Overexpression of CUGBP1 suppresses the FXTAS phenotype in Drosophila. Pur α, another protein found in intranuclear inclusions of FXTAS patients, plays a crucial role in DNA replication, neuronal mRNA transport, and translation. Pur α knockout mice show developmental delays and altered expression, and the distribution of axonal and dendritic proteins [123][124]. Overexpression of Pur α in a Drosophila model suppresses rCGG-mediated neurodegeneration in a dose-dependent manner. Sequestration of SAM68 in particular causes pre-mRNA alternative splicing misregulation in CGG-transfected cells and FXTAS patients, thus contributing to FXTAS pathogenesis via a splicing alteration mechanism [120]. TDP-43 (TAR DNA-binding protein 43), an ALS-associated RBP, has reduced association with ribosomes in the cerebellar Purkinje neurons of mice expressing 90 CGG repeats [125]. In the same study, the authors went on to find that, in the Drosophila model of FXTAS, wild-type TDP-43 expression leads to suppression of neurodegeneration, while the knockdown of the endogenous TDP-43 fly ortholog, TBPH, enhanced the eye phenotype.
Another study also independently reported that TDP-43 suppressed CGG-repeat-induced toxicity in a Drosophila model of FXTAS [126]. Interestingly, this suppression was shown to depend on HNRNP A2/B1, such that the deletion of the C-terminal domain of TDP-43, and thereby the prevention of interactions with HNRNP A2/B1, led to the abrogation of the TDP-43-dependent rescue of CGG repeat toxicity [126]. Finally, DGCR8, a protein binding to PM rCGG repeats, causes partial sequestration of DGCR8 and its partner, DROSHA (drosha, ribonuclease 3), within PM RNA aggregates. DGCR8 and DROSHA play a critical role in microRNA biogenesis. Sellier and colleagues found that the sequestration of DGCR8 and DROSHA precludes them from their normal functions, leading to reduced processing of pri-miRNAs in cells expressing expanded CGG repeats. Consequently, levels of mature miRNAs are also reduced in the brains of FXTAS patients [24].
Alternatively, the CGG repeats in 5′ UTR of FMR1 mRNA may be translated into toxic proteins through a process known as RAN translation. Initially described at CAG repeats in spinocerebellar ataxia type 8 (SCA8) and DM1 [127], noncanonical translation of short tandem repeats into proteins may occur in the absence of an AUG initiation codon when repeat-containing RNAs form stable secondary structures. RAN translation has been observed on repeats associated with ten disorders: SCA8, DM1, DM2, HD, FXTAS, C9orf72 amyotrophic lateral sclerosis and frontotemporal dementia (C9 ALS/FTD), FXPOI, SCA31, and Fuchs endothelial corneal dystrophy (FECD) (reviewed in [118][128]). In many of these diseases, RAN translation occurs in different reading frames on both sense and antisense transcripts, and the RAN products are detected in patient tissues.
In FXTAS, it is thought that CGG repeats form secondary structures that lead to the impairment of ribosomal scanning, reduced start codon fidelity, and, in consequence, aberrant translation initiation at near-cognate or noncognate codons located upstream or within the repeats [129]. Depending on the reading frame, different toxic proteins containing long mono-amino-acid tracts are produced: polyglycine (FMRpolyG), polyalanine (FMRpolyA), and polyarginine (FMRpolyR) [28][129]. Additionally, there is evidence that RAN translation also can occur on the CCG antisense transcript [130] to produce additional homopolymeric proteins. Translation through the repeat may also trigger frameshifting to produce chimeric RAN proteins [131]. The translation of FMRpolyG is the most efficient, and this protein is detected in FXTAS patient brains by both immunohistochemistry and mass spectrometry, colocalizing with p62 and ubiquitin-positive inclusions [11][18][28][119][130][132][133][134][135]. However, quantitation of this and other RAN-translation-generated proteins remains challenging due to their low abundance, solubility, multiple initiation sites, and early translation termination—all of which hamper its detection by antibodies targeting either the N- or C-terminus [21][134][135]. FMRpolyG was found to interact with the nuclear lamina protein LAP2β, leading to the impairment of the nuclear lamina architecture [132]. Additionally, FMRpolyG is capable of cell-to-cell propagation via exosomes in cell-culture studies and glia-to-neuronal propagation in mouse-model systems. Similar prion-like propagation is thought to play a central role in the pathogenic spread of alpha synuclein in Parkinson’s disease and of Tau in Alzheimer’s disease (PMID: 30917002). However, the role of this phenomenon in FXTAS pathogenesis remains unclear [134][136].
Whether RAN products generated from CGG repeats are drivers of toxicity or if there is instead a synergy between CGG-repeat RNA and RAN proteins remains unknown. Studies in overexpression systems in cells, flies, and mice suggest that near-cognate codons 5′ to the repeat that support the RAN translation of FMRpolyG are requisite to elicit maximal toxicity [28][132][137][138]. However, FMRpolyG inclusions can persist even as phenotypes resolve when the repeat is transcriptionally silenced [139]. Moreover, FMRpolyG production absent the repeat RNA is less toxic in neurons than is a RAN-competent CGG repeat [131].
The exact mechanism by which RAN translation occurs remains enigmatic, and may vary in different repeats (and even different reading frames of the same repeat). However, several recent studies reported modifiers of RAN translation that provide some clues. Unwinding the structured RNA is crucial for RAN translation, as it is shown that several RNA helicases, such as DDX3X (ATP-dependent RNA helicase DDX3X), DHX36 (ATP-dependent DNA/RNA helicase DHX36), eIF4A/B (eukaryotic initiation factor 4A-I/B), and H, are directly involved in the regulation of this process enabling proper ribosomal scanning [129][137][140]. In addition, the presence of RAN proteins, together with structured RNAs with CGG repeats, leads to the activation of the integrated stress response (ISR) and the phosphorylation of eIF2α, which, in a feed-forward-loop mechanism, shut down the global translation but selectively enhance RAN translation [141]. Proteins which interact with CGG-repeat RNAs may also influence the RAN translation, as SRSF1 (serine/arginine-rich splicing factor 1) mediates the nuclear retention of CGG-repeat RNAs to prevent these transcripts from becoming a template for RAN translation [118].
2.3. Therapeutic Perspectives to FXTAS from a RAN-Translation Perspective
There are currently no FDA (Food and Drug Administration Agency)-approved drugs to slow FXTAS progression or delay its onset. An emphasis point that was raised during the International Premutation Conference was that there is a critical need for the discovery of reliable, robust biomarkers to accurately understand predisease onset states and readouts for clinical progression. Some promising work suggests that metabolomic and/or proteomics biomarkers may serve this purpose [142][143][144]. Indeed, a small open-label pilot study in patients with validation studies in patient fibroblasts indicated that the mitochondrial activator sulforaphane showed some correction of these biomarkers that could serve as a precursor for a larger study [143].
Antisense oligonucleotides (ASOs) hold promise for FXTAS treatment by blocking RAN translation in neurons without degrading the FMR1 mRNA, enhancing FMRP expression, and improving neuron survival [138]. In rodent models, ASOs reduce FMRpolyG biosynthesis, correct disease-related traits, and normalize transcriptomic effects [145]. A recent study suggests that the ubiquitin–proteasome system may be an interesting therapeutic target based on the presence of PSMB5 (proteasome subunit beta type-5) polymorphisms as disease-onset modifiers in patients and the suppression of disease-relevant phenotypes in Drosophila with a genetic knockdown of this proteasomal subunit [146]. This factor also changes how CGG RAN translation happens in cell tests. For example, it affects how a molecule called CMBL4c can attach to CGG-repeat RNA structures and lower FMRpolyG levels [147]. The ISR targeted by protein kinase R (PKR) rescue in a mouse model of FXTAS [143], along with multiple CGG-repeat-associated RBPs, offer potential treatments targeting RAN-translation modifiers, although clinical application awaits further research due to the early stage of Drosophila, cell-based, and mouse experiments.
2.4. Genetic Modifiers in Fragile X-Associated Tremor Ataxia Syndrome (FXTAS)
The underlying neurobiological mechanisms of FXTAS are complex and not fully understood. As mentioned above, several mechanisms that have been proposed to explain the pathogenesis of FXTAS, including RNA toxicity, RAN translation producing the accumulation of the FMR PolyG polypeptide, and damage response, are linked to white-matter-tract connectivity in the brain, called white-matter hyperintensities, and strongly associated to the clinical impairment observed in FXTAS [6][28][148]. However, not all individuals who carry a PM allele will develop PM conditions, including FXTAS, in their older adulthood, which indicates the incomplete penetrance pattern of the disease. Therefore, nowadays, some studies have been dedicated to a plausible mechanism and exploring predisposing factors, including genetic modifiers, that may contribute to the occurrence of FXPAC. Investigations of genetic modifiers of clinical manifestation of diseases have also become a new research interest in FXTAS. They sought to provide an answer to the wide diversity and severity of clinical major criteria (intention tremor and gait ataxia) and minor criteria (cognitive impairment) [33][146][149]. Various genetic variants may contribute to cognitive impairment, including the APOe4 allelic variant, which represents the strongest risk factor of late-onset Alzheimer’s disease (AD), the most common type of dementia, in all ethnic groups [150]. The prevalence of the APOe4 allele is 13.7% in the general population; having one copy of the APOe4 allele increases the risk by around 3 times compared to individuals without the APOe4 allele, while having two copies boosts the risk of AD by 8–12 times [151].
APOE is an important cholesterol and lipid transporter that plays a critical role in a variety of signaling pathways in the development, maintenance, and repair mechanisms of the central nervous system (CNS) [152]. The APOe4 allele triggers β-amyloid (Aβ) accumulation/amyloidosis in oligodendrocytes and their myelin that leads to the slowing of brain electrical signaling, which is associated with cognitive impairment [153]. Postmortem examination of FXTAS brain tissue showed the presence of cortical amyloid plaques and neurofibrillary tangles, combined with presence of intranuclear inclusions in those with FXTAS and AD, which is additional evidence of the involvement of other genes that may modify the FXTAS phenotype [154]. Among the FMR1 PM carriers, the APOe4 allele frequency is higher (31.8%) in patients with FXTAS compared to the general population and increases the risk by more than 12 times to develop the disease [155]. During the International Premutation Conference, data on 180 PM males, aged over 50 years, were presented which showed that the APOe4/APOe2 and APOe4/APOe3 genotypes were more frequent in PM males with FXTAS compared to those without FXTAS (2% vs. 0% and 10.6% vs. 2.4%, respectively).
Recently, to identify the genetic modifiers of FXTAS, a large number of PM carriers were recruited for whole-genome sequencing (WGS), which was further combined with Drosophila genetic screening. It was demonstrated that using FXTAS Drosophila as a genetic screening tool can be powerful in the validation of candidate genes from WGS. Eighteen genes were identified as potential genetic modifiers of FXTAS. One of such candidate genes is the proteasome subunit beta-5 (PSMB5), which genetically modulates CGG-associated neurotoxicity in Drosophila as a strong suppressor of CGG-associated neurodegeneration. PM individuals who carry the variant PSMB5rs11543947-A, which is associated with decreased expression of PSMB5 mRNA, may be protected against FXTAS. In addition, there is a strong suppression of CGG-associated neurodegeneration through diminishing RAN translation in the Drosophila knockdown of PSMB5 [146]. The metabolomic approach to determine a genetic modifier in an FXTAS mouse model found metabolic changes and demonstrated that Schlank (ceramide synthase), Sk2 (sphingosine kinase), and Ras (IMP dehydrogenase), which encode enzymes in the sphingolipid and purine metabolism, respectively, were significantly related with FXTAS-CGG-associated neurodegeneration pathogenesis [149].
Finally, more studies are needed to identify possible genetic modifiers associated with FXTAS development and progression for better management of the disease and for the development of therapeutic strategies.
2.5. The Use of Human Pluripotent-Stem-Cell-Based Neurodevelopmental Models for FXTAS
Human models of FXPAC are essential tools for studying disease-specific mechanisms, such as RNA toxicity, RAN translation, and CGG somatic instability. However, generating improved model systems for all these pathologies requires patients’ disease-relevant cell cultures. In the case of FXTAS, this is especially challenging because postmortem brain samples are rarely available, limited to a small amount of biological material, and represent only the final stage of the disease.
Overcoming these limitations can be achieved by utilizing mutant human pluripotent stem cells (hPSCs) in conjunction with in vitro differentiation towards affected tissues (neurons). This approach provides a powerful tool for both fundamental and applied research, offering an excellent opportunity to investigate the disease’s pathogenic mechanisms and to identify potential targets for therapeutic intervention.
There are two types of pluripotent stem cell lines that can be utilized for FXTAS disease modeling: human embryonic stem cell (hESC) lines derived from genetically affected embryos that can be obtained by preimplantation genetic diagnosis (PGD) procedures [156], and patient-derived induced pluripotent stem cells (iPSCs), established by reprogramming somatic cells obtained from patients (e.g., blood, skin fibroblasts) [157]. Both PGD-derived hESCs and patient-derived iPSCs carry the disease-causing PM and can reproduce disease cellular phenotypes in vitro, and allow following dynamic processes that are misregulated during development and aging in patients.
The first in vitro model of FXTAS using pluripotent stem cells (PSCs) showed that differentiated neurons from iPSCs recapitulate the cellular phenotypes of FXTAS, including reduced synaptic puncta density, neurite length, and increased calcium transients [157]. FXTAS iPSCs were also used to discover a toxic mechanism linked to FMRPolyG proteins via RAN translation [132]. Additionally, human neurons derived from patient iPSCs were used to validate a therapeutic approach that selectively blocked CGG RAN initiation sites using noncleaving ASOs. ASO blockade improved endogenous FMRP expression, suppressed repeat toxicity, and prolonged survival in human neurons, showing the therapeutic potential of modulating RAN translation in FXTAS [138].
Nevertheless, despite recent progress, the currently available human iPSC-based models for FXTAS are insufficient in reproducing the full complexity of the disease. This is because these models are based on monolayer cell cultures, which restrict the analysis to less-mature and single-cell types. To gain a comprehensive understanding of the interactions between various cell populations in the brain, and to examine the contribution of each pathogenic mechanism associated with FXTAS during early brain development, a higher level of complexity than monolayer cell cultures, such as brain organoids, would be necessary.
Brain organoids are three-dimensional mini-organs derived from PSCs that mimic the cellular composition and architecture of specific brain regions [158]. As such, they are expected to provide a powerful tool for identifying critical molecular events in the development of FXTAS, much before the clinical signs appear in patients. Moreover, brain organoids could extend our knowledge on other aspects of the disease, such as CGG somatic instability and the generation of mosaicisms for expansion size and/or methylation, in a multicellular setting that more closely resembles the developing human brain.
2.6. Shared Molecular Mechanism with Other Repeat Expansion Disorders
FXTAS is a repeat expansion disorder that displays clinical symptoms similar to those observed in other diseases caused by repeat expansions. Parkinsonism, a varied array of cognitive impairments that can progress to dementia, and amyotrophic lateral sclerosis (ALS)-like phenotypes, including frontotemporal dementia and progressive supranuclear palsy, have all been reported in FXTAS [159]. Tremor and ataxia, which are also hallmark symptoms of other repeat expansion disorders, such as spinocerebellar ataxias, are commonly observed in FXTAS.
The genetic basis of the FXTAS repeat expansion is similar to other repeat expansions observed in several diseases, including C9orf72 ALS/frontotemporal dementia (GGGGCC-repeat), myotonic dystrophy type 1 (CTG-repeat), NOTCH2NLC (CGG-repeat), Huntington’s disease (CAG-repeat), and spinocerebellar ataxias (SCA-CAG-repeat). Regional aggregation of cytosolic, nuclear, or extracellular proteins is a common feature observed in these diseases and disrupts neuronal function [160]. Intranuclear eosinophilic ubiquitin-positive inclusions in neurons and astrocytes are characteristic of FXTAS pathology and have been observed in other trinucleotide disorders [161]. TDP-43 in ALS/frontotemporal dementia and poly (amino acid)/polypeptides in FXTAS, Huntington’s disease, and spinocerebellar ataxias are examples of the types of aggregates that result from the expansion of trinucleotide repeats [162].
The most common genetic cause of ALS/frontotemporal dementia is an expanded GGGGCC-repeat in the C9orf72 gene. Similar to FXTAS, RAN translation and the accumulation of toxic peptides in neurons and astrocytes (TDP-43) are the main pathological mechanisms in C9orf72 ALS/frontotemporal dementia [163]. The accumulation of toxic polypeptides resulting from expanded trinucleotide repeats is also observed in Huntington’s disease (CAG-repeat) and spinocerebellar ataxias (CAG-repeat) [164].
The NOTCH2NLC pathogenic CGG-expansions, located in the 5′ UTR (66-517) and having GGA or AGC interruptions, are particularly similar to those observed in FXTAS. They cause a late-onset disorder with a clinical variability that includes muscle weakness, dementia, parkinsonism, tremor, and ataxia. The molecular mechanisms of OTCH2NLC lead to neuronal intranuclear eosinophilic inclusions, and the antisense isoform has been hypothesized to be a pathological mechanism [165].
Anticipation, somatic instability, and clinical severity associated with the number of repeats has been described in many repeat expansion disorders, including, HD, DM1, FXTAS, ALS, and others [166].
Aside from these, FXTAS resembles DM1 in many respects. Firstly, because the primary mechanism for both pathologies is RNA toxicity [25][26][132][167][168][169][170]. Secondly, and as mentioned above, both affected loci exhibit RAN translation potential, leading to the production of toxic polyglycine, polyalanine and polyarginine containing proteins by CGG expansion in the PM range in FMR1 [28][132][171], and polyalanine- and polyserine-containing proteins by CTG expansions in DM1-affected cells [127][172]. To add further complexity, both disorders exhibit a decrease in protein levels, albeit through distinct mechanisms [6][76][173]. Lastly, both expansions in FMR1 and DM1 display maternal anticipation/expansion, giving rise to distinct phenotypes (namely, FXS in FMR1 and congenital myotonic dystrophy type 1 in DMPK) and to DNA hypermethylation. Altogether, the clinical presentation of individuals carrying the FMR1 PM is highly heterogeneous and shares similarities with the phenotypic heterogeneity observed in DM1 and other nucleotide repeat disorders. This variability likely results from the involvement of the multiple mechanisms that, together with modifier genes and environmental factors, contribute to disease pathology to varying degrees.
2.7. Mitochondrial Dysfunction in PM Carriers
Recently, studies on cultured cell lines, animal models, and human subjects have implicated mitochondrial dysregulation in the pathogenesis and progression of FXTAS. Using magnetic resonance imaging (MRI), Rizzo et al. (2006) [174] first described lactate accumulation in the lateral ventricles, as well as decreased ATP levels in the calf muscles of a patient with FXTAS. Subsequent studies on cultured fibroblasts from PM carriers and mouse models have confirmed impaired ATP production and the pathogenic role of expanded CGG repeats on mitochondrial functions [175][176]. Finally, clinical studies on living patients with FXTAS and postmortem brain tissues with the disease have showed altered Krebs-cycle intermediates, neurotransmitters, and neurodegeneration markers, as well as reduced mitochondrial DNA copy numbers in specific brain regions, such as the cerebellar vermis, parietal cortex, and hippocampus [32][177]. Finally, unlike the earlier results from human brain tissue, studies in Epstein–Barr-virus (EBV)-transformed blood lymphoblasts showed that mitochondrial respiratory activity was significantly elevated in FXTAS compared with controls. Specifically, altered complex I activity and ATP synthesis, accompanied by an altered mitochondrial mass and membrane potential, were observed, and were significantly associated with the white-matter-hyperintensity (WMH) scores in the supratentorial regions [178]. In addition, an elevation of AMP combined with the reduction of TORC in both FXTAS and non-FXTAS categories of PM carriers was reported [179]. In the later study, correlations between measures of mitochondrial and nonmitochondrial respiratory activity, AMPK, and TORC1 cellular protein kinases, and the scores representing motor, cognitive, and neuropsychiatric impairments, were found with the CGG repeat size, and a hyperactivity of cellular bioenergetic components was significantly associated with motor-impairment measures, including tremor–ataxia, parkinsonism, and neuropsychiatric changes, predominantly in the FXTAS subgroup [180]. Moreover, an elevation of AMPK activity and a decrease in TORC levels were significantly related to the size of the CGG expansion. All the above studies have suggested that the bioenergetic changes in blood lymphoblasts are biomarkers of the clinical status of FMR1 carriers. Furthermore, a decreased level of TORC1—the mechanistic target of the rapamycin complex—suggested a possible future approach to therapy in FXTAS.
Several molecular mechanisms have been proposed as mediators of abnormal mitochondrial function in FXTAS. RNA toxicity was the first model described, according to which the expanded CGG repeats in FMR1 mRNA binds and titrates specific RNA-binding proteins, resulting in loss of their normal functions [26]. Among these proteins, the pre-mRNA splicing factor TRA2A has gained significant attention, since it is also present in the pathognomonic ubiquitin inclusions of FXTAS [181]. Additionally, miRNAs are increasingly recognized as major determinants of normal mitochondrial function. One of their biogenesis regulators, the DROSHA/DGCR8 enzymatic complex, was found sequestered within the expanded CGG RNA foci, leading ultimately to the loss of its normal function [24][182][183]. Moreover, altered zinc and iron metabolism, a pivotal neuromodulator, and an essential element in maintaining mitochondrial physiology, respectively, may be additional contributing factors in FXTAS pathogenesis. Fibroblasts from PM carriers have been shown to express abnormal zinc transporter levels, thereby leading to altered zinc homeostasis [30], whereas increased iron levels were also observed in neurons and oligodendrocytes of the putamen of carriers of a PM [19]. Finally, among the functions of FMRP, the product of the FMR1 gene, is the binding to superoxide dismutase 1 (SOD) mRNA and the regulation of its levels. Consequently, lower expression of FMRP may result in decreased levels of SOD1, thereby leading to increased reactive oxygen species (ROS) levels and impaired oxidative phosphorylation [184].
More recently, emerging evidence has implicated the role of abnormal electron-transport-chain enzyme complexes in FXTAS pathogenesis. Gohel et al. had first observed defective complex activity in human cell lines and a transgenic mouse model [185]. Additionally, a recent study, presented at the International Premutation Conference, utilizing brain-derived extracellular vesicles, a novel and powerful platform for biomarker development for brain diseases, from plasma and from postmortem brain tissues from patients with FXTAS, found a decreased quantity and activity of complex IV and V, thus further validating this pathogenic process [143].
2.8. Omics Studies (Metabolomics and Proteomics) in PM Carriers
The development of targeted therapeutics for rare age-dependent neurodegenerative disorders encounters numerous challenges, encompassing the absence of biomarkers for early diagnosis and disease progression, intricate underlying molecular mechanisms, heterogeneous phenotypes, limited historical data, and the difficulties posed by conducting clinical trials with small patient populations, which restrict enrollment. In this context, contemporary omics studies, including metabolomics and proteomics, have emerged as promising tools for investigating global changes within a given sample, employing extensive data mining and bioinformatic analysis [186]. Recent advancements in metabolomic- and proteomic-profiling technologies and processing have enabled the efficient and precise analysis of several hundred metabolites/proteins, facilitating the identification of biomarkers associated with disease development and progression [187].
Giulivi et al. (2016) conducted a comprehensive analysis of the plasma metabolic profile in human PM carriers with FXTAS, comparing them to healthy noncarrier controls. Their findings identified a panel of four core serum metabolites (phenethylamine, oleamide, aconitate, and isocitrate) that exhibited high sensitivity and specificity in diagnosing PM carriers with and without FXTAS. Notably, the presence of oleamide/isocitrate was identified as a specific biomarker for FXTAS. Moreover, based on these plasma metabolic profiles, the researchers reported evidence of mitochondrial dysfunction, neurodegeneration markers, and proinflammatory damage in FXTAS PM carriers [32]. In a separate investigation, Song et al. (2016) reported increased mitochondrial oxidative stress in primary fibroblasts obtained from PM carriers compared to age- and sex-matched controls [46]. Napoli et al. (2016) examined peripheral blood mononuclear cells (PBMCs) derived from controls and carriers of a PM allele, with and without FXTAS, to investigate the presence of the Warburg effect. Their study revealed alterations in glycolysis and oxidative phosphorylation, indicating the involvement of the Warburg effect in FXTAS [188]. Using a PM murine model, Kong et al. (2019) investigated metabolic changes associated with FXTAS in the cerebellum. Their findings demonstrated significant alterations in sphingolipid and purine metabolism in the cerebellum of the mice. Furthermore, they identified genetic modifiers (Cers5, Sphk1, and Impdh1) of CGG toxicity in Drosophila [149]. In a 12-week open-label intervention study involving six males with FXTAS, Napoli et al. (2019) evaluated the effect of allopregnanolone on lymphocytic bioenergetics and plasma pharmacometabolomics. They observed the significant impact of allopregnanolone treatment on oxidative stress, the GABA metabolism, and certain mitochondria-related outcomes. These findings suggested the potential therapeutic use of allopregnanolone for improving cognitive function and the GABA metabolism in patients with FXTAS [189]. A more recent study by Zafarullah et al. (2020) aimed to identify metabolic biomarkers for early diagnosis and disease progression in FXTAS. Through characterization of individuals who developed FXTAS symptoms over time, alterations in the lipid metabolism, particularly in mitochondrial-bioenergetics-related pathways, were identified as significant contributors to FXTAS [88]. Subsequently, Zafarullah et al. (2021) established a significant correlation between the identified metabolic biomarkers and the area of the pons in individuals who developed FXTAS over time. They also demonstrated a notable association between these biomarkers and disease progression, highlighting their role within the context of the dysregulated lipid and sphingolipid metabolism [142].
In addition, the effort to identify the metabolic changes associated with FXPOI is ongoing, and preliminary data of a nontargeted metabolomic profiling of FXPOI patient plasma by LC/MS were presented during the International Premutation Conference. Initial differential abundance analyses revealed the altered abundance of compounds in the omega-6 fatty-acid (n-6 FA) metabolism and arachidonic acid formation between females with a FXPOI diagnosis and female carriers of a PM without POI across both cohorts. Pathways downstream of FA and the arachidonate metabolism were also identified, including prostaglandin synthesis and the formation of proinflammatory metabolites from the AA. Further investigation of metabolic changes associated with FXPOI is likely to provide critical information about the mechanism of dysfunction in PM ovaries.
In recent years, Ma et al. (2019) conducted an LC-MS/MS-based proteomics analysis of intranuclear inclusions isolated from the postmortem brain tissue of individuals with FXTAS. Their findings revealed the presence of over 200 proteins within the inclusions, with significant abundance of SUMO2 and p62/sequestosome-1 (p62/SQSTM1). These results support a model where inclusion formation is a consequence of increased protein loads and heightened oxidative stress [134]. Subsequently, Holm et al. (2020) characterized the proteomic profile of the FXTAS cortex compared to that of healthy controls (HCs). They observed a notable decrease in the abundance of proteins, such as tenascin-C (TNC), cluster of differentiation 38 (CD38), and phosphoserine aminotransferase 1 (PSAT1), in the FXTAS samples. Additionally, the authors confirmed a significantly elevated abundance of novel neurodegeneration-related proteins and small ubiquitin-like modifier 1/2 (SUMO1/2) in the FXTAS cortex compared to HCs [27]. Furthermore, Abbasi et al. (2022) reported changes in the level of multiple proteins, including amyloid-like protein 2, contactin-1, afamin, cell-adhesion molecule 4, NPC intracellular cholesterol transporter 2, and cathepsin, by comparing the cerebrospinal fluid (CSF) proteome of FXTAS patients with HCs. Alterations in acute-phase-response signaling, liver X receptor/retinoid X receptor (LXR/RXR) activation, and farnesoid X receptor (FXR)/RXR activation pathways were also observed [190]. In an ongoing study, the Tassone lab performed blood proteome profiling of PM-allele carriers who developed FXTAS over time and compared it to HC samples. Through this analysis, they identified potential proteomic biomarkers for early diagnosis and reported altered protein pathways between the groups, suggesting their involvement in the pathogenesis of the disorder [144]. However, due to the limitations of a small sample size, further studies with larger cohorts are necessary to validate the initial findings and elucidate the role of the identified markers and pathways.
2.9. CGG Short Tandem Repeat (STR) Expansions
It has been outlined that the molecular cause of FXTAS is the presence of a PM ranged (55–200 units) expansion of the CGG short-tandem-repeat (STR) locus located within the 5′-UTR of the FMR1 gene [7]. In recent years, several other neurodegenerative disorders have been associated with a PM ranged CGG STR expansion as their genetic cause [191][192][193][194][195]. These diseases include neuronal intranuclear inclusion disease (NIID), oculopharyngodistal myopathy (OPDM), and oculopharyngeal myopathy with leukoencephalopathy (OPML). These PM expansion loci are localized within the following genes and ncRNA: LRP12 (OPDM type 1), GIPC1 (OPDM type 2), NOTCH2NLC (OPDM type 3/NIID), RILPL1 (OPDM type 4), and LOC642361 (OPML). All of these disorders share a striking level of clinical similarity with FXTAS, suggesting a shared or similar molecular mechanism of pathology leading to a neurodegenerative phenotype. In search of potential additional disease loci, Annear and colleagues (2021) performed a bioinformatic in silico analysis of the reference genome and identified approximately 6000 additional CGG STR loci. When large population datasets were analyzed (n > 12,000), 99% of these novel loci were demonstrated as displaying at least some degree of polymorphism across the human population, and approximately 15% of all CGG loci were observed to expand up to or beyond the 55-unit PM breakpoint [196]. How many of these loci may be involved in neurodegenerative disease remains an enigma. While the repeat length is unlikely the only factor affecting the pathogenic potential of a given repeat, it is no doubt a core component. Moreover, half of these CGG STRs displayed characteristics similar to the known disease-linked repeats [196]. This included high rates of polymorphism and a genetic localization within the 5′ UTR and gene promoter regions, a typical characteristic of disease-linked CGG STRs. However, there may be further factors at play, such as cis elements flanking the repeat and the reading frame of the repeat in reference to the localized gene [132][197]. In each case, it cannot be excluded that additional expansions of CGG STRs may play a role in progressive neurodegeneration disorders with FXTAS and FXPOI-like phenotypes. While additional expansions are not detected in routine diagnostics using current short-read-based detection methods, the future introduction of long-read sequencing may expose potential additional loci in the clinic.
Fragile X-premutation-associated-condition involvement across the lifespan is presented in Figure 2.
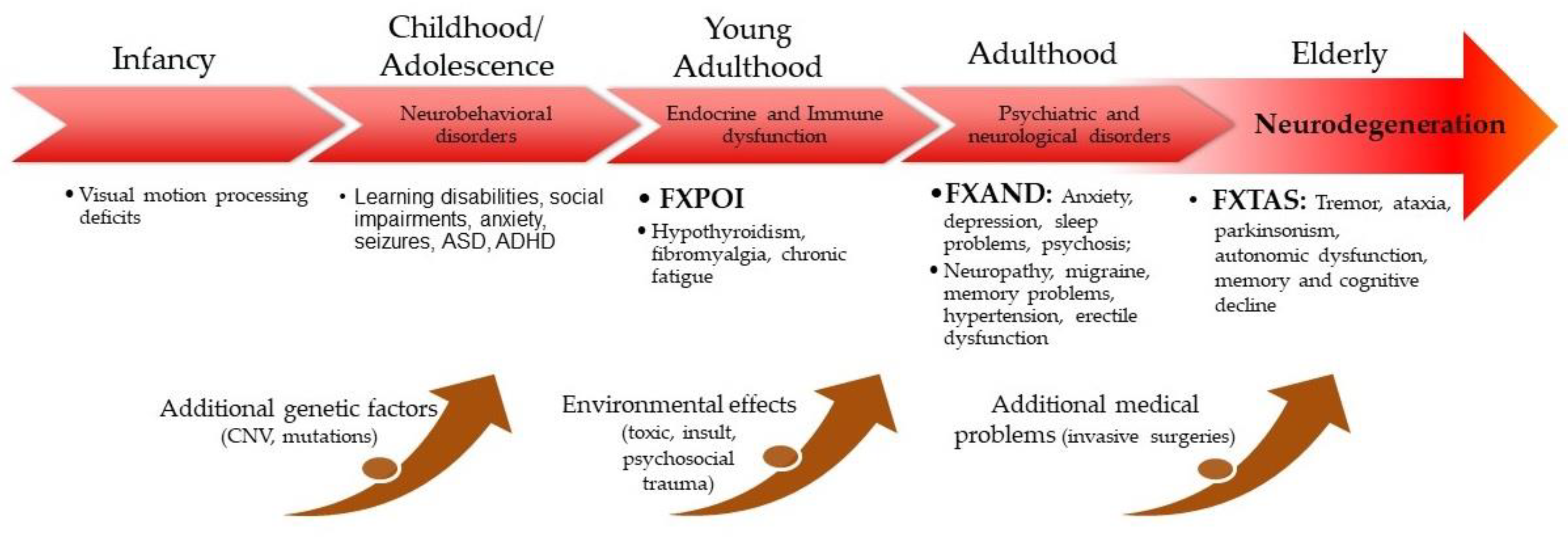
Figure 2. FXPAC involvement across the lifespan.
References
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.P.; et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65, 905–914.
- Cronister, A.; Schreiner, R.; Wittenberger, M.; Amiri, K.; Harris, K.; Hagerman, R.J. Heterozygous fragile X female: Historical, physical, cognitive, and cytogenetic features. Am. J. Med. Genet. 1991, 38, 269–274.
- Sherman, S.L. Premature ovarian failure in the fragile X syndrome. Am. J. Med. Genet. 2000, 97, 189–194.
- Mailick, M.R.; Hong, J.; Greenberg, J.; Smith, L.; Sherman, S. Curvilinear association of CGG repeats and age at menopause in women with FMR1 premutation expansions. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2014, 165, 705–711.
- Sullivan, A.K.; Marcus, M.; Epstein, M.P.; Allen, E.G.; Anido, A.E.; Paquin, J.J.; Yadav-Shah, M.; Sherman, S.L. Association of FMR1 repeat size with ovarian dysfunction. Hum. Reprod. 2005, 20, 402–412.
- Tassone, F.; Hagerman, R.J.; Taylor, A.K.; Gane, L.W.; Godfrey, T.E.; Hagerman, P.J. Elevated levels of FMR1 mRNA in carrier males: A new mechanism of involvement in the fragile-X syndrome. Am. J. Hum. Genet. 2000, 66, 6–15.
- Hagerman, R.J.; Leehey, M.; Heinrichs, W.; Tassone, F.; Wilson, R.; Hills, J.; Grigsby, J.; Gage, B.; Hagerman, P.J. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology 2001, 57, 127–130.
- Jacquemont, S.; Hagerman, R.J.; Leehey, M.; Grigsby, J.; Zhang, L.; Brunberg, J.A.; Greco, C.; Des Portes, V.; Jardini, T.; Levine, R.; et al. Fragile X premutation tremor/ataxia syndrome: Molecular, clinical, and neuroimaging correlates. Am. J. Hum. Genet. 2003, 72, 869–878.
- Jacquemont, S.; Hagerman, R.J.; Leehey, M.A.; Hall, D.A.; Levine, R.A.; Brunberg, J.A.; Zhang, L.; Jardini, T.; Gane, L.W.; Harris, S.W.; et al. Penetrance of the fragile X-associated tremor/ataxia syndrome in a premutation carrier population. JAMA 2004, 291, 460–469.
- Hall, D.A.; Birch, R.C.; Anheim, M.; Jønch, A.E.; Pintado, E.; O’Keefe, J.; Trollor, J.N.; Stebbins, G.T.; Hagerman, R.J.; Fahn, S.; et al. Emerging topics in FXTAS. J. Neurodev. Disord. 2014, 6, 31.
- Greco, C.M.; Berman, R.F.; Martin, R.M.; Tassone, F.; Schwartz, P.H.; Chang, A.; Trapp, B.D.; Iwahashi, C.; Brunberg, J.; Grigsby, J.; et al. Neuropathology of fragile X-associated tremor/ataxia syndrome (FXTAS). Brain 2006, 129 Pt 1, 243–255.
- Cabal-Herrera, A.M.; Tassanakijpanich, N.; Salcedo-Arellano, M.J.; Hagerman, R.J. Fragile X-Associated Tremor/Ataxia Syndrome (FXTAS): Pathophysiology and Clinical Implications. Int. J. Mol. Sci. 2020, 21, 4391.
- Aydin, E.Y.; Schneider, A.; Protic, D.; Wang, J.Y.; Martínez-Cerdeño, V.; Tassone, F.; Tang, H.T.; Perlman, S.; Hagerman, R.J. Rapidly Progressing Neurocognitive Disorder in a Male with FXTAS and Alzheimer’s Disease. Clin. Intervig. Aging 2020, 15, 285–292.
- Martínez-Cerdeño, V.; Wang, J.Y.; Grigsby, J.; Hall, D.; Hagerman, R.J. FXTAS New Advances and Treatments. In Fragile X Syndrome and Premutation Disorders; Hagerman, R.J., Hagerman, P.J., Eds.; Mac Keith Press: London, UK, 2020; pp. 83–96.
- Famula, J.; Ferrer, E.; Hagerman, R.J.; Tassone, F.; Schneider, A.; Rivera, S.M.; Hessl, D. Neuropsychological changes in FMR1 premutation carriers and onset of fragile X-associated tremor/ataxia syndrome. J. Neurodev. Disord. 2022, 14, 23.
- Tassone, F.; Hall, D.A. FXTAS, FXPOI, and Other Premutation Disorders; Springer International Publishing: Cham, Switzerland, 2016.
- Wang, J.; Napoli, E.; Kim, K.; McLennan, Y.A.; Hagerman, R.J.; Giulivi, C. Brain Atrophy and White Matter Damage Linked to Peripheral Bioenergetic Deficits in the Neurodegenerative Disease FXTAS. Int. J. Mol. Sci. 2021, 22, 9171.
- Greco, C.M.; Hagerman, R.J.; Tassone, F.; Chudley, A.E.; Del Bigio, M.R.; Jacquemont, S.; Leehey, M.; Hagerman, P.J. Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain 2002, 125 Pt 8, 1760–1771.
- Ariza, J.; Steward, C.; Rueckert, F.; Widdison, M.; Coffman, R.; Afjei, A.; Noctor, S.C.; Hagerman, R.; Hagerman, P.; Martínez-Cerdeño, V. Dysregulated iron metabolism in the choroid plexus in fragile X-associated tremor/ataxia syndrome. Brain Res. 2015, 1598, 88–96.
- Salcedo-Arellano, M.J.; Wang, J.Y.; McLennan, Y.A.; Doan, M.; Cabal-Herrera, A.M.; Jimenez, S.; Wolf-Ochoa, M.W.; Sanchez, D.; Juarez, P.; Tassone, F.; et al. Cerebral Microbleeds in Fragile X-Associated Tremor/Ataxia Syndrome. Mov. Disord. 2021, 36, 1935–1943.
- Salcedo-Arellano, M.J.; Wolf-Ochoa, M.W.; Hong, T.; Amina, S.; Tassone, F.; Lechpammer, M.; Hagerman, R.; Martínez-Cerdeño, V. Parkinsonism Versus Concomitant Parkinson’s Disease in Fragile X-Associated Tremor/Ataxia Syndrome. Mov. Disord. Clin. Pract. 2020, 7, 413–418.
- Seritan, A.L.; Kim, K.; Benjamin, I.; Seritan, I.; Hagerman, R.J. Risk Factors for Cognitive Impairment in Fragile X-Associated Tremor/Ataxia Syndrome. J. Geriatr. Psychiatry Neurol. 2016, 29, 328–337.
- Schneider, A.; Summers, S.; Tassone, F.; Seritan, A.; Hessl, D.; Hagerman, P.; Hagerman, R. Women with Fragile X-Associated Tremor/Ataxia Syndrome. Mov. Disord. Clin. Pract. 2020, 7, 910–919.
- Sellier, C.; Freyermuth, F.; Tabet, R.; Tran, T.; He, F.; Ruffenach, F.; Alunni, V.; Moine, H.; Thibault, C.; Page, A.; et al. Sequestration of DROSHA and DGCR8 by expanded CGG RNA repeats alters microRNA processing in fragile X-associated tremor/ataxia syndrome. Cell Rep. 2013, 3, 869–880.
- Jin, P.; Duan, R.; Qurashi, A.; Qin, Y.; Tian, D.; Rosser, T.C.; Liu, H.; Feng, Y.; Warren, S.T. Pur alpha binds to rCGG repeats and modulates repeat-mediated neurodegeneration in a Drosophila model of fragile X tremor/ataxia syndrome. Neuron 2007, 55, 556–564.
- Sofola, O.A.; Jin, P.; Qin, Y.; Duan, R.; Liu, H.; de Haro, M.; Nelson, D.L.; Botas, J. RNA-binding proteins hnRNP A2/B1 and CUGBP1 suppress fragile X CGG premutation repeat-induced neurodegeneration in a Drosophila model of FXTAS. Neuron 2007, 55, 565–571.
- Holm, K.N.; Herren, A.W.; Taylor, S.L.; Randol, J.L.; Kim, K.; Espinal, G.; Martiínez-Cerdeño, V.; Pessah, I.N.; Hagerman, R.J.; Hagerman, P.J. Human Cerebral Cortex Proteome of Fragile X-Associated Tremor/Ataxia Syndrome. Front. Mol. Biosci. 2020, 7, 600840.
- Todd, P.K.; Oh, S.Y.; Krans, A.; He, F.; Sellier, C.; Frazer, M.; Renoux, A.J.; Chen, K.C.; Scaglione, K.M.; Basrur, V.; et al. CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron 2013, 78, 440–455.
- Rosario, R.; Stewart, H.L.; Choudhury, N.R.; Michlewski, G.; Charlet-Berguerand, N.; Anderson, R.A. Evidence for a fragile X messenger ribonucleoprotein 1 (FMR1) mRNA gain-of-function toxicity mechanism contributing to the pathogenesis of fragile X-associated premature ovarian insufficiency. Faseb J. 2022, 36, e22612.
- Napoli, E.; Ross-Inta, C.; Wong, S.; Omanska-Klusek, A.; Barrow, C.; Iwahashi, C.; Garcia-Arocena, D.; Sakaguchi, D.; Berry-Kravis, E.; Hagerman, R.; et al. Altered zinc transport disrupts mitochondrial protein processing/import in fragile X-associated tremor/ataxia syndrome. Hum. Mol. Genet. 2011, 20, 3079–3092.
- Napoli, E.; Song, G.; Wong, S.; Hagerman, R.; Giulivi, C. Altered Bioenergetics in Primary Dermal Fibroblasts from Adult Carriers of the FMR1 Premutation before the Onset of the Neurodegenerative Disease Fragile X-Associated Tremor/Ataxia Syndrome. Cerebellum 2016, 15, 552–564.
- Giulivi, C.; Napoli, E.; Tassone, F.; Halmai, J.; Hagerman, R. Plasma metabolic profile delineates roles for neurodegeneration, pro-inflammatory damage and mitochondrial dysfunction in the FMR1 premutation. Biochem. J. 2016, 473, 3871–3888.
- Loesch, D.Z.; Duffy, D.L.; Martin, N.G.; Tassone, F.; Atkinson, A.; Storey, E. ‘Essential Tremor’ Phenotype in FMR1 Premutation/Gray Zone Sibling Series: Exploring Possible Genetic Modifiers. Twin Res. Hum. Genet. 2021, 24, 95–102.
- Johnson, D.; Santos, E.; Kim, K.; Ponzini, M.D.; McLennan, Y.A.; Schneider, A.; Tassone, F.; Hagerman, R.J. Increased Pain Symptomatology among Females vs. Males with Fragile X-Associated Tremor/Ataxia Syndrome. Front. Psychiatry 2021, 12, 762915.
- Coffey, S.M.; Cook, K.; Tartaglia, N.; Tassone, F.; Nguyen, D.V.; Pan, R.; Bronsky, H.E.; Yuhas, J.; Borodyanskaya, M.; Grigsby, J.; et al. Expanded clinical phenotype of women with the FMR1 premutation. Am. J. Med. Genet. A 2008, 146, 1009–1016.
- Cordeiro, L.; Abucayan, F.; Hagerman, R.; Tassone, F.; Hessl, D. Anxiety disorders in fragile X premutation carriers: Preliminary characterization of probands and non-probands. Intractable Rare Dis. Res. 2015, 4, 123–130.
- Farzin, F.; Perry, H.; Hessl, D.; Loesch, D.; Cohen, J.; Bacalman, S.; Gane, L.; Tassone, F.; Hagerman, P.; Hagerman, R. Autism spectrum disorders and attention-deficit/hyperactivity disorder in boys with the fragile X premutation. J. Dev. Behav. Pediatr. 2006, 27 (Suppl. S2), S137–S144.
- Hunter, J.E.; Leslie, M.; Novak, G.; Hamilton, D.; Shubeck, L.; Charen, K.; Abramowitz, A.; Epstein, M.P.; Lori, A.; Binder, E.; et al. Depression and anxiety symptoms among women who carry the FMR1 premutation: Impact of raising a child with fragile X syndrome is moderated by CRHR1 polymorphisms. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2012, 159, 549–559.
- Aishworiya, R.; Protic, D.; Tang, S.J.; Schneider, A.; Tassone, F.; Hagerman, R. Fragile X-Associated Neuropsychiatric Disorders (FXAND) in Young Fragile X Premutation Carriers. Genes 2022, 13, 2399.
- Clifford, S.; Dissanayake, C.; Bui, Q.M.; Huggins, R.; Taylor, A.K.; Loesch, D.Z. Autism spectrum phenotype in males and females with fragile X full mutation and premutation. J. Autism Dev. Disord. 2007, 37, 738–747.
- Bailey, D.B., Jr.; Raspa, M.; Olmsted, M.; Holiday, D.B. Co-occurring conditions associated with FMR1 gene variations: Findings from a national parent survey. Am. J. Med. Genet. A 2008, 146, 2060–2069.
- Aziz, M.; Stathopulu, E.; Callias, M.; Taylor, C.; Turk, J.; Oostra, B.; Willemsen, R.; Patton, M. Clinical features of boys with fragile X premutations and intermediate alleles. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2003, 121, 119–127.
- Chonchaiya, W.; Au, J.; Schneider, A.; Hessl, D.; Harris, S.W.; Laird, M.; Mu, Y.; Tassone, F.; Nguyen, D.V.; Hagerman, R.J. Increased prevalence of seizures in boys who were probands with the FMR1 premutation and co-morbid autism spectrum disorder. Hum. Genet. 2012, 131, 581–589.
- Lozano, R.; Hagerman, R.J.; Duyzend, M.; Budimirovic, D.B.; Eichler, E.E.; Tassone, F. Genomic studies in fragile X premutation carriers. J. Neurodev. Disord. 2014, 6, 27.
- Chen, H.; Chan, D.C. Mitochondrial dynamics—Fusion, fission, movement, and mitophagy—In neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, R169–R176.
- Song, G.; Napoli, E.; Wong, S.; Hagerman, R.; Liu, S.; Tassone, F.; Giulivi, C. Altered redox mitochondrial biology in the neurodegenerative disorder fragile X-tremor/ataxia syndrome: Use of antioxidants in precision medicine. Mol. Med. 2016, 22, 548–559.
- Ligsay, A.; El-Deeb, M.; Salcedo-Arellano, M.J.; Schloemerkemper, N.; Grayson, J.S.; Hagerman, R. General Anesthetic Use in Fragile X Spectrum Disorders. J. Neurosurg. Anesth. 2019, 31, 285–290.
- Muzar, Z.; Adams, P.E.; Schneider, A.; Hagerman, R.J.; Lozano, R. Addictive substances may induce a rapid neurological deterioration in fragile X-associated tremor ataxia syndrome: A report of two cases. Intractable Rare Dis. Res. 2014, 3, 162–165.
- Muzar, Z.; Lozano, R.; Schneider, A.; Adams, P.E.; Faradz, S.M.; Tassone, F.; Hagerman, R.J. Methadone use in a male with the FMRI premutation and FXTAS. Am. J. Med. Genet. A 2015, 167, 1354–1359.
- Keil Stietz, K.P.; Sethi, S.; Klocke, C.R.; de Ruyter, T.E.; Wilson, M.D.; Pessah, I.N.; Lein, P.J. Sex and Genotype Modulate the Dendritic Effects of Developmental Exposure to a Human-Relevant Polychlorinated Biphenyls Mixture in the Juvenile Mouse. Front. Neurosci. 2021, 15, 766802.
- Saldarriaga, W.; Salcedo-Arellano, M.J.; Rodriguez-Guerrero, T.; Ríos, M.; Fandiño-Losada, A.; Ramirez-Cheyne, J.; Lein, P.J.; Tassone, F.; Hagerman, R.J. Increased severity of fragile X spectrum disorders in the agricultural community of Ricaurte, Colombia. Int. J. Dev. Neurosci. 2019, 72, 1–5.
- Saldarriaga, W.; Lein, P.; González Teshima, L.Y.; Isaza, C.; Rosa, L.; Polyak, A.; Hagerman, R.; Girirajan, S.; Silva, M.; Tassone, F. Phenobarbital use and neurological problems in FMR1 premutation carriers. Neurotoxicology 2016, 53, 141–147.
- Sodhi, D.K.; Hagerman, R. Fragile X Premutation: Medications, Therapy and Lifestyle Advice. Pharmgenom. Pers. Med. 2021, 14, 1689–1699.
- Kaplan, E.S.; Cao, Z.; Hulsizer, S.; Tassone, F.; Berman, R.F.; Hagerman, P.J.; Pessah, I.N. Early mitochondrial abnormalities in hippocampal neurons cultured from FMR1 pre-mutation mouse model. J. Neurochem. 2012, 123, 613–621.
- Cao, Z.; Hulsizer, S.; Tassone, F.; Tang, H.T.; Hagerman, R.J.; Rogawski, M.A.; Hagerman, P.J.; Pessah, I.N. Clustered burst firing in FMR1 premutation hippocampal neurons: Amelioration with allopregnanolone. Hum. Mol. Genet. 2012, 21, 2923–2935.
- Aishworiya, R.; Protic, D.; Hagerman, R. Autism spectrum disorder in the fragile X premutation state: Possible mechanisms and implications. J. Neurol. 2022, 269, 4676–4683.
- Summers, S.M.; Cogswell, J.; Goodrich, J.E.; Mu, Y.; Nguyen, D.V.; Brass, S.D.; Hagerman, R.J. Fatigue and body mass index in the Fragile X premutation carrier. Fatigue Biomed. Health Behav. 2014, 2, 64–72.
- Summers, S.M.; Cogswell, J.; Goodrich, J.E.; Mu, Y.; Nguyen, D.V.; Brass, S.D.; Hagerman, R.J. Prevalence of restless legs syndrome and sleep quality in carriers of the fragile X premutation. Clin. Genet. 2014, 86, 181–184.
- Hunter, J.E.; Wheeler, A.C.; Allen, E.G.; Wald, K.; Rajkovic, A.; Hagerman, R.J.; Sherman, S.L. Fragile X Syndrome and Premutation Disorders: New Developments and Treatments; Hagerman, R.J., Hagerman, P.J., Eds.; Mac Keith Press: London, UK, 2020; pp. 75–82.
- Hagerman, R.J.; Protic, D.; Rajaratnam, A.; Salcedo-Arellano, M.J.; Aydin, E.Y.; Schneider, A. Fragile X-Associated Neuropsychiatric Disorders (FXAND). Front. Psychiatry 2018, 9, 564.
- Winarni, T.I.; Chonchaiya, W.; Sumekar, T.A.; Ashwood, P.; Morales, G.M.; Tassone, F.; Nguyen, D.V.; Faradz, S.M.; Van de Water, J.; Cook, K.; et al. Immune-mediated disorders among women carriers of fragile X premutation alleles. Am. J. Med. Genet. A 2012, 158, 2473–2481.
- Hamlin, A.A.; Sukharev, D.; Campos, L.; Mu, Y.; Tassone, F.; Hessl, D.; Nguyen, D.V.; Loesch, D.; Hagerman, R.J. Hypertension in FMR1 premutation males with and without fragile X-associated tremor/ataxia syndrome (FXTAS). Am. J. Med. Genet. A 2012, 158, 1304–1309.
- Au, J.; Akins, R.S.; Berkowitz-Sutherland, L.; Tang, H.T.; Chen, Y.; Boyd, A.; Tassone, F.; Nguyen, D.V.; Hagerman, R. Prevalence and risk of migraine headaches in adult fragile X premutation carriers. Clin. Genet. 2013, 84, 546–551.
- Tassanakijpanich, N.; McKenzie, F.J.; McLennan, Y.A.; Makhoul, E.; Tassone, F.; Jasoliya, M.J.; Romney, C.; Petrasic, I.C.; Napalinga, K.; Buchanan, C.B.; et al. Hypermobile Ehlers-Danlos syndrome (hEDS) phenotype in fragile X premutation carriers: Case series. J. Med. Genet. 2022, 59, 687–690.
- McKenzie, F.J.; Tassankijpanich, N.; Epps, K.C.; March, S.K.; Hagerman, R.J. Spontaneous Coronary Artery Dissection in Females with the Fragile X FMR1 Premutation. JACC Case Rep. 2020, 2, 40–44.
- Hunsaker, M.R.; Greco, C.M.; Spath, M.A.; Smits, A.P.; Navarro, C.S.; Tassone, F.; Kros, J.M.; Severijnen, L.A.; Berry-Kravis, E.M.; Berman, R.F.; et al. Widespread non-central nervous system organ pathology in fragile X premutation carriers with fragile X-associated tremor/ataxia syndrome and CGG knock-in mice. Acta Neuropathol. 2011, 122, 467–479.
- Bourgeois, J.A.; Coffey, S.M.; Rivera, S.M.; Hessl, D.; Gane, L.W.; Tassone, F.; Greco, C.; Finucane, B.; Nelson, L.; Berry-Kravis, E.; et al. A review of fragile X premutation disorders: Expanding the psychiatric perspective. J. Clin. Psychiatry 2009, 70, 852–862.
- Bourgeois, J.A.; Seritan, A.L.; Casillas, E.M.; Hessl, D.; Schneider, A.; Yang, Y.; Kaur, I.; Cogswell, J.B.; Nguyen, D.V.; Hagerman, R.J. Lifetime prevalence of mood and anxiety disorders in fragile X premutation carriers. J. Clin. Psychiatry 2011, 72, 175–182.
- Losh, M.; Klusek, J.; Martin, G.E.; Sideris, J.; Parlier, M.; Piven, J. Defining genetically meaningful language and personality traits in relatives of individuals with fragile X syndrome and relatives of individuals with autism. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2012, 159, 660–668.
- Roberts, J.E.; Tonnsen, B.L.; McCary, L.M.; Ford, A.L.; Golden, R.N.; Bailey, D.B., Jr. Trajectory and Predictors of Depression and Anxiety Disorders in Mothers with the FMR1 Premutation. Biol. Psychiatry 2016, 79, 850–857.
- Gossett, A.; Sansone, S.; Schneider, A.; Johnston, C.; Hagerman, R.; Tassone, F.; Rivera, S.M.; Seritan, A.L.; Hessl, D. Psychiatric disorders among women with the fragile X premutation without children affected by fragile X syndrome. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2016, 171, 1139–1147.
- Kraan, C.M.; Hocking, D.R.; Georgiou-Karistianis, N.; Metcalfe, S.A.; Archibald, A.D.; Fielding, J.; Trollor, J.; Bradshaw, J.L.; Cohen, J.; Cornish, K.M. Impaired response inhibition is associated with self-reported symptoms of depression, anxiety, and ADHD in female FMR1 premutation carriers. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2014, 165, 41–51.
- Movaghar, A.; Page, D.; Brilliant, M.; Baker, M.W.; Greenberg, J.; Hong, J.; DaWalt, L.S.; Saha, K.; Kuusisto, F.; Stewart, R.; et al. Data-driven phenotype discovery of FMR1 premutation carriers in a population-based sample. Sci. Adv. 2019, 5, eaaw7195.
- Loesch, D.Z.; Bui, M.Q.; Hammersley, E.; Schneider, A.; Storey, E.; Stimpson, P.; Burgess, T.; Francis, D.; Slater, H.; Tassone, F.; et al. Psychological status in female carriers of premutation FMR1 allele showing a complex relationship with the size of CGG expansion. Clin. Genet. 2015, 87, 173–178.
- Johnson, K.; Herring, J.; Richstein, J. Fragile X Premutation Associated Conditions (FXPAC). Front. Pediatr. 2020, 8, 266.
- Kenneson, A.; Zhang, F.; Hagedorn, C.H.; Warren, S.T. Reduced FMRP and increased FMR1 transcription is proportionally associated with CGG repeat number in intermediate-length and premutation carriers. Hum. Mol. Genet. 2001, 10, 1449–1454.
- Allen, E.G.; He, W.; Yadav-Shah, M.; Sherman, S.L. A study of the distributional characteristics of FMR1 transcript levels in 238 individuals. Hum. Genet. 2004, 114, 439–447.
- Tassone, F.; Beilina, A.; Carosi, C.; Albertosi, S.; Bagni, C.; Li, L.; Glover, K.; Bentley, D.; Hagerman, P.J. Elevated FMR1 mRNA in premutation carriers is due to increased transcription. Rna 2007, 13, 555–562.
- Primerano, B.; Tassone, F.; Hagerman, R.J.; Hagerman, P.; Amaldi, F.; Bagni, C. Reduced FMR1 mRNA translation efficiency in fragile X patients with premutations. Rna 2002, 8, 1482–1488.
- Peprah, E.; He, W.; Allen, E.; Oliver, T.; Boyne, A.; Sherman, S.L. Examination of FMR1 transcript and protein levels among 74 premutation carriers. J. Hum. Genet. 2010, 55, 66–68.
- Yrigollen, C.M.; Martorell, L.; Durbin-Johnson, B.; Naudo, M.; Genoves, J.; Murgia, A.; Polli, R.; Zhou, L.; Barbouth, D.; Rupchock, A.; et al. AGG interruptions and maternal age affect FMR1 CGG repeat allele stability during transmission. J. Neurodev. Disord. 2014, 6, 24.
- Nolin, S.L.; Glicksman, A.; Ersalesi, N.; Dobkin, C.; Brown, W.T.; Cao, R.; Blatt, E.; Sah, S.; Latham, G.J.; Hadd, A.G. Fragile X full mutation expansions are inhibited by one or more AGG interruptions in premutation carriers. Genet. Med. 2015, 17, 358–364.
- Yrigollen, C.M.; Tassone, F.; Durbin-Johnson, B.; Tassone, F. The role of AGG interruptions in the transcription of FMR1 premutation alleles. PLoS ONE 2011, 6, e21728.
- Ludwig, A.L.; Raske, C.; Tassone, F.; Garcia-Arocena, D.; Hershey, J.W.; Hagerman, P.J. Translation of the FMR1 mRNA is not influenced by AGG interruptions. Nucleic Acids Res. 2009, 37, 6896–6904.
- Ladd, P.D.; Smith, L.E.; Rabaia, N.A.; Moore, J.M.; Georges, S.A.; Hansen, R.S.; Hagerman, R.J.; Tassone, F.; Tapscott, S.J.; Filippova, G.N. An antisense transcript spanning the CGG repeat region of FMR1 is upregulated in premutation carriers but silenced in full mutation individuals. Hum. Mol. Genet. 2007, 16, 3174–3187.
- Hwang, Y.H.; Hayward, B.E.; Zafarullah, M.; Kumar, J.; Durbin Johnson, B.; Holmans, P.; Usdin, K.; Tassone, F. Both cis and trans-acting genetic factors drive somatic instability in female carriers of the FMR1 premutation. Sci. Rep. 2022, 12, 10419.
- Zafarullah, M.; Li, J.; Tseng, E.; Tassone, F. Structure and Alternative Splicing of the Antisense FMR1 (ASFMR1) Gene. Mol. Neurobiol. 2023, 60, 2051–2061.
- Zafarullah, M.; Palczewski, G.; Rivera, S.M.; Hessl, D.R.; Tassone, F. Metabolic profiling reveals dysregulated lipid metabolism and potential biomarkers associated with the development and progression of Fragile X-Associated Tremor/Ataxia Syndrome (FXTAS). Faseb J. 2020, 34, 16676–16692.
- Vittal, P.; Pandya, S.; Sharp, K.; Berry-Kravis, E.; Zhou, L.; Ouyang, B.; Jackson, J.; Hall, D.A. ASFMR1 splice variant: A predictor of fragile X-associated tremor/ataxia syndrome. Neurol. Genet. 2018, 4, e246.
- Pretto, D.I.; Mendoza-Morales, G.; Lo, J.; Cao, R.; Hadd, A.; Latham, G.J.; Durbin-Johnson, B.; Hagerman, R.; Tassone, F. CGG allele size somatic mosaicism and methylation in FMR1 premutation alleles. J. Med. Genet. 2014, 51, 309–318.
- Aishworiya, R.; Hwang, Y.H.; Santos, E.; Hayward, B.; Usdin, K.; Durbin-Johnson, B.; Hagerman, R.; Tassone, F. Clinical implications of somatic allele expansion in female FMR1 premutation carriers. Sci. Rep. 2023, 13, 7050.
- Dobkin, C.S.; Nolin, S.L.; Cohen, I.; Sudhalter, V.; Bialer, M.G.; Ding, X.H.; Jenkins, E.C.; Zhong, N.; Brown, W.T. Tissue differences in fragile X mosaics: Mosaicism in blood cells may differ greatly from skin. Am. J. Med. Genet. 1996, 64, 296–301.
- Maddalena, A.; Yadvish, K.N.; Spence, W.C.; Howard-Peebles, P.N. A fragile X mosaic male with a cryptic full mutation detected in epithelium but not in blood. Am. J. Med. Genet. 1996, 64, 309–312.
- Taylor, A.K.; Tassone, F.; Dyer, P.N.; Hersch, S.M.; Harris, J.B.; Greenough, W.T.; Hagerman, R.J. Tissue heterogeneity of the FMR1 mutation in a high-functioning male with fragile X syndrome. Am. J. Med. Genet. 1999, 84, 233–239.
- Fernández, E.; Gennaro, E.; Pirozzi, F.; Baldo, C.; Forzano, F.; Turolla, L.; Faravelli, F.; Gastaldo, D.; Coviello, D.; Grasso, M.; et al. FXS-Like Phenotype in Two Unrelated Patients Carrying a Methylated Premutation of the FMR1 Gene. Front. Genet. 2018, 9, 442.
- Jiraanont, P.; Sweha, S.R.; AlOlaby, R.R.; Silva, M.; Tang, H.T.; Durbin-Johnson, B.; Schneider, A.; Espinal, G.M.; Hagerman, P.J.; Rivera, S.M.; et al. Clinical and molecular correlates in fragile X premutation females. eNeurologicalSci 2017, 7, 49–56.
- Del Hoyo Soriano, L.; Thurman, A.J.; Harvey, D.J.; Ted Brown, W.; Abbeduto, L. Genetic and maternal predictors of cognitive and behavioral trajectories in females with fragile X syndrome. J. Neurodev. Disord. 2018, 10, 22.
- Lyon, M.F. Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature 1961, 190, 372–373.
- Sun, Z.; Fan, J.; Wang, Y. X-Chromosome Inactivation and Related Diseases. Genet. Res. 2022, 2022, 1391807.
- Devys, D.; Lutz, Y.; Rouyer, N.; Bellocq, J.P.; Mandel, J.L. The FMR-1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nat. Genet. 1993, 4, 335–340.
- Franke, P.; Leboyer, M.; Hardt, J.; Sohne, E.; Weiffenbach, O.; Biancalana, V.; Cornillet-Lefebre, P.; Delobel, B.; Froster, U.; Schwab, S.G.; et al. Neuropsychological profiles of FMR-1 premutation and full-mutation carrier females. Psychiatry Res. 1999, 87, 223–231.
- Loesch, D.Z.; Huggins, R.M.; Hagerman, R.J. Phenotypic variation and FMRP levels in fragile X. Ment. Retard. Dev. Disabil. Res. Rev. 2004, 10, 31–41.
- Godler, D.E.; Slater, H.R.; Bui, Q.M.; Ono, M.; Gehling, F.; Francis, D.; Amor, D.J.; Hopper, J.L.; Hagerman, R.; Loesch, D.Z. FMR1 intron 1 methylation predicts FMRP expression in blood of female carriers of expanded FMR1 alleles. J. Mol. Diagn. 2011, 13, 528–536.
- Berry-Kravis, E.; Potanos, K.; Weinberg, D.; Zhou, L.; Goetz, C.G. Fragile X-associated tremor/ataxia syndrome in sisters related to X-inactivation. Ann. Neurol. 2005, 57, 144–147.
- Hall, D.A.; Robertson-Dick, E.E.; O’Keefe, J.A.; Hadd, A.G.; Zhou, L.; Berry-Kravis, E. X-inactivation in the clinical phenotype of fragile X premutation carrier sisters. Neurol. Genet. 2016, 2, e45.
- Abrams, M.T.; Reiss, A.L.; Freund, L.S.; Baumgardner, T.L.; Chase, G.A.; Denckla, M.B. Molecular-neurobehavioral associations in females with the fragile X full mutation. Am. J. Med. Genet. 1994, 51, 317–327.
- Hessl, D.; Dyer-Friedman, J.; Glaser, B.; Wisbeck, J.; Barajas, R.G.; Taylor, A.; Reiss, A.L. The influence of environmental and genetic factors on behavior problems and autistic symptoms in boys and girls with fragile X syndrome. Pediatrics 2001, 108, E88.
- Heine-Suñer, D.; Torres-Juan, L.; Morlà, M.; Busquets, X.; Barceló, F.; Picó, G.; Bonilla, L.; Govea, N.; Bernués, M.; Rosell, J. Fragile-X syndrome and skewed X-chromosome inactivation within a family: A female member with complete inactivation of the functional X chromosome. Am. J. Med. Genet. Part. A 2003, 122, 108–114.
- Talebizadeh, Z.; Bittel, D.C.; Veatch, O.J.; Kibiryeva, N.; Butler, M.G. Brief report: Non-random X chromosome inactivation in females with autism. J. Autism Dev. Disord. 2005, 35, 675–681.
- Stembalska, A.; Łaczmańska, I.; Gil, J.; Pesz, K.A. Fragile X syndrome in females—A familial case report and review of the literature. Dev. Period. Med. 2016, 20, 99–104.
- Sobesky, W.E.; Taylor, A.K.; Pennington, B.F.; Bennetto, L.; Porter, D.; Riddle, J.; Hagerman, R.J. Molecular/clinical correlations in females with fragile X. Am. J. Med. Genet. 1996, 64, 340–345.
- Tassone, F.; Pan, R.; Amiri, K.; Taylor, A.K.; Hagerman, P.J. A rapid polymerase chain reaction-based screening method for identification of all expanded alleles of the fragile X (FMR1) gene in newborn and high-risk populations. J. Mol. Diagn. 2008, 10, 43–49.
- Godler, D.E.; Tassone, F.; Loesch, D.Z.; Taylor, A.K.; Gehling, F.; Hagerman, R.J.; Burgess, T.; Ganesamoorthy, D.; Hennerich, D.; Gordon, L.; et al. Methylation of novel markers of fragile X alleles is inversely correlated with FMRP expression and FMR1 activation ratio. Hum. Mol. Genet. 2010, 19, 1618–1632.
- Hadd, A.G.; Filipovic-Sadic, S.; Zhou, L.; Williams, A.; Latham, G.J.; Berry-Kravis, E.; Hall, D.A. A methylation PCR method determines FMR1 activation ratios and differentiates premutation allele mosaicism in carrier siblings. Clin. Epigenetics 2016, 8, 130.
- Protic, D.; Polli, R.; Hwang, Y.H.; Mendoza, G.; Hagerman, R.; Durbin-Johnson, B.; Hayward, B.E.; Usdin, K.; Murgia, A.; Tassone, F. Activation Ratio Correlates with IQ in Female Carriers of the FMR1 Premutation. Cells 2023, 12, 1711.
- Hagerman, R.J.; Hagerman, P. Fragile X-associated tremor/ataxia syndrome—Features, mechanisms and management. Nat. Rev. Neurol. 2016, 12, 403–412.
- Loomis, E.W.; Sanz, L.A.; Chédin, F.; Hagerman, P.J. Transcription-associated R-loop formation across the human FMR1 CGG-repeat region. PLoS Genet. 2014, 10, e1004294.
- Malik, I.; Kelley, C.P.; Wang, E.T.; Todd, P.K. Author Correction: Molecular mechanisms underlying nucleotide repeat expansion disorders. Nat. Rev. Mol. Cell Biol. 2021, 22, 644.
- Iwahashi, C.K.; Yasui, D.H.; An, H.J.; Greco, C.M.; Tassone, F.; Nannen, K.; Babineau, B.; Lebrilla, C.B.; Hagerman, R.J.; Hagerman, P.J. Protein composition of the intranuclear inclusions of FXTAS. Brain 2006, 129 Pt 1, 256–271.
- Sellier, C.; Rau, F.; Liu, Y.; Tassone, F.; Hukema, R.K.; Gattoni, R.; Schneider, A.; Richard, S.; Willemsen, R.; Elliott, D.J.; et al. Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. Embo J. 2010, 29, 1248–1261.
- Qurashi, A.; Liu, H.; Ray, L.; Nelson, D.L.; Duan, R.; Jin, P. Chemical screen reveals small molecules suppressing fragile X premutation rCGG repeat-mediated neurodegeneration in Drosophila. Hum. Mol. Genet. 2012, 21, 2068–2075.
- Tan, H.; Poidevin, M.; Li, H.; Chen, D.; Jin, P. MicroRNA-277 modulates the neurodegeneration caused by Fragile X premutation rCGG repeats. PLoS Genet. 2012, 8, e1002681.
- Khalili, K.; Del Valle, L.; Muralidharan, V.; Gault, W.J.; Darbinian, N.; Otte, J.; Meier, E.; Johnson, E.M.; Daniel, D.C.; Kinoshita, Y.; et al. Puralpha is essential for postnatal brain development and developmentally coupled cellular proliferation as revealed by genetic inactivation in the mouse. Mol. Cell Biol. 2003, 23, 6857–6875.
- Hokkanen, S.; Feldmann, H.M.; Ding, H.; Jung, C.K.; Bojarski, L.; Renner-Müller, I.; Schüller, U.; Kretzschmar, H.; Wolf, E.; Herms, J. Lack of Pur-alpha alters postnatal brain development and causes megalencephaly. Hum. Mol. Genet. 2012, 21, 473–484.
- Galloway, J.N.; Shaw, C.; Yu, P.; Parghi, D.; Poidevin, M.; Jin, P.; Nelson, D.L. CGG repeats in RNA modulate expression of TDP-43 in mouse and fly models of fragile X tremor ataxia syndrome. Hum. Mol. Genet. 2014, 23, 5906–5915.
- He, F.; Krans, A.; Freibaum, B.D.; Taylor, J.P.; Todd, P.K. TDP-43 suppresses CGG repeat-induced neurotoxicity through interactions with HnRNP A2/B1. Hum. Mol. Genet. 2014, 23, 5036–5051.
- Zu, T.; Gibbens, B.; Doty, N.S.; Gomes-Pereira, M.; Huguet, A.; Stone, M.D.; Margolis, J.; Peterson, M.; Markowski, T.W.; Ingram, M.A.; et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. USA 2011, 108, 260–265.
- Banez-Coronel, M.; Ranum, L.P.W. Repeat-associated non-AUG (RAN) translation: Insights from pathology. Lab. Investig. 2019, 99, 929–942.
- Kearse, M.G.; Green, K.M.; Krans, A.; Rodriguez, C.M.; Linsalata, A.E.; Goldstrohm, A.C.; Todd, P.K. CGG Repeat-Associated Non-AUG Translation Utilizes a Cap-Dependent Scanning Mechanism of Initiation to Produce Toxic Proteins. Mol. Cell 2016, 62, 314–322.
- Krans, A.; Skariah, G.; Zhang, Y.; Bayly, B.; Todd, P.K. Neuropathology of RAN translation proteins in fragile X-associated tremor/ataxia syndrome. Acta Neuropathol. Commun. 2019, 7, 152.
- Wright, S.E.; Rodriguez, C.M.; Monroe, J.; Xing, J.; Krans, A.; Flores, B.N.; Barsur, V.; Ivanova, M.I.; Koutmou, K.S.; Barmada, S.J.; et al. CGG repeats trigger translational frameshifts that generate aggregation-prone chimeric proteins. Nucleic Acids Res. 2022, 50, 8674–8689.
- Sellier, C.; Buijsen, R.A.M.; He, F.; Natla, S.; Jung, L.; Tropel, P.; Gaucherot, A.; Jacobs, H.; Meziane, H.; Vincent, A.; et al. Translation of Expanded CGG Repeats into FMRpolyG Is Pathogenic and May Contribute to Fragile X Tremor Ataxia Syndrome. Neuron 2017, 93, 331–347.
- Buijsen, R.A.; Visser, J.A.; Kramer, P.; Severijnen, E.A.; Gearing, M.; Charlet-Berguerand, N.; Sherman, S.L.; Berman, R.F.; Willemsen, R.; Hukema, R.K. Presence of inclusions positive for polyglycine containing protein, FMRpolyG, indicates that repeat-associated non-AUG translation plays a role in fragile X-associated primary ovarian insufficiency. Hum. Reprod. 2016, 31, 158–168.
- Ma, L.; Herren, A.W.; Espinal, G.; Randol, J.; McLaughlin, B.; Martinez-Cerdeño, V.; Pessah, I.N.; Hagerman, R.J.; Hagerman, P.J. Composition of the Intranuclear Inclusions of Fragile X-associated Tremor/Ataxia Syndrome. Acta Neuropathol. Commun. 2019, 7, 143.
- Zhang, Y.; Glineburg, M.R.; Basrur, V.; Conlon, K.; Wright, S.E.; Krans, A.; Hall, D.A.; Todd, P.K. Mechanistic convergence across initiation sites for RAN translation in fragile X associated tremor ataxia syndrome. Hum. Mol. Genet. 2022, 31, 2317–2332.
- Asamitsu, S.; Yabuki, Y.; Ikenoshita, S.; Kawakubo, K.; Kawasaki, M.; Usuki, S.; Nakayama, Y.; Adachi, K.; Kugoh, H.; Ishii, K.; et al. CGG repeat RNA G-quadruplexes interact with FMRpolyG to cause neuronal dysfunction in fragile X-related tremor/ataxia syndrome. Sci. Adv. 2021, 7, eabd9440.
- Linsalata, A.E.; He, F.; Malik, A.M.; Glineburg, M.R.; Green, K.M.; Natla, S.; Flores, B.N.; Krans, A.; Archbold, H.C.; Fedak, S.J.; et al. DDX3X and specific initiation factors modulate FMR1 repeat-associated non-AUG-initiated translation. EMBO Rep. 2019, 20, e47498.
- Rodriguez, C.M.; Wright, S.E.; Kearse, M.G.; Haenfler, J.M.; Flores, B.N.; Liu, Y.; Ifrim, M.F.; Glineburg, M.R.; Krans, A.; Jafar-Nejad, P.; et al. A native function for RAN translation and CGG repeats in regulating fragile X protein synthesis. Nat. Neurosci. 2020, 23, 386–397.
- Haify, S.N.; Mankoe, R.S.D.; Boumeester, V.; van der Toorn, E.C.; Verhagen, R.F.M.; Willemsen, R.; Hukema, R.K.; Bosman, L.W.J. Lack of a Clear Behavioral Phenotype in an Inducible FXTAS Mouse Model Despite the Presence of Neuronal FMRpolyG-Positive Aggregates. Front. Mol. Biosci. 2020, 7, 599101.
- Tseng, Y.J.; Sandwith, S.N.; Green, K.M.; Chambers, A.E.; Krans, A.; Raimer, H.M.; Sharlow, M.E.; Reisinger, M.A.; Richardson, A.E.; Routh, E.D.; et al. The RNA helicase DHX36-G4R1 modulates C9orf72 GGGGCC hexanucleotide repeat-associated translation. J. Biol. Chem. 2021, 297, 100914.
- Green, K.M.; Glineburg, M.R.; Kearse, M.G.; Flores, B.N.; Linsalata, A.E.; Fedak, S.J.; Goldstrohm, A.C.; Barmada, S.J.; Todd, P.K. RAN translation at C9orf72-associated repeat expansions is selectively enhanced by the integrated stress response. Nat. Commun. 2017, 8, 2005.
- Zafarullah, M.; Durbin-Johnson, B.; Fourie, E.S.; Hessl, D.R.; Rivera, S.M.; Tassone, F. Metabolomic Biomarkers Are Associated with Area of the Pons in Fragile X Premutation Carriers at Risk for Developing FXTAS. Front. Psychiatry 2021, 12, 691717.
- In Proceedings of the 5th International Conference on FMR1 Premutation: Molecular Mechanism, Clinical Involvements and Target. Waitangi, New Zealand, 27 February–3 March 2023.
- Zafarullah, M.; Li, J.; Salemi, M.; Phinney, B.; Durbin-Johnson, B.P.; Hagerman, R.; Hessl, D.; Rivera, S.M.; Tassone, F. Blood proteome profiling reveals biomarkers and pathways alterations in Fragile X premutation carriers at risk for developing FXTAS. Int. J. Mol. Biol. 2023, 24, 13477.
- Derbis, M.; Kul, E.; Niewiadomska, D.; Sekrecki, M.; Piasecka, A.; Taylor, K.; Hukema, R.K.; Stork, O.; Sobczak, K. Short antisense oligonucleotides alleviate the pleiotropic toxicity of RNA harboring expanded CGG repeats. Nat. Commun. 2021, 12, 1265.
- Kong, H.E.; Lim, J.; Linsalata, A.; Kang, Y.; Malik, I.; Allen, E.G.; Cao, Y.; Shubeck, L.; Johnston, R.; Huang, Y.; et al. Identification of PSMB5 as a genetic modifier of fragile X-associated tremor/ataxia syndrome. Proc. Natl. Acad. Sci. USA 2022, 119, e2118124119.
- Konieczny, P.; Mukherjee, S.; Stepniak-Konieczna, E.; Taylor, K.; Niewiadomska, D.; Piasecka, A.; Walczak, A.; Baud, A.; Dohno, C.; Nakatani, K.; et al. Cyclic mismatch binding ligands interact with disease-associated CGG trinucleotide repeats in RNA and suppress their translation. Nucleic Acids Res. 2021, 49, 9479–9495.
- Filley, C.M.; Brown, M.S.; Onderko, K.; Ray, M.; Bennett, R.E.; Berry-Kravis, E.; Grigsby, J. White matter disease and cognitive impairment in FMR1 premutation carriers. Neurology 2015, 84, 2146–2152.
- Kong, H.E.; Lim, J.; Zhang, F.; Huang, L.; Gu, Y.; Nelson, D.L.; Allen, E.G.; Jin, P. Metabolic pathways modulate the neuronal toxicity associated with fragile X-associated tremor/ataxia syndrome. Hum. Mol. Genet. 2019, 28, 980–991.
- Fan, J.; Tao, W.; Li, X.; Li, H.; Zhang, J.; Wei, D.; Chen, Y.; Zhang, Z. The Contribution of Genetic Factors to Cognitive Impairment and Dementia: Apolipoprotein E Gene, Gene Interactions, and Polygenic Risk. Int. J. Mol. Sci. 2019, 20, 1177.
- Liu, C.C.; Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118.
- Raulin, A.C.; Doss, S.V.; Trottier, Z.A.; Ikezu, T.C.; Bu, G.; Liu, C.C. ApoE in Alzheimer’s disease: Pathophysiology and therapeutic strategies. Mol. Neurodegener. 2022, 17, 72.
- Blanchard, J.W.; Akay, L.A.; Davila-Velderrain, J.; von Maydell, D.; Mathys, H.; Davidson, S.M.; Effenberger, A.; Chen, C.Y.; Maner-Smith, K.; Hajjar, I.; et al. APOE4 impairs myelination via cholesterol dysregulation in oligodendrocytes. Nature 2022, 611, 769–779.
- Tassone, F.; Greco, C.M.; Hunsaker, M.R.; Seritan, A.L.; Berman, R.F.; Gane, L.W.; Jacquemont, S.; Basuta, K.; Jin, L.W.; Hagerman, P.J.; et al. Neuropathological, clinical and molecular pathology in female fragile X premutation carriers with and without FXTAS. Genes. Brain Behav. 2012, 11, 577–585.
- Silva, F.; Rodriguez-Revenga, L.; Madrigal, I.; Alvarez-Mora, M.I.; Oliva, R.; Milà, M. High apolipoprotein E4 allele frequency in FXTAS patients. Genet. Med. 2013, 15, 639–642.
- Avitzour, M.; Mor-Shaked, H.; Yanovsky-Dagan, S.; Aharoni, S.; Altarescu, G.; Renbaum, P.; Eldar-Geva, T.; Schonberger, O.; Levy-Lahad, E.; Epsztejn-Litman, S.; et al. FMR1 epigenetic silencing commonly occurs in undifferentiated fragile X-affected embryonic stem cells. Stem Cell Rep. 2014, 3, 699–706.
- Liu, J.; Koscielska, K.A.; Cao, Z.; Hulsizer, S.; Grace, N.; Mitchell, G.; Nacey, C.; Githinji, J.; McGee, J.; Garcia-Arocena, D.; et al. Signaling defects in iPSC-derived fragile X premutation neurons. Hum. Mol. Genet. 2012, 21, 3795–3805.
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science 2014, 345, 1247125.
- Kraff, J.; Tang, H.T.; Cilia, R.; Canesi, M.; Pezzoli, G.; Goldwurm, S.; Hagerman, P.J.; Tassone, F. Screen for excess FMR1 premutation alleles among males with parkinsonism. Arch. Neurol. 2007, 64, 1002–1006.
- Popovic, D.; Vucic, D.; Dikic, I. Ubiquitination in disease pathogenesis and treatment. Nat. Med. 2014, 20, 1242–1253.
- Wenzel, H.J.; Hunsaker, M.R.; Greco, C.M.; Willemsen, R.; Berman, R.F. Ubiquitin-positive intranuclear inclusions in neuronal and glial cells in a mouse model of the fragile X premutation. Brain Res. 2010, 1318, 155–166.
- Vardinon, N.; Spirer, Z.; Goldhar, J.; Kacevman, B.; Eylan, E. Human milk anti-E. coli antibodies: Relationship to maternal parity. Eur. J. Pediatr. 1979, 130, 173–180.
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133.
- Gutekunst, C.A.; Li, S.H.; Yi, H.; Mulroy, J.S.; Kuemmerle, S.; Jones, R.; Rye, D.; Ferrante, R.J.; Hersch, S.M.; Li, X.J. Nuclear and neuropil aggregates in Huntington’s disease: Relationship to neuropathology. J. Neurosci. 1999, 19, 2522–2534.
- Ng, A.S.L.; Xu, Z.; Chen, Z.; Tan, Y.J.; Lim, W.K.; Ting, S.K.S.; Yu, W.Y.; Cheng, Q.H.; Foo, J.N.; Tan, E.K.; et al. NOTCH2NLC-linked neuronal intranuclear inclusion body disease and fragile X-associated tremor/ataxia syndrome. Brain 2020, 143, e69.
- Malik, I.; Kelley, C.P.; Wang, E.T.; Todd, P.K. Molecular mechanisms underlying nucleotide repeat expansion disorders. Nat. Rev. Mol. Cell Biol. 2021, 22, 589–607.
- Todd, P.K.; Paulson, H.L. RNA-mediated neurodegeneration in repeat expansion disorders. Ann. Neurol. 2010, 67, 291–300.
- Mankodi, A.; Logigian, E.; Callahan, L.; McClain, C.; White, R.; Henderson, D.; Krym, M.; Thornton, C.A. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science 2000, 289, 1769–1773.
- Brook, J.D.; McCurrach, M.E.; Harley, H.G.; Buckler, A.J.; Church, D.; Aburatani, H.; Hunter, K.; Stanton, V.P.; Thirion, J.P.; Hudson, T.; et al. Molecular basis of myotonic dystrophy: Expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 1992, 69, 385.
- Mahadevan, M.S.; Yadava, R.S.; Yu, Q.; Balijepalli, S.; Frenzel-McCardell, C.D.; Bourne, T.D.; Phillips, L.H. Reversible model of RNA toxicity and cardiac conduction defects in myotonic dystrophy. Nat. Genet. 2006, 38, 1066–1070.
- Oh, S.Y.; He, F.; Krans, A.; Frazer, M.; Taylor, J.P.; Paulson, H.L.; Todd, P.K. RAN translation at CGG repeats induces ubiquitin proteasome system impairment in models of fragile X-associated tremor ataxia syndrome. Hum. Mol. Genet. 2015, 24, 4317–4326.
- Koehorst, E.; Núñez-Manchón, J.; Ballester-López, A.; Almendrote, M.; Lucente, G.; Arbex, A.; Chojnacki, J.; Vázquez-Manrique, R.P.; Gómez-Escribano, A.P.; Pintos-Morell, G.; et al. Characterization of RAN Translation and Antisense Transcription in Primary Cell Cultures of Patients with Myotonic Dystrophy Type 1. J. Clin. Med. 2021, 10, 5520.
- Furling, D.; LaM, L.T.; Agbulut, O.; Butler-Browne, G.S.; Morris, G.E. Changes in myotonic dystrophy protein kinase levels and muscle development in congenital myotonic dystrophy. Am. J. Pathol. 2003, 162, 1001–1009.
- Rizzo, G.; Pizza, F.; Scaglione, C.; Tonon, C.; Lodi, R.; Barbiroli, B.; Ambrosetto, P.; Martinelli, P. A case of fragile X premutation tremor/ataxia syndrome with evidence of mitochondrial dysfunction. Mov. Disord. 2006, 21, 1541–1542.
- Ross-Inta, C.; Omanska-Klusek, A.; Wong, S.; Barrow, C.; Garcia-Arocena, D.; Iwahashi, C.; Berry-Kravis, E.; Hagerman, R.J.; Hagerman, P.J.; Giulivi, C. Evidence of mitochondrial dysfunction in fragile X-associated tremor/ataxia syndrome. Biochem. J. 2010, 429, 545–552.
- Chen, Y.; Tassone, F.; Berman, R.F.; Hagerman, P.J.; Hagerman, R.J.; Willemsen, R.; Pessah, I.N. Murine hippocampal neurons expressing FMR1 gene premutations show early developmental deficits and late degeneration. Hum. Mol. Genet. 2010, 19, 196–208.
- Alvarez-Mora, M.I.; Podlesniy, P.; Gelpi, E.; Hukema, R.; Madrigal, I.; Pagonabarraga, J.; Trullas, R.; Mila, M.; Rodriguez-Revenga, L. Fragile X-associated tremor/ataxia syndrome: Regional decrease of mitochondrial DNA copy number relates to clinical manifestations. Genes. Brain Behav. 2019, 18, e12565.
- Loesch, D.Z.; Annesley, S.J.; Trost, N.; Bui, M.Q.; Lay, S.T.; Storey, E.; De Piazza, S.W.; Sanislav, O.; Francione, L.M.; Hammersley, E.M.; et al. Novel Blood Biomarkers Are Associated with White Matter Lesions in Fragile X-Associated Tremor/Ataxia Syndrome. Neurodegener. Dis. 2017, 17, 22–30.
- Fisher, P.R.; Allan, C.Y.; Sanislav, O.; Atkinson, A.; Ngoei, K.R.W.; Kemp, B.E.; Storey, E.; Loesch, D.Z.; Annesley, S.J. Relationships between Mitochondrial Function, AMPK, and TORC1 Signaling in Lymphoblasts with Premutation Alleles of the FMR1 Gene. Int. J. Mol. Sci. 2021, 22, 10393.
- Loesch, D.Z.; Kemp, B.E.; Bui, M.Q.; Fisher, P.R.; Allan, C.Y.; Sanislav, O.; Ngoei, K.R.W.; Atkinson, A.; Tassone, F.; Annesley, S.J.; et al. Cellular Bioenergetics and AMPK and TORC1 Signalling in Blood Lymphoblasts Are Biomarkers of Clinical Status in FMR1 Premutation Carriers. Front. Psychiatry 2021, 12, 747268.
- Cid-Samper, F.; Gelabert-Baldrich, M.; Lang, B.; Lorenzo-Gotor, N.; Ponti, R.D.; Severijnen, L.; Bolognesi, B.; Gelpi, E.; Hukema, R.K.; Botta-Orfila, T.; et al. An Integrative Study of Protein-RNA Condensates Identifies Scaffolding RNAs and Reveals Players in Fragile X-Associated Tremor/Ataxia Syndrome. Cell Rep. 2018, 25, 3422–3434.e7.
- Hu, Y.; Deng, H.; Xu, S.; Zhang, J. MicroRNAs Regulate Mitochondrial Function in Cerebral Ischemia-Reperfusion Injury. Int. J. Mol. Sci. 2015, 16, 24895–24917.
- Han, J.; Lee, Y.; Yeom, K.H.; Kim, Y.K.; Jin, H.; Kim, V.N. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004, 18, 3016–3027.
- Tafuri, F.; Ronchi, D.; Magri, F.; Comi, G.P.; Corti, S. SOD1 misplacing and mitochondrial dysfunction in amyotrophic lateral sclerosis pathogenesis. Front. Cell Neurosci. 2015, 9, 336.
- Gohel, D.; Sripada, L.; Prajapati, P.; Singh, K.; Roy, M.; Kotadia, D.; Tassone, F.; Charlet-Berguerand, N.; Singh, R. FMRpolyG alters mitochondrial transcripts level and respiratory chain complex assembly in Fragile X associated tremor/ataxia syndrome . Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1379–1388.
- Jové, M.; Portero-Otín, M.; Naudí, A.; Ferrer, I.; Pamplona, R. Metabolomics of human brain aging and age-related neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2014, 73, 640–657.
- Peng, B.; Li, H.; Peng, X.X. Functional metabolomics: From biomarker discovery to metabolome reprogramming. Protein Cell 2015, 6, 628–637.
- Napoli, E.; Song, G.; Schneider, A.; Hagerman, R.; Eldeeb, M.A.; Azarang, A.; Tassone, F.; Giulivi, C. Warburg effect linked to cognitive-executive deficits in FMR1 premutation. Faseb J. 2016, 30, 3334–3351.
- Napoli, E.; Schneider, A.; Wang, J.Y.; Trivedi, A.; Carrillo, N.R.; Tassone, F.; Rogawski, M.; Hagerman, R.J.; Giulivi, C. Allopregnanolone Treatment Improves Plasma Metabolomic Profile Associated with GABA Metabolism in Fragile X-Associated Tremor/Ataxia Syndrome: A Pilot Study. Mol. Neurobiol. 2019, 56, 3702–3713.
- Abbasi, D.A.; Nguyen, T.T.A.; Hall, D.A.; Robertson-Dick, E.; Berry-Kravis, E.; Cologna, S.M. Characterization of the Cerebrospinal Fluid Proteome in Patients with Fragile X-Associated Tremor/Ataxia Syndrome. Cerebellum 2022, 21, 86–98.
- Deng, J.; Yu, J.; Li, P.; Luan, X.; Cao, L.; Zhao, J.; Yu, M.; Zhang, W.; Lv, H.; Xie, Z.; et al. Expansion of GGC Repeat in GIPC1 Is Associated with Oculopharyngodistal Myopathy. Am. J. Hum. Genet. 2020, 106, 793–804.
- Ishiura, H.; Shibata, S.; Yoshimura, J.; Suzuki, Y.; Qu, W.; Doi, K.; Almansour, M.A.; Kikuchi, J.K.; Taira, M.; Mitsui, J.; et al. Noncoding CGG repeat expansions in neuronal intranuclear inclusion disease, oculopharyngodistal myopathy and an overlapping disease. Nat. Genet. 2019, 51, 1222–1232.
- Sone, J.; Mitsuhashi, S.; Fujita, A.; Mizuguchi, T.; Hamanaka, K.; Mori, K.; Koike, H.; Hashiguchi, A.; Takashima, H.; Sugiyama, H.; et al. Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nat. Genet. 2019, 51, 1215–1221.
- Yu, X.; Wang, Y. Tonotopic differentiation of presynaptic neurotransmitter-releasing machinery in the auditory brainstem during the prehearing period and its selective deficits in FMR1 knockout mice. J. Comp. Neurol. 2022, 530, 3248–3269.
- Zeng, Y.H.; Yang, K.; Du, G.Q.; Chen, Y.K.; Cao, C.Y.; Qiu, Y.S.; He, J.; Lv, H.D.; Qu, Q.Q.; Chen, J.N.; et al. GGC Repeat Expansion of RILPL1 is Associated with Oculopharyngodistal Myopathy. Ann. Neurol. 2022, 92, 512–526.
- Annear, D.J.; Vandeweyer, G.; Elinck, E.; Sanchis-Juan, A.; French, C.E.; Raymond, L.; Kooy, R.F. Abundancy of polymorphic CGG repeats in the human genome suggest a broad involvement in neurological disease. Sci. Rep. 2021, 11, 2515.
- Pearson, C.E.; Nichol Edamura, K.; Cleary, J.D. Repeat instability: Mechanisms of dynamic mutations. Nat. Rev. Genet. 2005, 6, 729–742.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.4K
Entry Collection:
Neurodegeneration
Revisions:
2 times
(View History)
Update Date:
28 Sep 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No